

Abstract
Skeletal muscle regeneration and work-induced hypertrophy rely on molecular events responsible for activation and quiescence of resident myogenic stem cells, satellite cells. Recent studies demonstrated that hepatocyte growth factor (HGF) triggers activation and entry into the cell cycle in response to mechanical perturbation, and that subsequent expression of myostatin may signal a return to cell quiescence. However, mechanisms responsible for coordinating expression of myostatin after an appropriate time lag following activation and proliferation are not clear. Here we address the possible role of HGF in quiescence through its concentration-dependent negative-feedback mechanism following satellite cell activation and proliferation. When activated/proliferating satellite cell cultures were treated for 24 h beginning 48-h postplating with 10–500 ng/ml HGF, the percentage of bromodeoxyuridine-incorporating cells decreased down to a baseline level comparable to 24-h control cultures in a HGF dose-dependent manner. The high level HGF treatment did not impair the cell viability and differentiation levels, and cells could be reactivated by lowering HGF concentrations to 2.5 ng/ml, a concentration that has been shown to optimally stimulate activation of satellite cells in culture. Coaddition of antimyostatin neutralizing antibody could prevent deactivation and abolish upregulation of cyclin-dependent kinase (Cdk) inhibitor p21. Myostatin mRNA expression was upregulated with high concentrations of HGF, as demonstrated by RT-PCR, and enhanced myostatin protein expression and secretion were revealed by Western blots of the cell lysates and conditioned media. These results indicate that HGF could induce satellite cell quiescence by stimulating myostatin expression. The HGF concentration required (over 10–50 ng/ml), however, is much higher than that for activation, which is initiated by rapid release of HGF from its extracellular association. Considering that HGF is produced by satellite cells and spleen and liver cells in response to muscle damage, local concentrations of HGF bathing satellite cells may reach a threshold sufficient to induce myostatin expression. This time lag may delay action of the quiescence signaling program in proliferating satellite cells during initial phases of muscle regeneration followed by induction of quiescence in a subset of cells during later phases.
Keywords: activation, hepatocyte growth factor, muscle regeneration, myostatin, quiescence, satellite cells, time lag
skeletal muscle satellite cells, resident myogenic stem cells interposed between the overlying external lamina and the sarcolemma of a subjacent mature muscle fiber, are normally found in a mitotically and metabolically quiescent or dormant state during most of adult postnatal life. When muscle is injured, overused, or mechanically stretched, satellite cells activate to enter the cell cycle from a protracted G1 state (often referred to as G0) in a hepatocyte growth factor (HGF)/nitric oxide (NO) radical-dependent manner (7, 80–83, 85, 86, 88, 103, 104; reviewed in Refs. 9, 10, 87, 90, 101). Activated satellite cells have been shown to migrate to the damaged site where they replicate DNA, divide, differentiate, and fuse with the adjacent muscle fiber (or form new fibers) to repair damaged regions and to enhance hypertrophy of muscle fibers (4, 13, 32, 47, 68, 70, 87, 94, 96, 101). After cell proliferation, some cells in the population withdraw from the cell cycle again and reenter mitotic quiescence, which is an essential event for self-renewal or maintenance of the myogenic stem cells responsible for sustainable muscle regeneration and hypertrophy (19, 23). Despite rapid advances in our understanding of the molecules and mechanisms involved in regulating satellite cell activation as described below, relatively little is known about control of the satellite cell quiescence program that is initiated following a period of satellite cell activation and proliferation during successful regeneration and work-induced hypertrophy of functional muscle fibers.
Of all growth factors studied thus far, only myostatin (also called growth differentiation factor 8), a secreted factor of the transforming growth factor (TGF)-β superfamily (50) and TGF-βs, have been well documented to provide a powerful signal that can stimulate satellite cell quiescence (or block activation, proliferation, and differentiation to maintain quiescence) in cultures (1, 48, 91) and in vivo (95), and these factors may be an important signaling molecules for imposing quiescence on myogenic precursors during chick embryonic and fetal development (5). Gene mutations that impair myostatin functions in mice, cattle, and sheep result in a dramatic increase in muscle mass (18, 30, 37, 49, 50), and similar mutation was recently identified in humans, which leads to a decrease in myostatin levels and a phenotype similar to that observed in myostatin-null mice (67). Myostatin protein is synthesized in myogenic cells and a few other cell types as a 52-kDa pro-form (49, 61, 72) and proteolytically processed at the RSRR site to give rise to a 26-kDa active form (11, 43, 72), which has been shown to associate with activin receptor type II family to elicit biological functions (42, 62). Therefore, myostatin expression and secretion followed by presentation to the receptor may be a crucial step of the autonomical quiescence of proliferating satellite cells, although the molecular mechanism has not been elucidated.
Another key factor that regulates the cell proliferation activity of satellite cells is HGF, which is a heparin-binding protein localized in the extracellular domain of uninjured muscle fibers. HGF can provide a signal that activates quiescent satellite cells in vitro with a peak concentration as low as 2.5 ng/ml in primary cultures, and HGF also activates satellite cells in vivo (2, 3, 80, 85). The intracellular signaling receptor having a high-affinity specific for HGF is the c-met proto-oncogene, a membrane-bound, disulphide-linked heterodimer with an intracellular tyrosine kinase domain (27–29, 34), and its message and protein have been found in quiescent and activated satellite cells (2, 21, 80). In previous works, we employed a FlexerCell system (Flexcell International, McKeesport, PA) to apply cyclic stretch to isolated rat satellite cells and found that mechanical stretch triggers satellite cell activation by rapid release of HGF from its extracellular tethering and subsequent presentation to the c-met receptor (81–83, 88, 103, 104). This phenomenon is relevant to satellite cells in living skeletal muscle as revealed by an in vivo muscle stretch model (65, 85).
Additional important observations related to these studies are that extracellular HGF concentrations increase in response to muscle injury, mechanical stretch, exercise, and NO radical treatments, possibly because of the release of HGF from the extracellular matrix of muscle fibers and satellite cells (36, 75, 76, 79, 80, 84, 85, 101) and to HGF synthesis by activated satellite cells (6, 73). In addition, Suzuki et al. (76) demonstrated that spleen and liver cells respond to muscle injury to produce HGF message and protein, resulting in increased serum levels of HGF that may be delivered via blood flow to the extracellular space in regions of damage. Early in these situations, satellite cells can be rapidly activated to enter the cell proliferation cycle through a low-level HGF ligand/high-affinity receptor c-met pathway as shown by Allen's and Anderson's groups (2, 3, 7, 8, 80, 81, 83, 85, 88, 98–104; reviewed in Refs. 9, 10, 87, 90). However, the physiological significance of further increases in the extracellular HGF level in the later phase (at the cell proliferation and differentiation stages) has not been examined.
Experiments in the present study were designed to test the hypothesis that increasing concentrations of HGF in regenerating muscle may be involved in driving cells back into quiescence. This novel idea is based on the unexpected outcome of cell activation experiments (83), in which the HGF dose-dependence curve showed a drastic decrease in an activation index in cultures treated with over 50 ng/ml HGF for 24 h (preliminary results presented in Ref. 90a). Similar results have been published very recently by Johnson's group, who described that treatment of mouse satellite cells with high concentrations of HGF (50 ng/ml) inhibits their proliferation (44).
As a first step toward understanding the mechanism through which high-level HGF may promote the quiescence of proliferating satellite cells, we examined whether satellite cells can produce myostatin in response to the growth factor. Results clearly demonstrated that myostatin expression and secretion were significantly upregulated in proliferating satellite cells in response to HGF at concentrations over 10–50 ng/ml in vitro in primary tissue cultures. Furthermore, immunoneutralization of myostatin reversed the inhibition of proliferation observed in response to high concentrations of HGF. Therefore, we propose a possible role for HGF in reestablishing quiescence through a negative feedback mechanism following satellite cell activation and proliferation.
MATERIALS AND METHODS
Materials.
The following materials were obtained for primary cell culture experiments: Dulbecco's modified Eagle's medium (DMEM, low glucose type, 31600-034), normal horse serum (HS, 16050-122), antibiotic-antimycotic (15240-062), and gentamicin (15710-064) from Invitrogen (Grand Island, NY); poly-l-lysine (P9155), bovine plasma fibronectin (F1141), protease type XIV (P5147), and 5-bromo-2′-deoxyuridine (BrdU, B5002) from Sigma (St. Louis, MO); recombinant human HGF (294-HG) from R&D Systems (Minneapolis, MN); SuperScript III reverse transcriptase (18080-044) and TRIzol reagent (15596-018) from Invitrogen; RNeasy Micro kit (74004) and QIAshredder homogenizer spin column (79654) from Qiagen (Hilden, Germany); LightCycler TaqMan Master (04735536001), Universal ProbeLibrary probes, and oligo(dT) primer (H09876) from Roche (Mannheim, Germany); and ExTaq DNA polymerase (RR001A) from Takara Bio (Otsu, Japan).
For the immunodetection of target proteins in satellite cells, the following materials were additionally used: antigen-affinity-purified goat polyclonal anti-mouse myostatin neutralizing antibody (AF788), rat monoclonal anti-mouse myostatin antibody (MAB788) and goat polyclonal anti-mouse c-met antibody (AF527) from R&D Systems; D3 mouse monoclonal antidesmin and G3G4 mouse monoclonal anti-BrdU antibodies from the Developmental Studies Hybridoma Bank (Iowa City, IA); SX118 mouse monoclonal anti-p21 (556430) and 5.8A mouse anti-MyoD antibodies (554130) from BD Biosciences (San Jose, CA); DM1A mouse monoclonal anti-chick α-tubulin antibody (T9026) from Sigma; affinity-purified biotinylated rabbit anti-goat IgG (BA-5000), horse anti-mouse IgG (BA-2000), rabbit anti-rat IgG (BA-4000) and horseradish peroxidase (HRPO)-labeled avidin kit (PK-6100) from Vector Laboratories (Burlingame, CA); affinity-purified HRPO-conjugated goat anti-mouse IgG (A-4416) and 3,3′-diaminobenzidine (DAB) from Sigma; enhanced chemiluminescence (ECL) detection kit (PRN2106) and nitrocellulose membranes (Hybond ECL, RPN2020D) from GE Healthcare (Little Chalfont, UK); biotinylated molecular weight standards (161-0319) from Bio-Rad (Hercules, CA); BIOMAX-XAR X-ray film (166-0760) from Eastman Kodak (Rochester, NY). Other materials were as reported (89).
Animal care and use.
Experiments involving animals were conducted according to institutional guideline and with the approval of Kyushu University Institutional Review Board and the University of Arizona Institutional Animal Care and Use Committee.
Satellite cell isolation and primary culture.
Satellite cells were isolated from 9-mo-old male Sprague-Dawley rats according to Allen et al. (3), with slight modifications (86). Briefly, muscle groups from the upper hindlimb and back were excised, trimmed of fat and connective tissue, hand minced with scissors, and digested for 1 h at 37°C with 1.25 mg/ml protease type XIV. Cells were separated from muscle-fiber fragments and tissue debris by differential centrifugation and plated on poly-l-lysine and fibronectin-coated dishes in DMEM containing 10% HS, 1% antibiotic-antimycotic mixture, and 0.5% gentamicin (DMEM-10% HS, pH 7.2). Cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C. In experiments in which conditioned medium was assayed for the presence of secreted myostatin, cultures were washed with serum-free DMEM at 66-h postplating, and treatments were imposed for the next 6 h in the DMEM; conditioned media were collected, centrifuged for 3 min at 1,300 g, filtered through 0.22-μm filter, and stored at −30°C until use. In addition, companion satellite cell cultures were immunostained for the presence of c-met or desmin at 30 h after plating using a polyclonal anti-mouse c-met antibody, D3 monoclonal anti-desmin antibody, biotinylated anti-goat/mouse IgG antibodies, and HRPO-labeled avidin to determine the percentage of myogenic cells present. Cultures with <95% DAB-positive cells were not used for experiments.
In vitro activation assay.
Cultures were pulse-labeled with 10 μM BrdU in DMEM-10% HS for the final 2 h at each time point from 24 to 120-h postplating, followed by immunocytochemistry for detection of BrdU using a G3G4 anti-BrdU monoclonal antibody [1:100 dilution in 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS)] and a HRPO-conjugated anti-mouse IgG antibody (1:500 dilution) according to Tatsumi et al. (80). The percentage of BrdU-labeled cells is an indicator of activation (entry into the cell cycle) and the subsequent proliferation of satellite cells.
ECL-Western blotting and immunocytochemistry.
Whole cell lysates and conditioned media (serum-free DMEM) from satellite cell cultures with and without HGF were subjected to 12.5% polyacrylamide gel electrophoresis under reducing conditions (41) and transferred to nitrocellulose membranes using an electrode buffer of 25 mM Tris, 0.192 M glycine, 0.1% SDS, and 20% ethanol. Blots were blocked with 10% powdered milk in 0.1% polyethylene sorbitan monolaurate (Tween 20)-Tris buffered saline (TTBS) before incubation with antimyostatin, anti-MyoD, anti-cyclin-dependent kinase (Cdk) inhibitor p21, or anti-α-tubulin antibodies (1:1,000 dilution in 1% powdered milk-TTBS additionally containing 0.05% NaN3) overnight at room temperature. Blots were subsequently treated for 1 h with biotinylated secondary antibody at 1:5,000 dilution in 1% powdered milk-TTBS, then with HRPO-labeled avidin at a 1:500 dilution in TTBS for 30 min at room temperature, followed by ECL detection onto Kodak BIOMAX-XAR X-ray films.
For immunolocalization of myostatin and MyoD, satellite cell cultures were fixed at each time point from 24 to 120-h postplating for 10 min in 3.7% paraformaldehyde-PBS, and blocked with 1% goat serum in PBS for 15 min before incubation overnight at 4°C in primary antibody to myostatin and c-met (1:2,000 dilution in 0.1% BSA-PBS). Cells were subsequently incubated with biotinylated anti-goat or anti-mouse IgG secondary antibodies (1:10,000 dilution in 0.1% BSA-PBS) and HRPO-avidin (1:500 dilution in PBS) for 2 h at an ambient temperature, followed determination of the percentage of DAB-stained cells.
Reverse transcription-polymerase chain reaction.
Total RNA was isolated from cultured satellite cells using TRIzol reagent. cDNA was synthesized from 0.4 μg of total RNA by a reverse-transcriptase SuperScript III using oligo(dT) primer. PCR was performed using ExTaq polymerase on the mRNA expression level of myostatin (accession no. NM_019151) standardized with the expression of hypoxanthine guanine phosphoribosyl transferase [HPRT, accession no. NM_012583.2 for rat cDNA cloning and sequence analysis originally determined by Chiaverotti et al. (17)]. The intron-spanning primer sets used are as follows: for rat myostatin, forward 5′-GCTTTGGATGAGAATGGGCA-3′, reverse 5′-TGCACAAGATGAGTATGCGG-3′, annealing temperature 55°C, 35 cycles, amplicon 299 nucleotides (nt); for murine HPRT, forward 5′-GCTGGTGAAAAGGACCTCT-3′, reverse 5′-CACAGGACTAGAACRYCTGC-3′, annealing temperature 58°C, 35 cycles, amplicon 249 nt. Annealing temperatures and cycle numbers were optimized so that the amplification reactions are within the linear range. The PCR products were visualized with ethidium bromide after agarose gel electrophoresis.
Myogenin mRNA expression (accession no. NM_017115.2) was also monitored by the real-time quantitative PCR using Roche LightCycler1.5 run under the TaqMan probe detection format standardized with HPRT expression. Total RNA was isolated from 72-h cultured satellite cells with and without HGF (2.5 and 500 ng/ml) using RNeasy Micro kit according to the manufacturer's recommendation, and the primer sets were designed by the ProbeFinder (version 2.35 for rat, Roche) with an intron-spanning assay: for rat myogenin, forward 5′-CCTTGCTCAGCTCCCTCA-3′, reverse 5′-TGGGAGTTGCATTCACTGG-3′, amplicon 94 nt; for murine HPRT, forward 5′-GACCGGTTCTGTCATGTCG-3′, reverse 5′-ACCTGGTTCATCATCACTAATCAC-3′, amplicon 61 nt. Annealing temperature was set to 60°C in both cases.
Statistical analysis.
Analysis of variance procedures were employed to analyze experimental results using general linear model procedures of SRISTAT2 for Windows software (Social Survey Research Information, Tokyo, Japan). Least-squares means for each treatment were separated on the basis of least significant differences. Data are represented as means ± SE for four cultures per treatment and were considered significantly different from the mean in control cultures when P < 0.05.
RESULTS
HGF may induce satellite cell quiescence.
The purpose of this study was to examine if high concentrations of HGF could induce proliferating satellite cells to return to quiescence. Satellite cells prepared from adult rat skeletal muscles were stimulated for activation during 24 h from 24- to 48-h postplating by 2.5 ng/ml HGF, a treatment that has been shown to peak activation of the cells in our culture system (83). Following activation, cultures were incubated with higher concentrations of HGF for the next 24-h period (Fig. 1A). BrdU-incorporation assay at 72-h postplating demonstrated that HGF concentrations greater than 10 ng/ml decreased the activation index relative to cultures receiving 2.5–10 ng/ml HGF. The BrdU-labeling index at 250 ng/ml was significantly lower than that of the starting activation-culture receiving 2.5 ng/ml HGF (b, assayed at 48-h postplating), and the BrdU-labeling index in cultures receiving 500 ng/ml HGF was comparable to the control culture receiving no HGF (a, assayed at 24-h postplating).
Fig. 1.
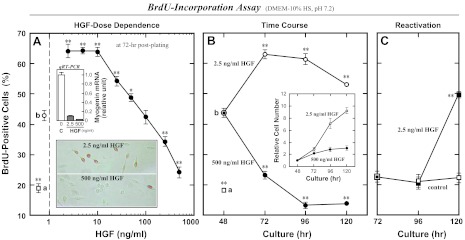
High-concentration hepatocyte growth factor (HGF) treatments reduce the proliferation activity of satellite cells in cultures. Rat satellite cells were stimulated for activation for 24 h from 24- to 48-h postplating by 2.5 ng/ml human recombinant HGF in DMEM-10% normal horse serum (HS) (b), then incubated with higher concentrations of HGF for the next 72-h period followed by 5-bromo-2′-deoxyuridine (BrdU)-incorporation assay at 24-h intervals of time. A: HGF dose dependence monitored at 72-h postplating by the proliferation index decreasing down to a baseline level comparable to the 24-h control culture not receiving 2.5 ng/ml HGF (a). Typical immunomicrographs are shown in bottom inset with positive (brown) and negative cells. Cell lysates of companion cultures were analyzed for the mRNA expression of a differentiation marker myogenin at 72-h postplating by real-time quantitative (q)RT-PCR run under the TaqMan Probe assay standardized with hypoxanthine guanine phosphoribosyl transferase (HPRT; top inset); bar C, control without HGF; bars HGF, with 2.5 and 500 ng/ml HGF. B: time courses of the BrdU-incorporation activity and the relative cell density (inset) in cultures treated with 2.5 ng/ml (○) and 500 ng/ml HGF (●) from 48- to 120-h postplating. C: reactivation activity of cells that received 500 ng/ml HGF for 24-h period from 48- to 72-h postplating and then maintained in the presence (■) or absence (□) of 2.5 ng/ml HGF in DMEM-10% HS for the next 48 h. Data points represent means ± SE for four cultures per treatment; significant differences from starting culture means (data point b in A and B and the 72-h data point in C) are indicated (*P < 0.05; **P < 0.01).
This issue was further examined by assessing the time course of deactivation of satellite cell cultures with 500 ng/ml HGF in the media (Fig. 1B). The proliferating cell frequency was further decreased in the 72- to 120-h cultures (closed circle) in contrast to a higher level maintained in cultures with 2.5 ng/ml HGF (open circle), consistent with the observation that the 500 ng/ml HGF did not stimulate an increase in cell number during the culture period from 72- to 120-h postplating, in contrast to cultures receiving 2.5 ng/ml HGF (inset in Fig. 1B). To determine whether this deactivation phenomenon represents quiescence or differentiation of satellite cells, reactivation activity of the high-level HGF-deactivated cells was examined. In experiments displayed in Fig. 1C, proliferating satellite cells were deactivated by 500 ng/ml HGF addition as shown in Fig. 1, A and B, and subsequently maintained in DMEM-10% HS with or without 2.5 ng/ml HGF for 48 h from 72- to 120-h postplating. The activation index was increased again in the 120-h culture with a 24-h time lag after lowering the HGF concentration (closed square); activation index remained low in the control cultures not receiving any HGF (open square). These data provide evidence that high-level HGF (over 10 ng/ml) can decrease the proliferation activity of satellite cells in vitro, which may represent satellite cell quiescence but not differentiation. This issue was further supported by the real-time RT-PCR analysis of myogenin mRNA expression (top inset in Fig. 1A); relative to control cultures (bar C), there was very little myogenin mRNA expression with either 2.5 or 500 ng/ml HGF (bars HGF).
Myostatin mediates HGF-induced deactivation of satellite cells.
Experiments were conducted to investigate the potential involvement of myostatin in satellite cell deactivation stimulated by high concentrations of HGF. In experiments described in Fig. 2, activated satellite cells were treated with 500 ng/ml HGF in the presence of various concentrations of antimyostatin neutralizing antibody for 24 h from 48- to 72-h postplating. With increasing concentrations of the neutralizing antibody in cultures, the BrdU-incorporation activity was rescued (Fig. 2A). The proliferation activity of cultures with 10 μg/ml neutralizing antibody was almost equivalent to the 72-h activation cultures with 2.5 ng/ml HGF (c), demonstrating that high-level HGF-induced satellite cell deactivation was cancelled by addition of antimyostatin neutralizing antibody to culture media. Experiments included a control culture treated with 500 ng/ml HGF plus 10 μg/ml of a nonneutralizing antibody (d″); cultures receiving nonneutralizing antibody did not show a reversal of the effect of 500 ng/ml HGF (leftmost data point). One of the targets of myostatin signaling is p21 (91), a Cdk inhibitor that regulates the G1/S and G2/M transitions during cell cycle progression (24, 74). The effect of antimyostatin neutralizing antibody on p21 expression level was examined by Western blotting of the HGF-treated cultures at 72-h postplating (Fig. 2B). The band corresponding to p21 was clearly detected in satellite cell lysates from the deactivation culture with 500 ng/ml HGF (lane d), but very little p21 was found in the activation culture with 2.5 ng/ml HGF (lane c). Upregulation of p21 protein in response to high concentrations of HGF was cancelled by adding antimyostatin neutralizing antibody to the 500 ng/ml HGF medium (lane d′), indicating that the p21 may be involved in high-level HGF-induced satellite cell deactivation downstream of myostatin activity.
Fig. 2.

Myostatin mediates high-level HGF-induced deactivation of satellite cells. Satellite cell activation was stimulated by 2.5 ng/ml HGF as shown in Fig. 1. Cultures were then treated with 500 ng/ml HGF for the next 24 h from 48- to 72-h postplating in the presence of various concentrations of polyclonal antimyostatin neutralizing antibody (0.1–10 μg/ml, plotted on a log scale as the x-axis) and evaluated for the BrdU-incorporation activity at 72-h postplating (A). Experiments included the following control cultures: c, with 2.5 ng/ml HGF; d″, with 500 ng/ml HGF plus 10 μg/ml control immunoglobulin (goat IgG). Data points depict the means ± SE for four cultures per treatment; significant differences from the control culture mean (leftmost data point without neutralizing antibody) are indicated (*P < 0.05; **P < 0.01). B: companion cultures were analyzed for p21 expression by enhanced chemiluminescence (ECL)-Western blotting of cell lysates; α-tubulin protein levels were included to standardize the cell loading. CNT, control blots of 500 ng/ml HGF-treated cells without primary antibody and with secondary reagents.
The role of HGF in myostatin expression was further examined by analyzing myostatin message and protein expression. Whole cell lysates and conditioned media from a constant number of satellite cells were analyzed for myostatin expression and secretion at 72-h postplating by Western blotting using antimyostatin antibody that recognizes the COOH-terminal mature bioactive region of the protein (Fig. 3A, top row). In cultures receiving 500 ng/ml HGF, a 52-kDa band corresponding to the promyostatin form was detected in cell lysates and conditioned media along with weak staining of an unknown 60-kDa band (lanes d), which was not observed in conditioned medium from a positive control culture of human embryonic kidney (HEK)293 cells transfected with myostatin-expressing plasmid (lane P1). By comparison with this deactivation culture, activated (lanes b and c, assayed at 48-h and 72-h postplating, respectively) and reactivated satellite cells (lane e, at 120-h) showed no myostatin expression, comparable to the starting control culture without HGF (lane a, at 24-h). Myostatin upregulation by 500 ng/ml HGF was also demonstrated by RT-PCR of the 72-h cultured-cells (Fig. 3C), since there was a large increase in the intensity of the 299-bp myostatin transcript standardized relative to the level of HPRT mRNA in the same samples (lane d); other cultures including activated/proliferating and reactivated cells did not show any response (lanes a–c and e), consistent with the protein expression profiles described above. Similar results were also observed by immunocytochemistry for the same series of the cells (Fig. 3B), with myostatin-positive cell percentages reaching 90% in the 500 ng/ml HGF culture (solid bar d, at 72-h). Myostatin was seen in ∼10% of the cells in both activation and reactivation cultures (solid bars b, c, and e, assayed at 48, 72, and 120-h, respectively). Myostatin expression was completely opposite to the expression pattern of MyoD protein (see Fig. 3A, bottom row, and B, light-grayed bars), which has been well documented to be upregulated in proliferating satellite cells and then downregulated in the quiescent cells (39, 105, 106). Together, these results highlight a prominent role for high-level HGF as a potent cue to upregulate myostatin expression and secretion and therefore to deactivate (not differentiate) proliferating satellite cells in culture.
Fig. 3.

High-level HGF stimulates myostatin expression and secretion in proliferating satellite cell cultures. Satellite cell cultures were activated by 2.5 ng/ml HGF as shown in Fig. 1 and incubated with 500 ng/ml HGF for the next 24 h from 48- to 72-h postplating. Cultures were evaluated for myostatin expression by ECL-Western blotting of whole cell lysates and conditioned media from a constant number of cells (A), indirect immunocytochemistry (B, solid bars, representing means ± SE), and RT-PCR standardized with internal HPRT (C). Counterpart expression levels of MyoD protein are included in A (bottom row) and B (light-grayed bars). STD, biotinylated molecular weight standards; a, culture before activation treatment at 24 h; b, 2.5 ng/ml HGF culture at 48 h; c, 2.5 ng/ml HGF culture at 72 h; d, 500 ng/ml HGF culture at 72 h; e, 2.5 ng/ml HGF reactivation culture at 120 h. CNT1, CNT2, and CNT3, control blots of the cell lysate (d), conditioned medium (d), and cell lysate (c) without primary antibody and with secondary reagents, respectively; P1, positive control [conditioned medium from human embryonic kidney (HEK)293 cells transfected with His-tagged myostatin-expressing plasmid]; N1, negative control [conditioned medium from HEK293 cells transfected with enhanced green fluorescent protein (EGFP)-expressing plasmid]; P, rat skeletal muscle cDNA; N, no template. *52-kDa pro-myostatin form.
These data do not necessarily prove that satellite cell deactivation responds to HGF in its physiological concentration range found in regenerating or growing muscle tissue, because the myostatin expression was demonstrated just at 500 ng/ml HGF, which was optimized for the in vitro culture assay that enables adequate visualization of the HGF effect within a short culture period of 24 h. It is possible that this HGF concentration may be beyond a physiological range of localized HGF concentrations in the extracellular compartment of damaged muscle tissue. Therefore, the final experiments were conducted to determine minimum concentrations of HGF required for myostatin synthesis and secretion in cultures (Fig. 4). Activation of satellite cells was stimulated by 2.5 ng/ml HGF for 24 h and then incubated with higher concentration of HGF for the next 24-h period as in Fig. 1A. Whole cell lysates and conditioned media were analyzed for myostatin expression by Western blot, standardized with α-tubulin protein in the cell lysates. Results showed significant upregulation of 52-kDa myostatin in the cell lysates (Fig. 4, bottom row) at concentrations greater than 50–100 ng/ml and at concentrations greater than 10–50 ng/ml in conditioned media (top row). HGF dose dependence of myostatin expression can account for the proliferation activity profile of satellite cells shown in Fig. 1A, in which the BrdU-incorporation index decreased at HGF concentrations over 10 ng/ml and reached to a level equivalent to the cultures at the beginning of activation-cultures with HGF concentrations of 50–100 ng/ml. These data indicate that 10–50 ng/ml HGF is enough to initiate myostatin synthesis and secretion that may mark a crucial step in the negative feedback following low-level HGF-triggered activation and the subsequent proliferation of satellite cells.
Fig. 4.

HGF dose dependence of myostatin expression and secretion. Activated satellite cell cultures were incubated with 2.5–500 ng/ml HGF for 24 h from 48- to 72-h postplating as shown in Fig. 1. Myostatin expression and secretion were evaluated by ECL-Western blotting of whole cell lysates (middle row) and conditioned media (top row) standardized relative to α-tubulin protein levels (bottom row). STD, biotinylated molecular weight standards; CNT, control blots of 500 ng/ml HGF culture without primary antibody and with secondary reagents.
DISCUSSION
The importance of satellite cell activity in skeletal muscle regeneration and hypertrophy has been studied for a considerable time. It is well documented that both phenomena are initiated by mechanical insult or other perturbation and one of the earliest events is triggering the activation of quiescent satellite cells, which enables satellite cells to migrate and enter the cell cycle. Allen's group first demonstrated that HGF is unique in its ability to activate satellite cell division in vitro and in living muscle (2, 80) and later verified that the growth factor is present in uninjured muscle and located primarily in the extracellular matrix of muscle fibers (80, 84, 85). Injection of recombinant HGF into muscle tissue resulted in an increase in the numbers of BrdU-positive or MyoD-expressing satellite cells and a reduction in fiber formation (51, 80), supporting widely documented in vitro evidence that HGF stimulates the cell activation and proliferation and inhibits differentiation. In further experiments, Tatsumi et al. (81–83, 85, 86) showed that satellite cells are stimulated to enter the cell cycle when subjected to mechanical stretch in culture and living muscle. These experiments also demonstrated that satellite cell activation is due to rapid release of HGF from its extracellular tethering and its subsequent presentation to c-met receptor. Furthermore, HGF release does not require new growth factor synthesis (see reviews of Refs. 87, 90). Although a significant body of work points to the importance of HGF for satellite cell activation, the contribution of HGF to cell deactivation and the return to G0 phase is a novel and intriguing idea. This hypothesis was evaluated in the present study and strongly supported by our in vitro observations that satellite cells respond to high concentrations of HGF (over 10–50 ng/ml) to generate myostatin protein expression and secretion along with decreased cell proliferation and MyoD expression in primary cultures (Figs. 1–3). These results are consistent with very recent observations by Li et al. (44) that treatment of mouse satellite cells with 50 ng/ml HGF inhibits their proliferation. Analysis of myogenin expression revealed no increase in differentiation in cultures treated with 500 ng/ml HGF (top inset in Fig. 1A), arguing that the high-level HGF-induced deactivation does not permit terminal differentiation but directs cells to reenter a mitotically quiescent phase. Cells may then be reactivated by low HGF concentrations (2.5 ng/ml, see Fig. 1C). This same model may be extrapolated to skeletal muscle regeneration and work/exercise-induced hypertrophy.
In the proposed scenario, the time-coordinated increase in extracellular concentrations of HGF is a key modulator for the two contrary pathways having low and high thresholds to initiate activation and the counterpart quiescence of satellite cells, respectively. Therefore, regulation of HGF concentration in muscle tissue is a key aspect of the proposed HGF activation/quiescence model. HGF is an autocrine/paracrine factor that accumulates in the extracellular matrix of uninjured muscle fibers and can be released by NO radical-activated matrix metalloproteinase activity in response to mechanical perturbation of muscle tissue (75, 76, 79, 80, 82–86, 101, 103, 104). In addition, proliferating satellite cells initiate synthesis of HGF (6, 73), and therefore, as the numbers of active satellite cells increase, the local concentration of HGF may increase. Jennische et al. (36) reported that mRNA for HGF is first expressed in regenerating muscle 3 days after injury, the earliest time measured, and declines again to barely detectable levels by a week after injury. Although concentrations of HGF were not measured, its location and time of expression are consistent with its function as a mitogen and motogen. Interestingly, spleen and liver cells also respond to muscle injury to produce HGF, which is potentially delivered to damaged regions via blood flow during the period of rapid activation, proliferation, and the subsequent early-differentiation of the cell population (76). Similar increases in serum HGF level was also reported in some abnormal (but physiological) conditions including obesity (Ref. 77; 2 ng/ml HGF versus 1 ng/ml in control humans) and spontaneous hypertension (Ref. 54; 7 ng/ml versus 2 ng/ml in control rats), in which the HGF production may be upregulated originally in the disease-related organs and distributed to blood. The combination of endogenous HGF secretion by activated satellite cells and HGF produced elsewhere may elevate local HGF concentration to a level sufficient to induce myostatin expression. It remains to be determined whether extracellular HGF concentrations reach a threshold (at least over 10 ng/ml); however, the measurement may be rather difficult in living animals and will require a novel in vivo method to monitor the time course of local extracellular HGF concentrations with adequate sensitivity. In this case, nondestructive procedures would be suitable such as an in vivo measurement reported by Cromack et al. (22), in which wound fluid was collected from chambers implanted subcutaneously in rats with minimal tissue damage and assayed for TGF-β. They reported TGF-β concentrations of about 1 ng/ml at 3 days and a peak of about 20 ng/ml at 7 days after muscle injury. Common destructive procedures that involve tissue homogenization followed by a centrifugation step to collect supernatants as injured muscle extracts may overestimate the local HGF concentrations because the mechanical perturbation (added to the tissue at the homogenization and spin steps) may liberate additional HGF from its extracellular tethering as described in the previous sections.
The ability of HGF to positively and negatively alter mitotic activity of satellite cells is supportive of a dual function for the growth factor that may be essential for successful muscle growth and regeneration. Similar observations were described by Bischoff (14) in demonstrating that HGF and TGF-β exhibited significant chemotactic activity in the 1–10 ng/ml range, but the dose-response curves for both of these factors were bell-shaped and activity declined at higher concentrations to a level equivalent to control satellite cell cultures without any growth factor treatment.
The mechanisms for divergent behavior of satellite cells in response to a common growth factor and the intracellular signaling intermediates are completely unknown at this time. A simple model may be represented as a consequence of different signal intensities generated from the high-affinity receptor c-met that has been known to be responsible for stimulating a cascade of signaling molecules including Gab1, SHP2, RhoA, phosphatidylinositol 3-kinase, Grb2, MAPK, Akt, ERK1/2, and p21 (31, 38, 59, 60, 66, 93; reviewed in Refs. 12, 92). Li et al. (44) demonstrated by using gene transfection techniques that a protein tyrosine phosphatase SHP2, which is recruited to the c-met kinase domain via a docking protein Gab1 (64), is involved in the 50 ng/ml HGF-induced deactivation of satellite cells in primary cultures, and that this inhibitory actions of HGF as mediated through SHP2 include an initial rise in sustained ERK1/2 phosphorylation that is absent within 20 min poststimulus, being in striking contrast to the prolonged ERK1/2 response in many cell types (53, 56). In addition, 24-h treatment with recombinant myostatin upregulates p21 protein and decreases the levels and activity of Cdk2 protein in C2C12 myoblast (91) and satellite cell cultures (48). Our present experiments confirmed these results by demonstrating the upregulation of p21 protein in high-level HGF cultures, which was cancelled by coaddition of antimyostatin neutralizing antibody (Fig. 2B). These data could be supportive of the single-receptor hypothesis to account for the contrary behavior of satellite cells through the HGF ligand association to c-met. The subsequent signal transduction cascade of common intracellular signaling intermediates with their different expression levels and activities could allow generation of different signal intensities responsible for the satellite cell activation and quiescence as a consequence. In this contrary regulation mechanism, the signal intensity could result from a balance between positive and negative modulation of the enzymatic activity of c-met. The differential regulation of the c-met β-chain (a larger membrane-passing subunit) may be determined by phosphorylation of two tyrosine residues within the catalytic domain (resulting in upregulation of kinase activity) or by phosphorylation of a serine residue in the juxtamembrane domain (downregulation of kinase activity). The COOH-terminal domain of c-met includes two critical tyrosine residues that together form a docking site for several signal transducers and adaptors when phosphorylated (15, 55; reviewed in Ref. 92), also providing regulatory points for the presumed divergent behavior of c-met signaling.
Alternatively, the other possible hypothesis may be that there are two classes of receptors having a high affinity (c-met) and low affinity for HGF (an unknown receptor) to generate signals for activation and quiescence of satellite cells, respectively. The nature of the alternate receptor system is unclear at this time, but additional evidence for an alternative pathway is emerging from experiments in which the HGF was shown to stimulate expression of neural chemorepellent semaphorin 3A (Sema3A) in cultured satellite cells through a pathway that does not involve c-met with a maximum induction at 10–25 ng/ml (89). Considering that the presence of c-met protein is drastically downregulated to barely detectable levels in our culture condition during 48–72 h culture time (see Fig. 5A inset of Ref. 89), which is the same time period at which Sema3A and myostatin expressions were demonstrated to respond to HGF, it is interesting to speculate on the original divergence in signaling between activation and return to the quiescence. Actually, the presence of low-affinity binding sites for HGF has been shown in both HGF-responsive epithelial cells and also in HGF-nonresponsive mesenchymal cells, although their physiological functions are not yet understood. Scatchard plot analysis of the specific binding of [125I]-labeled HGF on fibroblast-derived COS-7 cells revealed the dissociation constant (Kd) of 513 pM for low-affinity receptors (Bmax = 22,800 sites/cell) (34) compared with 20–30 pM for high-affinity receptor c-met in the COS-7 cells transfected with c-met plasmid (Bmax = 3,540 sites/cell), rat hepatocytes (33), and renal tubular epithelial cells (35). At the present, however, it is not known what protein(s) constitutes low-affinity receptor for HGF, although Higuchi et al. (34) have found that heparin dose-dependently replaced the binding of HGF to low-affinity sites and therefore suggested that the low-affinity HGF binding sites may be heparin-like extracellular matrixes. As a first step toward understanding this question, it may be invaluable to highlight the extracellular region of c-met β-chains that displays structural analogies with the extracellular domains of semaphorins and plexins, which together form a wide family of ligand-receptor pairs that were originally identified in the nervous system, but that are now known to be widely expressed in other cell types (reviewed in Refs. 78, 92). They all contain the so-called sema domain (a conserved sequence that encompasses about 500 amino acids) and comprise an eight-cysteine peptide module that is conventionally termed MRS (met-related sequence), together with three glycine-proline-rich (G-P) repeats. Considering that the MRS in c-met β-chain provides a HGF-binding site, plexins and a transmembrane-type of semaphorins might be plausible candidates for the HGF low-affinity receptor. In addition, α6/β4-integrin has been shown to interact with c-met to modify c-met-dependent responses, and E-cadherin, hyaluronan receptor CD44, neuropilins, and Fas can also interact with c-met (reviewed in Refs. 20, 92). These interactions might be hypothesized to contribute to the functional separation of the two signals for activation and the counterpart quiescence (myostatin expression and secretion) of satellite cells, according to their different binding affinity for the HGF ligand. Recently, we have found that the myostatin expression and deactivation activities of satellite cells can be abolished by coaddition of neutralizing antibody to neuropilin-1, which is known as a membrane-bound coreceptor protein of a tyrosine kinase receptor for both vascular endothelial growth factor (VEGF) and Sema3A (Yamada M and Tatsumi R, unpublished observations). These experiments also showed that the neuropilin-1 protein coimmunoprecipitates with HGF in the cell lysates, supporting the above coreceptor protein hypothesis of an unknown low-affinity receptor for HGF. In this case, because neuropilins do not possess the MRS module as revealed by the sequence homology analysis, it is speculated that neuropilin-1 might be a receptor partner with c-met, plexins, or semaphorins to constitute the functional low-affinity receptor; its detail, however, awaits further study. Nonetheless, in this model, the time-coordinated coexpression and formation of the functional receptor protein pair have an additional implication for understanding the delayed action of the quiescence signaling auto-program in proliferating satellite cells during initial phases of muscle growth and regeneration.
Fig. 5.
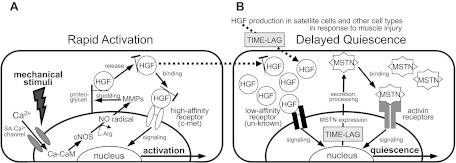
Delayed action model for satellite cell quiescence. HGF may play dual roles in cell activation and quiescence in a dose-dependent manner as follows. A: possible mechanism for the activation: quiescent satellite cells are activated to reenter the cell cycle in response to mechanical perturbation of muscle tissue through a molecular cascade of events including calcium ion influx from extracellular compartment through a stretch-activated (SA)-Ca2+ channel, calcium-calmodulin (Ca-CaM) formation, nitric oxide (NO) radical production by activated constitutive NO synthase (cNOS; neuronal NOS and endothelial NOS), matrix metalloproteinase (MMP) activation, rapid release of HGF from its extracellular tethering (possibly with associated extracellular segment of proteoglycans) to give rise to ng/ml level of the growth factor, and the subsequent presentation to the high-affinity receptor c-met to generate a signal for satellite cell activation. [From Tatsumi and Allen (87) and Tatsumi (90), with modification.] B: satellite cell quiescence through a negative feedback mechanism following satellite cell activation and proliferation. HGF synthesis is initiated in satellite cells and spleen and liver cells in response to muscle damage. Local concentrations of HGF bathing proliferating satellite cells reach a threshold after an appropriate time lag for activation and proliferation of satellite cells. HGF can bind to an unknown low-affinity receptor responsible for signaling myostatin expression. The myostatin protein (MSTN) is secreted and processed to the active form that associates with activin type IIAB receptors that signal satellite cell quiescence. l-Arg, l-arginine.
Finally, an important issue for discussion may be an extracellular regulation of secreted myostatin activity. It is worth noting again that a 52-kDa protein corresponding to the inactive promyostatin was a main form found in serum-free DMEM conditioned media from cultures treated with HGF over 10 ng/ml (top rows in Figs. 3A and 4); the active form, which is generated by proteolytic processing of the pro-form along with a NH2-terminal latency-associated peptide (LAP) (43), was barely detected in conditioned media or cell lysates by our ECL-Western blot analysis. Therefore, the activation of myostatin protein secreted to extracellular compartment is a crucial step for the high-level HGF-induced return to quiescence of proliferating satellite cells. It has been shown that the circulatory promyostatin is cleaved and activated by a bone morphogenetic protein-1 (BMP-1)/tolloid family of metalloproteinases (97). Anderson et al. (11) also demonstrated that myostatin is present extracellularly as uncleaved pro-form and can be processed there by a furin family pro-protein convertase. Although there is no evidence at this time implicating these extracellular proteases in our satellite cell culture phenomenon, it may be possible that an enzyme present in serum is essential for processing of the promyostatin. In the presence of 10% HS in culture media, the secreted protein actually displayed biological activities to downregulate the cell proliferation and upregulate p21 expression, which can be cancelled by coaddition of antimyostatin neutralizing antibody to levels equivalent to the positive control culture with 2.5 ng/ml HGF (Fig. 2). The importance of promyostatin activators may be emphasized by physiological situation specifically found in damaged muscle, in which blood is leaked out of damaged intramuscular capillaries in regions of injury and the processing enzymes present in sera may be potentially responsible for activating promyostatin protein secreted from proliferating satellite cells. After processing, the active myostatin COOH-terminal peptide forms the disulfide-linked homodimer and then binds to activin IIAB receptors to generate the cell quiescence signaling (42, 58) through phosphorylation of TGF-β-specific Smad2 and 3 to form a ternary complex with Smad4. The subsequent translocation of the Smad complex to the nucleus regulates expression of targeted genes such as MyoD, Pax7, and the other myogenic regulatory factors (40, 42, 46, 63). As a result, satellite cells return to quiescent, undifferentiated state, and activation is blocked to maintain quiescence (48, 57, 91, 95). In this cascade, there are some additional factors that have been known to regulate myostatin activity. LAP can form a noncovalent complex with the COOH-terminal active dimer, which maintains it in a latent/inactive state (42, 91), and follistatin, known as a negative regulator of myostatin, has been well documented to inhibit satellite cell proliferation by blocking myostatin and activin association with the receptor (26, 43). In the present study, however, the potential role of these inhibitory peptides in regulation of the high-level HGF-induced deactivation was not examined. Finally, it may be worthy to note that small natural splice variants of HGF, NK1, and NK2 composed of only the NH2-terminal hairpin loop and the subsequent first and second kringle domains, have been demonstrated to be expressed in many normal tissues including skeletal muscle, liver, mammary gland, and kidney (16, 45, 52, 69). It is conceivable that there is an important physiological role for these factors in coordinating with the full-length ligand to act as antagonists or agonists for the HGF/low-affinity receptor system.
In summary, the present experiments propose the possible role for the HGF in the quiescence through its concentration-dependent negative feedback mechanism following satellite cell activation and proliferation, as modeled in the low-affinity receptor hypothesis in Fig. 5. Briefly, initiation of satellite cell activation requires only ng/ml levels of HGF; thus the amount of HGF released from extracellular tethering may be sufficient for rapid activation through the growth factor ligand/high-affinity receptor c-met system (see reviews of Refs. 87 and 90 for more details). HGF synthesis is initiated in satellite cells and other cell types in response to muscle perturbations and causes an increase in local HGF concentrations bathing proliferating satellite cells. Satellite cells continue to proliferate during regeneration and hypertrophy (or hyperplasia), but when extracellular HGF concentrations reach a threshold high enough to associate with an unidentified low-affinity receptor, satellite cell-derived myoblasts start expressing myostatin. Active myostatin is then produced and secreted from satellite cells to signal the cell quiescence.
It should be noted that the present work does not determine whether high concentrations of HGF act on all myogenic cells or a subset of cells that are programmed to self-renew. Schultz (68) provided some of the original evidence for different populations of satellite cells. His work indicated that there was a large population that was active during growth and a smaller population that was not, and consequently, might represent the reserve population responsible for repair during adulthood. This later population might therefore have greater capabilities to move in and out of quiescence as the population is renewed. In contrast to most of the reported work on satellite cell self-renewal, the experiments reported here and with all of our previous work on activation were all done with 9-mo-old adult rats. Therefore, only the adult maintenance population of satellite cells should be present, and most of these cells might be expected to have the ability to return to quiescence after activation and proliferation.
According to this model, activation and quiescence of satellite cells may be a temporally coordinated sequence of events centering on the actions of HGF on myogenic cells. The role of HGF in muscle repair, however, may not be restricted to myogenesis; Tatsumi et al. (89) demonstrated that HGF (over 2.5 ng/ml) upregulates neurochemorepellent Sema3A expression in satellite cells at early-differentiation phase (not activation and proliferation stages) in primary cultures of the cells and muscle fibers and in vivo. The results lead to speculation that the expression and secretion of Sema3A from satellite and satellite-derived myogenic cells may mediate restoration or remodeling of the nerve-muscle connections through chemorepulsive events. HGF stimulation of Sema3A may be involved in coordination of a delay in sprouting and reattachment of motoneuron terminals onto damaged muscle fibers in synchrony with recovery of muscle fiber structures that enables persistent or restored contractile activity (see a model in Fig. 7, A1–A3, of Ref. 89). Sema3A expression may potentially also regulate vascular morphogenesis through Sema3A-VEGF competitive binding to receptor neuropilins, and integrin-mediated endothelial cell adhesion and migration (71; reviewed in Ref. 25), and muscle HGF may be directly involved in angiogenesis. Together, the previous reports and the present results lead to the idea that successful muscle regeneration, composed of myogenesis and intramuscular neuritogenesis and angiogenesis, may be a programmed sequence of events that respond to a mechanical insult or other perturbation in a synchronous, HGF-dependent, and time-coordinated manner.
GRANTS
This work was supported by Grants-in-Aid for Scientific Research (B) 19380152 and for Exploratory Research 20658067 from the Japan Society for the Promotion of Science (JSPS), and by grant funds from the Ito Foundation and the Uehara Memorial Foundation (all to R. Tatsumi). The research was also supported by funds from the Arizona Agriculture Experiment Station and grants from the U.S. Department of Agriculture National Research Initiative Competitive Grant 2005-35206-15255 and Muscular Dystrophy Association Grant MDA3685 (all to R. E. Allen). M. Yamada received a JSPS Pre-Doctoral Research fellowship for Young Scientists and Grant-in-Aid for JSPS Fellows 19-7723 during the course of this research.
DISCLOSURES
No conflicts of interest are declared by the author(s).
ACKNOWLEDGMENTS
The helpful discussions with Dr. Judy E. Anderson of University of Manitoba, Canada were invaluable. The mouse anti-BrdU monoclonal antibody developed by S. J. Kaufman and the antidesmin monoclonal antibody developed by D. A. Fischman were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by Department of Biological Sciences, The University of Iowa, Iowa City, IA 52242.
Present address: of T. Hosoyama: University of Texas Health Science Center at San Antonio, Greehey Children's Cancer Research Institute, San Antonio, TX 78229.
REFERENCES
- 1.Allen RE, Boxhorn LK. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J Cell Physiol 138: 311–315, 1989 [DOI] [PubMed] [Google Scholar]
- 2.Allen RE, Sheehan SM, Taylor RG, Kendall TL, Rice GM. Hepatocyte growth factor activates quiescent skeletal muscle satellite cells in vitro. J Cell Physiol 165: 307–312, 1995 [DOI] [PubMed] [Google Scholar]
- 3.Allen RE, Temm-Grove CJ, Sheehan SM, Rice GM. Skeletal muscle satellite cell cultures. Methods Cell Biol 52: 155–176, 1997 [DOI] [PubMed] [Google Scholar]
- 4.Allen RE, Goll DE. Cellular and developmental biology of skeletal muscle as related to muscle growth. In: Biology of Growth of Domestic Animals, edited by Scanes CG. Ames, IA: Iowa State Press, 2003, p. 148–169 [Google Scholar]
- 5.Amthor H, Otto A, Macharia R, McKinnell I, Patel K. Myostatin imposes reversible quiescence on embryonic muscle precursors. Dev Dyn 235: 672–680, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Anastasi S, Giordano S, Sthandier O, Gambarotta G, Maione R, Comoglio P, Amati P. A natural hepatocyte growth factor/scatter factor autocrine loop in myoblast cells and the effect of the constitutive Met kinase activation on myogenic differentiation. J Cell Biol 137: 1057–1068, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell 11: 1859–1874, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson JE, Pilipowicz O. Activation of muscle satellite cells in single-fiber cultures. Nitric Oxide 7: 36–41, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Anderson JE, Wozniak AC. Satellite cell activation on fibers: modeling events in vivo. Can J Physiol Pharmacol 82: 300–310, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Anderson JE. The satellite cell as a companion in skeletal muscle plasticity: currency, conveyance, clue, connector and colander. J Exp Biol 209: 2276–2292, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Anderson SB, Goldberg AL, Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem 283: 7027–7035, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Birchmeier C, Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol 8: 404–410, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Bischoff R. The satellite cell and muscle regeneration. In: Myology: Basic and Clinical (2nd ed.), edited by Engel AG, Franzini-Armstrong C. New York: McGraw-Hill, 1994, vol. 1, p. 97–118 [Google Scholar]
- 14.Bischoff R. Chemotaxis of skeletal muscle satellite cells. Dev Dyn 208: 505–515, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 251: 802–804, 1991 [DOI] [PubMed] [Google Scholar]
- 16.Chan AM, Rubin JS, Bottaro DP, Hirschfield DW, Chedid M, Aaronson SA. Identification of a competitive HGF antagonist encoded by an alternative transcript. Science 254: 1382–1385, 1991 [DOI] [PubMed] [Google Scholar]
- 17.Chiaverotti TA, Battula N, Monnat RJ., Jr Rat hypoxanthine phosphoribosyltransferase cDNA cloning and sequence analysis. Genomics 11: 1158–1160, 1991 [DOI] [PubMed] [Google Scholar]
- 18.Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet 38: 813–818, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Collins CA. Satellite cell self-renewal. Curr Opin Pharmacol 6: 301–306, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Comoglio PM, Boccaccio C, Trusolino L. Interactions between growth factor receptors and adhesion molecules: breaking the rules. Curr Opin Cell Biol 15: 565–571, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Cornelison DDW, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol 191: 270–283, 1997 [DOI] [PubMed] [Google Scholar]
- 22.Cromack DT, Sporn MB, Roberts AB, Merino MJ, Dart LL, Norton JA. Transforming growth factor-β levels in rat wound chambers. J Surg Res 42: 622–628, 1987 [DOI] [PubMed] [Google Scholar]
- 23.Dhawan J, Rando TA. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol 15: 666–673, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Dulic V, Stein GH, Far DF, Reed SI. Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Mol Cell Biol 18: 546–557, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellis LM. The role of neuropilins in cancer. Mol Cancer Ther 5: 1099–1107, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab 297: E157–E164, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Giordano S, Ponzetto C, Di Renzo MF, Cooper CS, Comoglio PM. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 339: 155–156, 1989 [DOI] [PubMed] [Google Scholar]
- 28.Giordano S, Di Renzo MF, Narsimhan RP, Cooper CS, Rosa C, Comoglio PM. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 4: 1383–1388, 1989 [PubMed] [Google Scholar]
- 29.Gonzatti-Haces M, Seth A, Park M, Copeland T, Oroszlan S, Vande Woude GF. Characterization of the TPR-MET oncogene p65 and the MET protooncogene p140 protein-tyrosine kinases. Proc Natl Acad Sci USA 85: 21–25, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grobet L, Martin LJR, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Menissier F, Massabanda J, Fries R, Hanset R, Georges M. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet 17: 71–74, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Halevy O, Cantley LC. Differential regulation of the phosphoinositide 3-kinase and MAP kinase pathways by hepatocyte growth factor vs. insulin-like growth factor-I in myogenic cells. Exp Cell Res 297: 224–234, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol 91: 534–551, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Higuchi O, Nakamura T. Identification and change in the receptor for hepatocyte growth factor in rat liver after partial hepatectomy or induced hepatitis. Biochem Biophys Res Commun 176: 599–607, 1991 [DOI] [PubMed] [Google Scholar]
- 34.Higuchi O, Mizuno K, VandeWoude GF, Nakamura T. Expression of c-met proto-oncogene in COS cells induces the signal transducing high-affinity receptor for hepatocyte growth factor. FEBS Lett 301: 282–286, 1992 [DOI] [PubMed] [Google Scholar]
- 35.Igawa T, Kanda S, Kanetake H, Saitoh Y, Ichihara A, Tomita Y, Nakamura T. Hepatocyte growth factor is a potent mitogen for cultured rabbit renal tubular epithelial cells. Biochem Biophys Res Commun 174: 831–838, 1991 [DOI] [PubMed] [Google Scholar]
- 36.Jennische E, Ekberg S, Matejka GL. Expression of hepatocyte growth factor in growing regenerating rat skeletal muscle. Am J Physiol Cell Physiol 265: C122–C128, 1993 [DOI] [PubMed] [Google Scholar]
- 37.Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res 7: 910–916, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Kontaridis MI, Eminaga S, Fornaro M, Zito CI, Sordella R, Settleman J, Bennett AM. SHP-2 positively regulates myogenesis by coupling to the Rho GTPase signaling pathway. Mol Cell Biol 24: 5340–5352, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuang S, Rudnicki MA. The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med 14: 82–91, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Langley B, Thomas M, Bishop A, Sharma M, Glimour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem 277: 49831–49840, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 277: 680–685, 1970 [DOI] [PubMed] [Google Scholar]
- 42.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA 98: 9306–9311, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol 20: 61–86, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Li J, Reed SA, Johnson SE. Hepatocyte growth factor (HGF) signals through SHP2 to regulate primary mouse myoblast proliferation. Exp Cell Res 315: 2284–2292, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lokker NA, Godowski PJ. Generation and characterization of a competitive antagonist of human hepatocyte growth factor, HGF/NK1. J Biol Chem 268: 17145–17150, 1993 [PubMed] [Google Scholar]
- 46.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 19: 1745–1754, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCormick KM, Schultz E. Mechanisms of nascent fiber formation during avian skeletal-muscle hypertrophy. Dev Biol 150: 319–334, 1992 [DOI] [PubMed] [Google Scholar]
- 48.McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self renewal. J Cell Biol 162: 1135–1147, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387: 83–90, 1997 [DOI] [PubMed] [Google Scholar]
- 50.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA 94: 12457–12461, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller KJ, Thaloor D, Matteson S, Pavlath GK. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. Am J Physiol Cell Physiol 278: C174–C181, 2000 [DOI] [PubMed] [Google Scholar]
- 52.Miyazawa K, Kitamura A, Naka D, Kitamura N. An alternatively processed mRNA generated from human hepatocyte growth factor gene. Eur J Biochem 197: 15–22, 1991 [DOI] [PubMed] [Google Scholar]
- 53.Mohi MG, Neel BG. The role of SHP2 (PTPN11) in cancer. Curr Opin Genet Dev 17: 23–30, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Nakano N, Moriguchi A, Morishita R, Kida I, Tomita N, Natsyniti K, Nakamura T, Higaki J, Ogihara T. Role of angiotensin II in the regulation of novel vascular modulator, hepatocyte growth factor (HGF), in experimental hypertensive rats. Hypertension 30: 1448–1454, 1997 [DOI] [PubMed] [Google Scholar]
- 55.Naldini L, Vigna E, Narshimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, Comoglio PM. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the protooncogene c-met. Oncogene 6: 501–504, 1991 [PubMed] [Google Scholar]
- 56.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28: 284–293, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Olguin HC, Olwin BB. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol 275: 375–388, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rabbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol 23: 7230–7242, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reuveny M, Heller H, Bengal E. RhoA controls myoblast survival by inducing the phosphatidylinositol 3-kinase-Akt signaling pathway. FEBS Lett 569: 129–134, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Ridley AJ, Comoglio PM, Hall A. Regulation of Scatter factor/Hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol Cell Biol 15: 1110–1122, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rios R, Carneiro I, Arce VM, Devesa J. Myostatin regulates cell survival during C2C12 myogenesis. Biochem Biophys Res Commun 280: 561–566, 2001 [DOI] [PubMed] [Google Scholar]
- 62.Rios R, Fernandez-Nocelos S, Carneiro I, Arce VM, Devesa JS. Differential response to exogenous and endogenous myostatin in myoblasts suggests that myostatin acts as an autocrine factor in vivo. Endocrinology 145: 2795–2803, 2004 [DOI] [PubMed] [Google Scholar]
- 63.Rodino-Klapac LR, Haidet AM, Kota J, Handy C, Kaspar BK, Mendell JR. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 39: 283–296, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosario M, Birchmeier W. How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol 13: 328–335, 2003 [DOI] [PubMed] [Google Scholar]
- 65.Sakata T, Tatsumi R, Yamada M, Shiratsuchi S, Okamoto S, Mizunoya W, Hattori A, Ikeuchi Y. Preliminary experiments on mechanical stretch-induced activation of skeletal muscle satellite cells in vivo. Anim Sci J 77: 518–525, 2006 [Google Scholar]
- 66.Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J Cell Biol 149: 1419–1432, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688, 2004 [DOI] [PubMed] [Google Scholar]
- 68.Schultz E. Satellite cell proliferative compartments in growing skeletal muscles. Dev Biol 175: 84–94, 1996 [DOI] [PubMed] [Google Scholar]
- 69.Schwall RH, Chang LY, Godowski PJ, Kahn DW, Hillan KJ, Bauer KD, Zioncheck TF. Heparin induces dimerization and confers proliferative activity onto the hepatocyte growth factor antagonists NK1 and NK2. J Cell Biol 133: 709–718, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seale P, Rudnicki MA. A new look at the origin, function, and “Stem-Cell” status of muscle satellite cells. Dev Biol 218: 115–124, 2000 [DOI] [PubMed] [Google Scholar]
- 71.Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, Tessier-Lavigne M, Taniguchi M, Püschel AW, Bussolino F. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424: 391–397, 2003 [DOI] [PubMed] [Google Scholar]
- 72.Sharma M, Kambadur R, Matthews KG, Somers WG, Devlin GP, Conaglen JV, Fowke PJ, Bass JJ. Myostatin, a transforming growth factor-β superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J Cell Physiol 180: 1–9, 1999 [DOI] [PubMed] [Google Scholar]
- 73.Sheehan SM, Tatsumi R, Temm-Grove CJ, Allen RE. HGF is an autocrine growth factor for skeletal muscle satellite cells in vitro. Muscle Nerve 23: 239–245, 2000 [DOI] [PubMed] [Google Scholar]
- 74.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512, 1999 [DOI] [PubMed] [Google Scholar]
- 75.Smith HK, Maxwell L, Rodgers CD, Mckee NH, Plyley MJ. Exercise-enhanced satellite cell proliferation and new myonuclear accretion in rat skeletal muscle. J Appl Physiol 90: 1407–1414, 2001 [DOI] [PubMed] [Google Scholar]
- 76.Suzuki S, Yamanouchi K, Soeta C, Katakai Y, Harada R, Naito K, Tojo H. Skeletal muscle injury induces hepatocyte growth factor expression in spleen. Biochem Biophys Res Commun 292: 709–714, 2002 [DOI] [PubMed] [Google Scholar]
- 77.Swierczynski J, Korczynska J, Goyke E, Adrych K, Raczynska S, Sledzinski Z. Serum hepatocyte growth factor concentration in obese women decreases after vertical banded gastroplasty. Obes Surg 15: 803–808, 2005 [DOI] [PubMed] [Google Scholar]
- 78.Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol 10: 377–383, 2000 [DOI] [PubMed] [Google Scholar]
- 79.Tanaka S, Tachino K, Kawahara E, Tanaka J, Funakoshi H, Nakamura T. Hepatocyte growth factor in mouse soleus muscle increases with reloading after unloading. J Phys Ther Sci 18: 33–41, 2006 [Google Scholar]
- 80.Tatsumi R, Anderson JE, Nevoret CJ, Halevy O, Allen RE. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev Biol 194: 114–128, 1998 [DOI] [PubMed] [Google Scholar]
- 81.Tatsumi R, Sheehan SM, Iwasaki H, Hattori A, Allen RE. Mechanical stretch induces activation of skeletal muscle satellite cells in vitro. Exp Cell Res 267: 107–114, 2001 [DOI] [PubMed] [Google Scholar]
- 82.Tatsumi R, Hattori A, Allen RE, Ikeuchi Y, Ito T. Mechanical stretch-induced activation of skeletal muscle satellite cells is dependent on nitric oxide production in vitro. Anim Sci J 73: 235–239, 2002 [Google Scholar]
- 83.Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE. Release of hepatocyte growth factor from mechanical stretched skeletal muscle satellite cells and the role of pH and nitric oxide. Mol Biol Cell 13: 2909–2918, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tatsumi R, Allen RE. Active hepatocyte growth factor is present in skeletal muscle extracellular matrix. Muscle Nerve 30: 654–658, 2004 [DOI] [PubMed] [Google Scholar]
- 85.Tatsumi R, Liu X, Pulido A, Morales M, Sakata T, Dial S, Hattori A, Ikeuchi Y, Allen RE. Satellite cell activation in stretched skeletal muscle and the role of nitric oxide and hepatocyte growth factor. Am J Physiol Cell Physiol 290: C1487–C1494, 2006 [DOI] [PubMed] [Google Scholar]
- 86.Tatsumi R, Yamada M, Katsuki Y, Okamoto S, Ishizaki J, Mizunoya W, Ikeuchi Y, Hattori A, Shimokawa H, Allen RE. Low-pH preparation of skeletal muscle satellite cells can be used to study activation in vitro. Int J Biochem Cell Biol 38: 1678–1685, 2006 [DOI] [PubMed] [Google Scholar]
- 87.Tatsumi R, Allen RE. Mechano-biology of resident myogenic stem cells: molecular mechanism of stretch-induced activation of satellite cells. Anim Sci J 79: 279–290, 2008 [DOI] [PubMed] [Google Scholar]
- 88.Tatsumi R, Wuollet AL, Tabata K, Nishimura S, Tabata S, Mizunoya W, Ikeuchi Y, Allen RE. A role for calcium-calmodulin in regulating nitric oxide production during skeletal muscle satellite cell activation. Am J Physiol Cell Physiol 296: C922–C929, 2009 [DOI] [PubMed] [Google Scholar]
- 89.Tatsumi R, Sankoda Y, Anderson JE, Sato Y, Mizunoya W, Shimizu N, Suzuki T, Yamada M, Rhoads RP, Jr, Ikeuchi Y, Allen RE. Possible implication of satellite cells in regenerative motoneuritogenesis: HGF up-regulates neural chemorepellent Sema3A during myogenic differentiation. Am J Physiol Cell Physiol 297: C238–C252, 2009 [DOI] [PubMed] [Google Scholar]
- 90.Tatsumi R. Mechano-biology of skeletal muscle hypertrophy and regeneration: possible mechanism of stretch-induced activation of resident myogenic stem cells. Anim Sci J In press. [DOI] [PubMed] [Google Scholar]
- 90a.Tatsumi R, Yamanouchi K, Hosoyama T, Shiratsuchi SI, Yamada M, Mizunoya W, Ikeuchi Y, Allen RE. A possible mechanism for muscle satellite cell quiescence: HGF induces myostatin expression and secretion (Abstract). FASEB Summer Research Conference on Skeletal Muscle and Stem Cells, Tucson, AZ, June 11–16, 2005 [Google Scholar]
- 91.Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem 275: 40235–40243, 2000 [DOI] [PubMed] [Google Scholar]
- 92.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signaling for invasive growth. Nat Rev 2: 289–300, 2002 [DOI] [PubMed] [Google Scholar]
- 93.Tsukada Y, Tanaka T, Miyazawa K, Kitamura N. Involvement of down-regulation of Cdk2 activity in hepatocyte growth factor-induced cell cycle arrest at G1 in the human hepatocellular carcinoma cell line HepG2. J Biochem (Tokyo) 186: 701–709, 2004 [DOI] [PubMed] [Google Scholar]
- 94.Wagers AJ, Conboy IM. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 122: 659–667, 2005 [DOI] [PubMed] [Google Scholar]
- 95.Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA 102: 2519–2524, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Winchester PK, Davis ME, Alway SE, Gonyea WJ. Satellite cell activation in the stretch-enlarged anterior latissimus-dorsi muscle of the adult quail. Am J Physiol Cell Physiol 260: C206–C212, 1991 [DOI] [PubMed] [Google Scholar]
- 97.Wolfman NM, Mcpherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, Wright JF, Zhao L, Sebald SM, Greenspspan DS, Leet SI. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci USA 100: 15842–15846, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wozniak AC, Pilipowicz O, Yablonka-Reuveni Z, Greenway S, Craven S, Scott E, Anderson JE. C-met expression and mechanical activation of satellite cells on cultured muscle fibers. J Histochem Cytochem 51: 1–9, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wozniak AC, Anderson JE. Single-fiber isolation and maintenance of satellite cell quiescence. Biochem Cell Biol 83: 674–676, 2005 [DOI] [PubMed] [Google Scholar]
- 100.Wozniak AC, Kong JM, Bock E, Pilipowicz O, Anderson JE. Signaling satellite-cell activation in skeletal muscle: markers, models, stretch, and potential alternate pathways. Muscle Nerve 31: 283–300, 2005 [DOI] [PubMed] [Google Scholar]
- 101.Wozniak AC, Anderson JE. Nitric oxide-dependence of satellite stem cell activation and quiescence on normal skeletal muscle fibers. Dev Dyn 236: 240–250, 2007 [DOI] [PubMed] [Google Scholar]
- 102.Wozniak AC, Anderson JE. The dynamics of the nitric oxide release-transient from stretched muscle cells. Int J Biochem Cell Biol 41: 625–631, 2009 [DOI] [PubMed] [Google Scholar]
- 103.Yamada M, Tatsumi R, Kikuiri T, Okamoto S, Nonoshita S, Mizunoya W, Ikeuchi Y, Shimokawa H, Sunagawa K, Allen RE. Matrix metalloproteinases are involved in mechanical stretch-induced activation of skeletal muscle satellite cells. Muscle Nerve 34: 313–319, 2006 [DOI] [PubMed] [Google Scholar]
- 104.Yamada M, Sankoda Y, Tatsumi R, Mizunoya W, Ikeuchi Y, Sunagawa K, Allen RE. Matrix metalloproteinase-2 mediates stretch-induced activation of skeletal muscle satellite cells in a nitric oxide dependent manner. Int J Biochem Cell Biol 40: 2183–2191, 2008 [DOI] [PubMed] [Google Scholar]
- 105.Yoshida N, Yoshida S, Koishi K, Masuda K, Nabeshima Y. Cell heterogeneity upon differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J Cell Sci 111: 769–779, 1998 [DOI] [PubMed] [Google Scholar]
- 106.Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol 166: 347–357, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]