

Main Text
To the Editor: Warby et al.1 present fascinating data on the haplotype background of chromosomes carrying the Huntington's disease (HD [MIM 143100]) mutation and the length distribution of the CAG repeat for different haplotypes within the general population. One of their conclusions is that cis-elements are likely to represent a major predisposing element in HD expansion. Here, I use evolutionary modeling of the CAG repeat length distribution within populations to argue that the distribution of CAG repeat length and disease incidence in different haplotypes can be explained by founder events, each of which involved expansion of repeats to lengths that are classified as normal by HD investigators (<28 repeats). There is therefore no need to invoke cis-element polymorphism within the human population.
Mutation of the HD CAG repeat is both upwardly biased (increases in repeat length are more frequent than decreases) and length dependent (longer repeats mutate more frequently than short ones). Based on sperm typing data, Falush et al.2 estimated that the mutation rate was proportional to the number of repeats to the power of eight, so that, for example, alleles with 23 copies of the repeat would be approximately 10 times more mutable than alleles with 17 repeats, and alleles with 32 repeats would be approximately 100 times more mutable. The strong length dependence of the mutation rate means that CAG length in itself is a powerful factor in determining the stability of the repeat. Additionally, beyond approximately 55 repeats, the HD mutation causes juvenile HD, which makes further transmission impossible. In fact, the data argue that in modern populations, selection acts strongly against repeat lengths of 44 or more.2
I simulated the repeat length distribution in an infinite population based on the mutational model in Falush et al. In order to simulate the effect of natural selection, I removed all repeats of length 50 or more from the population. Simulations show that the assumptions made in modeling selection against disease alleles of different lengths have a negligible effect on the repeat length distribution among normal chromosomes (data not shown). In small populations, e.g., the early settlers of Europe, particular haplotypes can drift to high frequency, also increasing the frequency of the CAG repeat that they carry. In order to investigate the effect of founder events, the population was initially started with three haplotypes each at 1/3 frequency and with initial repeat lengths of 17, 23, and 32 (Figure 1).
Figure 1.
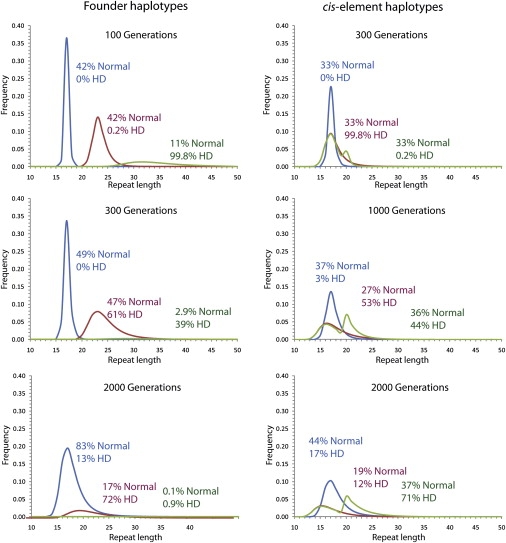
Repeat Length Distribution Evolving in a Simulated Large Population
In the simulations on the left-hand side, the population consists initially of three haplotypes, carrying repeats of length 17 (blue), 23 (red), and 32 (green). On the right-hand side, the population consists of three haplotypes with empirical mutation frequencies (blue), five times the mutation rate (red), and five times the mutation rate only for repeat lengths less than 20 (green). Chromosomes with 36 or more repeats are considered to have Huntington's disease (HD), but natural selection acts only on chromosomes with 50 repeats or more, which are removed from the population.
A repeat of length 17 has a <0.2% chance of mutating in each generation, so that after 100 generations, most repeats of this length remained unchanged. A repeat of length 32 has a 20% chance within each generation. After 100 generations, most of the repeats of length 32 have mutated at least once, with a majority expanding to length 50 and being removed by natural selection. Consequently, the frequency of the 32-repeat haplotype is only approximately 1/3 of its initial value. After 300 generations, the frequency of the 32-repeat haplotype has decreased sufficiently that it no longer causes the majority of disease in the population. After 2000 generations, its frequency is negligible and it causes less disease than the 17-repeat haplotype.
These simulations highlight counterintuitive patterns of repeat length evolution within populations caused by the combination of the length dependence of the mutation rate combined with natural selection against disease alleles. Notwithstanding an upward bias, in a large population, the average repeat length will stabilize and then progressively decrease. In these simulations, because of the high mutability of the CAG repeat carried by the 32-repeat haplotype, the mean length peaked after only 30 generations and reduced progressively thereafter. Furthermore, the frequency of alleles of length 36 or more peaked after 45 generations before also declining up to 300 generations. Disease incidence then rose modestly due to increasing disease incidence associated with the 23-repeat founder haplotype before peaking at 500 generations and decreasing progressively for the rest of the simulation. These results contradict a previous claim that disease incidence will increase indefinitely over time as a result of an upward bias in mutation.3
The simulation also shed light on the repeat length distribution and associated disease incidence observed by Warby et al. for different haplogroups. Warby et al. genotyped individuals for CAG repeat length and neighboring SNPs. Based on these SNPs, each CAG repeat was assigned to a haplogroup (A, B, or C). Within haplogroup A, repeats were assigned to variants A1–A5. The chromosomes genotyped by Warby et al. include chromosomes that were ascertained within HD pedigrees, meaning that the proportion of disease-causing alleles is much higher than in the population as a whole. In order to discuss the population repeat length distribution, I therefore ignore all repeats of length 36 or more in their Figures 3 and 4.
Within both haplogroups A and C, the modal repeat length is 17. However, haplogroup A has a second mode at 20 repeats. For haplotype variant A2, 20 is the modal repeat length. This variant is associated with a substantially higher disease incidence than variants A3, A4, and A5, which each have average repeat lengths of 17 or less. As the simulations illustrate, a haplotype seeded with a larger-than-average repeat length will in time be associated with a higher-than-average rate of disease. If the initial A2 haplotype had 20 repeats, then this can explain both the current mode of 20 repeats associated with the haplotype and also the high disease incidence associated with it compared both to haplogroup C and also to variants A3, A4, and A5.
The A1 variant has an even higher average repeat length than A2, with almost all alleles having 20 or more repeats and with all lengths between 20 and 35 observed at significant frequency. I propose that the founder A1 haplotype had 22 or more repeats. These large repeats are predicted by the mutational model to mutate quickly, leading to the frequency distribution becoming relatively flat, as is seen in the simulations for a modal repeat length of 32 after 100 generations or more and for a repeat of length 23 after 300 generations or more. The high mutation rate leads also to a very high rate of HD mutations associated with the background, as is observed. The simulations also predict that the frequency of A1 has been decreasing over time, but there is no empirical evidence on this.
The co-occurrence of two variants with high average CAG length, namely A1 and A2, within the A haplogroup is unlikely to be a coincidence. The two variants presumably shared a common ancestor with 20 repeats or more. As repeats become larger, they become more mutable, thus leading to the observation of multiple founder events on related backgrounds.
Warby et al. give three arguments against the importance of founder CAG expansions. First, unlike in other trinucleotide disease genes, HD mutations arise on multiple backgrounds, albeit at different rates. It is clear that mutational properties do differ between different trinucleotide repeats within the human genome and that some of this variation is due to cis-elements.3 Differences between loci, however, are not necessarily indicative of cis-element polymorphism within the human population. In addition to mutational properties, the pattern of disease incidence is determined by the repeat length distribution, which can differ between populations and species as a result of genetic drift. The modal repeat length at the locus in European and East Asian populations is 17.4 African populations have broader repeat length distributions, but with the great majority of chromosomes having 15 repeats or more.4 Within the chimpanzee, the modal CAG repeat length distribution is 9, with a secondary mode at 12.4 Given equivalent mutational properties of the repeat in the two species, these distributions would lead to a qualitatively lower expected disease incidence among chimpanzee populations and a narrower range of haplotypes associated with the disease. Thus, the high frequency of Huntington's disease and variety of haplotype backgrounds in humans compared to other CAG expansion-related diseases might be in part explained by genetic drift leading to chance fixation of mutations increasing CAG length in the ancestors of modern humans.
Second, Warby et al. note that the pattern of strong SNP linkages to CAG expansion is punctuated, rather than decaying as a function of genetic distance. A proportion of SNPs may fail to be associated strongly with each other or with CAG repeat length or disease incidence despite tight genetic linkage for a number of reasons. For example, they may have been involved in gene conversion events and thus be associated with more than one background. Within each haplogroup and haplogroup variant, mutation continuously generates variation in CAG length, and specific SNPs may arise on a background with a repeat length that is atypical of the haplotype as a whole.
Third, Warby et al. argue that the data are not consistent with a large-normal CAG founder because the disease-associated haplotypes are found at high frequency within the normal population. The frequency that haplotypes can reach among normal and disease chromosomes depends critically on the ancestral repeat length of the haplotype. As the simulation in Figure 1 illustrates, haplotypes descended from an allele with 32 repeats can cause a very high proportion of disease cases while forming a relatively small proportion of the overall population. Such a situation corresponds, for example, to the cluster of closely related HD chromosomes carried by several thousand individuals as observed in Venezuela.5 However, the simulations also illustrate that long repeats suffer extremely strong selection against them over the timescale of hundreds of years, leading to a rapid decrease in frequency both in the population and among HD cases. The cluster in Venezuela is probably only possible because of extremely rapid recent local population expansion within the region. The European population has been large for several thousand years, making such concentrated clusters unlikely. The simulation also illustrates that when the population has been large for hundreds of generations, a founder haplotype of length 23 can be responsible for a large proportion of disease cases while also forming a substantial proportion of the overall population, as observed for haplogroup variant A1 among those of European descent.
Based on these arguments, there is no need to invoke cis-elements that alter the mutational properties of the CAG repeat to explain the observed patterns. Is cis-element polymorphism at least consistent with the data? This depends on exactly what the elements do. I simulated a population with three haplotypes each of which had a starting repeat length of 17. The first had the mutational properties estimated from sperm typing data, the second had a 5-fold higher mutation rate for all repeat lengths, and the third had a 5-fold increased mutation rate for repeat lengths under 20 but an unchanged rate for larger repeat lengths. After 300 generations, the second haplotype was responsible for the vast majority of disease cases; however, the modal repeat length associated with the haplotype remained 17. After 1000 generations, the second and third haplotypes were together responsible for most of the disease. Within the general population, the second haplotype was associated with shorter repeat lengths, while the third haplotype showed a bimodal distribution. After 2000 generations, more than 2/3 of disease was associated with the third haplogroup, with the second haplogroup largely associated with shorter repeat lengths.
These results make it unlikely that common HD haplotypes are associated with cis-elements that cause a general increase in HD instability at all CAG repeat lengths. Such a cis-element would cause a higher rate of expansion into the disease range and therefore increase the effect of natural selection. If cis-elements of this sort did exist, it would therefore be most likely that they would be observed on haplotype backgrounds with a smaller-than-average number of repeats, even if they were associated with an average or higher-than-average disease incidence. This pattern is not consistent with the repeat length distribution for the A haplogroup or its variants. A cis-element with a specific effect increasing the mutation rate specifically for shorter alleles is more consistent with the data, because for example it can generate the bimodal distribution of repeat lengths within the general population observed for haplogroup A. However, invoking an element that induces peculiar mutational properties seems unparsimonious and does not on its own account for the difference in CAG length and disease incidence among the variants of the A haplogroup.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM/
Acknowledgments
S. Warby, S. Butland, and two anonymous reviewers provided numerous helpful comments. The author is funded by Science Foundation of Ireland grant 05/FE1/B882.
References
- 1.Warby S.C., Montpetit A., Hayden A.R., Carroll J.B., Butland S.L., Visscher H., Collins J.A., Semaka A., Hudson T.J., Hayden M.R. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 2009;84:351–366. doi: 10.1016/j.ajhg.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falush D., Almqvist E.W., Brinkmann R.R., Iwasa Y., Hayden M.R. Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial underascertainment of late-onset cases. Am. J. Hum. Genet. 2001;68:373–385. doi: 10.1086/318193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libby R.T., Hagerman K.A., Pineda V.V., Lau R., Cho D.H., Baccam S.L., Axford M.M., Cleary J.D., Moore J.M., Sopher B.L. CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: A novel basis for mutational hot spot determination. PLoS Genet. 2008;4:e1000257. doi: 10.1371/journal.pgen.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubinsztein D.C., Amos W., Leggo J., Goodburn S., Ramesar R.S., Old J., Bontrop R., McMahon R., Barton D.E., Ferguson-Smith M.A. Mutational bias provides a model for the evolution of Huntington's disease and predicts a general increase in disease prevalence. Nat. Genet. 1994;7:525–530. doi: 10.1038/ng0894-525. [DOI] [PubMed] [Google Scholar]
- 5.Paradisi I., Hernández A., Arias S. Huntington disease mutation in Venezuela: Age of onset, haplotype analyses and geographic aggregation. J. Hum. Genet. 2008;53:127–135. doi: 10.1007/s10038-007-0227-1. [DOI] [PubMed] [Google Scholar]