

Abstract
Background and purpose:
Bradykinin (BK) and B2 receptors have been implicated in the pathophysiology of osteoarthritis (OA), and synovitis is one of its hallmarks. Here, the selective B2 receptor antagonists MEN16132 and icatibant have been pharmacologically characterized in human synovial cells.
Experimental approach:
Radioligand and functional studies (inositol phosphate (IP) accumulation, interleukin (IL)-6 and IL-8 release) were performed in cultured synoviocytes.
Key results:
[3H]-BK saturation studies indicated receptor density (Bmax) and Kd values of 121 550 sites per cell and 1.14 nM respectively. In synoviocytes, MEN16132 (pKi 8.9) was threefold more potent than icatibant (pKi 8.4). Both antagonists showed competitive antagonism in the BK-induced IP assay (control EC50 0.45 nM), with pKB values of 9.9 (MEN16132) and 8.1 (icatibant). 24h incubation with BK induced IL-6 (EC50 216 nM) and IL-8 (EC50 53 nM) release. Both MEN16132 (IL-6: pIC50 8.1; IL-8: pIC50 8.4) and icatibant (IL-6: pIC50 6.6; IL-8: pIC50 6.7) completely prevented this BK-induced release. Indomethacin did not affect the basal or the IL-6/IL-8 release induced by BK, whereas nordihydroguaiaretic acid decreased the basal release, although BK still increased IL-6 and IL-8 production. BK-induced IL-8 release was attenuated by inhibitors of phospholipase C (U73122), p38 (SB203580), JNK (SP600125), ERK 1/2 (PD98059) MAPKs, phosphoinositide 3-kinase (LY294002), NF-κb (BAY-117085) and by the glucocorticoid dexamethasone.
Conclusions and implications:
Bradykinin via B2 receptors can participate in inflammatory events in synovitis. MEN16132 is a highly potent B2 receptor antagonist capable of blocking pro-inflammatory responses to BK evoked in human synoviocytes.
Keywords: MEN16132, icatibant, osteoarthritis, knee, synovium, interleukin-6, interleukin-8, cyclooxygenase, lipoxygenase
Introduction
Bradykinin (BK, H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH) is a peptide which is known to participate both in acute and chronic inflammation. BK exerts its actions by selectively activating the kinin B2 receptor (nomenclature follows Alexander et al., 2008), which belongs to the superfamily of seven transmembrane, G-protein coupled receptors. Its activation leads to Gq/11 protein coupling and consequent triggering of phospholipase C (PLC), thus producing inositol 3-phosphate and diacylglycerol (Leeb-Lundberg et al., 2005). BK is enzymatically generated from kininogen precursors circulating in plasma and is also present in interstitial fluid. Over 30 years ago, BK was detected in knee synovial fluid of patients with knee osteoarthritis (OA) (Melmon et al., 1967; Eisen, 1970), and the possibility of its involvement in the pathophysiology of knee OA has been recently reviewed (Meini and Maggi, 2008). So far, the two main actions of BK that suggest B2 receptor blockade as a therapeutically relevant target for local treatment of OA are its algogenic effect by activating nociceptors which innervate the capsule and the synovium, and its inflammatory effects, actions that are involved in producing the pain and synovitis of knee OA respectively. Very recently a study conducted with the peptidic B2 receptor antagonist icatibant (previously known as HOE140, Hock et al., 1991) administered intra-articularly in patients with knee OA has been released (Song et al., 2009) and indicated that this antagonist is capable of producing an analgesic effect in OA patients.
In OA, synovitis appears involved and correlated to the other signs of this pathology, such as cartilage volume loss, bone oedema and pain (Nishimura et al., 2002; Bonnet and Walsh, 2005; Loeuille et al., 2005). Fibroblast-like synoviocytes in the synovium are mainly responsible for the homeostasis of extracellular matrix components and the synthesis of hyaluronan, an important constituent of synovial fluid (Pap and Gay, 2009).
Several studies reported the presence of kinin B2 receptors in human synoviocytes (reviewed in Meini and Maggi, 2008), and indicated BK as capable not only of activating intracellular calcium signalling, as a consequence of PLC activation, but also of evoking activation of phospholipase A2 (PLA2), as shown by the augmented release of arachidonic acid or its metabolites such as prostaglandin E2 (PGE2) after incubation of these cells with BK (Newcombe et al., 1977; Bathon et al., 1992a; Uhl et al., 1992).
The aim of the present study was to evaluate the pharmacological profile of the recently disclosed non-peptide B2 receptor antagonist, MEN16132, in comparison with the reference compound icatibant, and to explore new pro-inflammatory actions by BK in human synovial cells. We have previously shown that MEN16132 is a selective kinin B2 receptor antagonist in human and several animal species in vitro (Cucchi et al., 2005; Bellucci et al., 2007; Meini et al., 2007; 2009;). This compound also exhibited long lasting antagonist properties in different in vivo preclinical models (Valenti et al., 2005; 2008;).
To pharmacologically characterize the two antagonists, we carried out radioligand binding inhibition experiments, using tritiated BK to measure their affinity, whereas the antagonist potencies were evaluated in assays of the concentration-dependent accumulation of inositol phosphates (IP) induced by BK, as an index of PLC activation and an early step of receptor activation.
Cytokines and chemokines, such as interleukin (IL)-6 and IL-8, have been described as playing a relevant role in OA pathophysiology. They can be released by several cell types, such as osteoblasts, chondrocytes and synoviocytes by pro-inflammatory stimuli (Sanchez et al., 2002; Bendre et al., 2003; Wang et al., 2006; Sakao et al., 2008), and have been detected in the synovial fluid of patients with knee OA (Bianchi et al., 2006). IL-6 and IL-8 have been shown to be associated with inflammation, and to be involved in the processes of cartilage degradation, as well as being able to sensitize primary afferent neurons (Fernandes et al., 2002; White et al., 2000). In the present study, the ability of BK to release IL-6 and IL-8 has been evaluated and the potency of the two kinin B2 receptor antagonists to prevent that release tested. Moreover, the participation of cyclooxygenase (COX) and lipoxygenase (LOX) products in IL-6 and IL-8 production induced by BK has been assessed by means of appropriate inhibitors, and the mechanism of BK-induced B2 receptor dependent release of IL-8 investigated by using a panel of different intracellular inhibitors for different possible downstream signalling pathways.
Our results suggest that BK is likely to participate in mechanisms involving inflammation of the synovium and confirm the high potency of MEN16132 in blocking the responses to BK, both at early and late steps of B2 receptor activation.
Methods
Cell culture conditions
Human fibroblast-like synoviocytes derived from normal synovial tissue were purchased as frozen vials (catalog No. 408-05a: lots No. 1828 and 1798) from European Collection of Cell Cultures (ECACC, Salisbury, UK). Cells were handled as recommended by the producer: the seeding density for attachment was 7000 cells·cm−2 on plastic wells, and the culture medium was Synoviocyte Growth Medium (ECACC, catalog No. 06091516, CAI No. 415-500) supplemented with L-glutamine (Gln, 2 mM), penicillin (50 µg·mL−1), streptomycin (50 µg·mL−1) and amphotericin B (15 µg·mL−1). When cells reached 90% of confluency, they were split by using a subculture reagent kit according to the manufacturer's instructions (Lonza, catalog No. CC3233) and seeded in 75 cm2 flasks (7000 cells·cm−2) for cell culture maintenance, and in 12-well plates or 24-well plates (10–12 000 cells·cm−2) for binding or functional experiments respectively. Cells were used between two and 10 passages.
Radioligand binding
Binding assay was performed in Nutrient mixture F-12 Ham (F12, Sigma-Aldrich, N4888) medium containing BSA (0.1 wt·vol−1), NaN3 (0.1%) to prevent receptor internalization (Bathon et al., 1992b), and captopril (1 µM), bacitracin (140 µg·mL−1) and 1,10-phenanthroline (1 mM) (pH 7.4 at 4°C) to inhibit enzyme activities. In saturation experiments, cells were incubated with different [3H]-BK concentrations (100 pM–30 nM) in the absence (total binding) or presence of un-labelled BK (1 µM, to determine non-specific binding), whereas competition experiments were performed with 1 nM of [3H]-BK and varying concentrations of ligands. Subsequent experimental steps were as follows: the culture medium was aspirated and the binding medium was added to wells (800 µL), then the un-labelled ligand (10×) and the radioligand (10×) were added in sequence. BK, MEN16132 and icatibant were tested in a range of concentrations between 10 pM and 1 µM. After an incubation of 2 h on ice (4°C), the reaction was stopped by rapid aspiration of the medium and three consecutive washings (3 × 1 mL per well) with ice-cold binding buffer. Cells were lysed with 0.5 mL per well NaOH (0.3 M) for 30 min. The content of each well was transferred to ponyvials and 10 mL of Cytoscint (MP Biomedicals, code 0188245301) was added to each vial. Radioactivity was determined by a liquid β-scintillation counter (2200 CA, Packard).
Inositol phosphate (IP) accumulation
Cells at confluence were preincubated with myo-[1,2-3H(N)]inositol (1 µCi·mL−1) for 48 h in Dulbecco's modified Eagle's medium (Sigma-Aldrich, D5671) and F12 medium (1:1) supplemented with foetal bovine serum (FBS, 1%), penicillin (50 µg·mL−1), streptomycin (50 µg·mL−1), amphotericin B (15 µg·mL−1) and Gln (2 mM) (0.5 mL final volume). On the day of the experiments the medium was removed and the antagonist was added at the indicated concentrations in IP buffer (final volume 0.5 mL; IP buffer composition (mM): PBS Ca2+ and Mg2+ free (135), Na HEPES (20), CaCl22H2O (2), MgSO47H2O (1.2), EGTA (1), glucose (11.1), LiCl (25), added with BSA (0.05%), captopril (1 µM), thiorphan (10 µM), bacitracin (2 µg·mL−1). Peptidase inhibitors were included in order to prevent BK degradation (Bathon et al., 1992b). Preliminary experiments performed with both MEN16132 and icatibant indicated overlapping results by using an antagonist equilibration time of 15 or 60 min; therefore, a 15-min antagonist pre-incubation time was chosen. After the antagonist preincubation, the appropriate concentration of agonist was added and the assay mixture incubated for 60 min at 37°C. The reaction was stopped on ice, IP buffer was removed and cells were lysed with MeOH and HCl (1:1; 1 mL per well, 4°C). Total IP levels were determined as previously described (Bellucci et al., 2004).
Determination of IL-6, IL-8, PGE2 and leukotrienes
Cells were incubated for 24 h at 37°C with a range of concentrations of BK (1 nM–10 µM) in F12 medium supplemented with FBS (1%), penicillin (50 µg·mL−1), streptomycin (50 µg·mL−1), amphotericin B (0.75 µg·mL−1), Gln (2 mM) and captopril (1 µM) (0.8 mL final volume). Antagonists or inhibitors were pre-incubated for 30 min, before the addition of BK (1 µM), except for indomethacin, nordihydroguaiaretic acid (NDGA) and dexamethasone which were pre-incubated for 60 min. At the end of agonist incubation, the medium of stimulation was removed and stored at −80°C until assay.
The content of IL-6, IL-8, PGE2 and leukotrienes in the supernatant was assayed by commercially available enzyme immunoassay kits, according to the procedure described by the manufacturer, as follows (the sensitivity of each test is shown in parentheses): IL-6 (PK-EL-61606; 15.6–100 pg·mL−1) and IL-8 (PK-EL-61806; 15.6–1000 pg·mL−1) from Promokine (Heidelberg, Germany), PGE2 (514010; 7.8–1000 pg·mL−1) from Cayman (Ann Arbor, MI, USA), and leukotrienes C4, D4 and E4 (LTC4/D4/E4) (RPN224; 15.4–990 pg·mL−1) from Amersham (Buckinghamshire, UK).
Analysis of data
Each value in the text represents the mean ± SEM or the mean and 95% confidence limits (c.l.) in parentheses, of the reported number of independent experiments (n), each one performed in triplicate.
Binding data were fitted by the appropriate nonlinear regression using GraphPad Prism 4.0 (San Diego, CA, USA), in order to determine (i) the maximum binding site density (Bmax) and the equilibrium dissociation constant (Kd) from saturation experiments; and (ii) the ligand concentration inhibiting the radioligand binding of 50% (IC50) from competition experiments. Inhibitory affinity constant values (Ki), and their negative logarithm (pKi), were obtained by normalizing the obtained IC50 by the radioligand dissociation constant (Kd) and its concentration ([L*]), according to the Cheng-Prusoff relationship Ki= IC50/(1 +[L*]/Kd) (Cheng and Prusoff, 1973).
Functional data were fitted by sigmoidal nonlinear regression (GraphPad Prism 4.0) to determine the agonist concentration producing 50% (EC50) of the maximal response (Emax) from the concentration-response curves, and the antagonist concentration producing 50% inhibition (IC50) of the control agonist response.
In IP accumulation experiments the responses to BK either in the absence (control) or presence of antagonist were normalized towards the Emax of control BK. The antagonist affinity was expressed as apparent pKB calculated from the equation: pKB= log [CR – 1]– log [antagonist concentration], where CR is the ratio of equieffective concentrations (EC50) of BK in the presence and absence of antagonist (Kenakin, 1997).
The nature of the antagonist interaction was checked using Schild regression as follows: estimates of log [CR – 1] were plotted against log [antagonist concentration] (Arunlakshana and Schild, 1959). Data were fitted by linear regression and a slope value not significantly different from unity was considered indicative of competitive antagonism.
Data obtained from IL-6 and IL-8 release experiments are shown as pg·mL−1, or normalized towards the basal release or towards the BK control response, obtained in untreated cells (in the absence of agonist or inhibitor respectively) in each experimental session.
Statistical analysis was performed using GraphPad Prism 4.0 and GB-STAT (Dynamic Microsystems Inc., Silver Spring, MD, USA) and comparisons were made using one-way or two-way analysis of variance (anova), followed by Bonferroni's or Dunnett's post hoc tests,as indicated in the text.
Materials
[3H]-BK was from GE Healthcare (Europe GmbH, TRK943, specific activity 54 Ci·mmol−1) and PerkinElmer (Boston, MA, USA, NET706, specific activity 80 Ci·mmol−1), myo-[1,2-3H(N)]inositol was from PerkinElmer (NET906, specific activity 60 Ci·mmol−1). The kinin B2 receptor agonist BK was obtained from Neosystem (Strasbourg, France), the aminopeptidase inhibitor bestatin from Peninsula (Cheshire, UK), the neutral endopeptidase inhibitor thiorphan was from Bachem (Essex, UK), the cytokine tumour necrosis factor α (TNFα), the angiotensin converting enzyme inhibitor captopril, the protease inhibitor 1,10-phenantroline, the non-selective COX inhibitor indomethacin, the synthetic glucocorticoid dexamethasone, the NF-kB inhibitor BAY-117085, the PLC inhibitor U73122 and its inactive isomer U73343 were all from Sigma-Aldrich (Dorset, UK). The p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580 and the c-Jun N (JNK) terminal MAPK inhibitor SP600125 were from Tocris Bioscience (Ellisville, MO, USA). The ERK 1/2 MAPK inhibitor PD98059 and the phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 were purchased from Calbiochem (San Diego, CA, USA). The non-selective LOX inhibitor NDGA was from Cayman (Ann Arbor, MI, USA). All salts used were purchased from Merck (Darmstadt, Germany). Kinin B2 receptor antagonists were synthesized at Menarini Ricerche (Chemistry Departments of Florence and Pomezia, Italy). Icatibant (Hock et al., 1991; DArg[Hyp3,Thi5,DTic7,Oic8]BK) (batches 254-21, 30061-3 and 30058-2) and MEN16132 (4-(S)-Amino-5-(4-{4-[2,4-dichloro-3-(2,4-dimethyl-8-quinolyloxymethyl) phenylsulphonamido]-tetrahydro-2H-4-pyranylcarbonyl}piperazino)-5-oxopentyl](trimethyl)ammonium chloride hydrochloride) (batches 3, 6 and 7), were dissolved in distilled water at 10 mM and 1 mM concentrations (stock solutions) stored at −25°C. All other compounds were dissolved at 10 mM concentration (stock solutions) in water or DMSO as indicated by data sheets and stored at −25°C. Further working dilutions were made in the appropriate buffer.
Results
Affinity of BK, MEN16132 and icatibant in radioligand binding experiments
Saturation experiments with [3H]-BK were carried out with a range of radioligand concentrations (100 pM–30 nM): the calculated Kd value was 1.14 ± 0.06 nM and the Bmax was 121 550 ± 45 550 sites per cell (n= 3).
The affinity values of the B2 receptor antagonists MEN16132 and icatibant were evaluated by means of inhibition curves at the [3H]-BK binding sites (Figure 1). BK and the two antagonists concentration-dependently (10 pM–1 µM) inhibited the specific binding of [3H]-BK with IC50 values of 2.1 nM (1.6–2.6, 95% c.l.) for BK, 2.5 nM (2.0–3.2, 95% c.l.) for MEN16132 and 10.7 nM (8.4–13.6, 95% c.l.) for icatibant. The calculated inhibitory constant affinity values for the two antagonists, or pKi values are reported in Table 1 and indicated that MEN16132 was about threefold more potent than icatibant as an antagonist at the BK B2 receptor.
Figure 1.
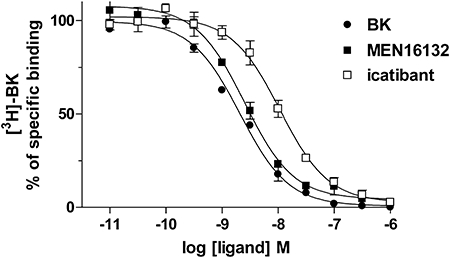
Bradykinin (BK), MEN16132 and icatibant inhibit [3H]-BK specific binding to human synoviocytes. Cells were incubated for 2 h at 4°C with [3H]-BK (1 nM) and varying concentrations of competing ligands as described in Methods. Data are expressed as mean ± SEM of three independent experiments, each one performed in triplicate.
Table 1.
Affinity (pKi) and antagonist potency (pKB, pIC50) values of MEN16132 and icatibant in human synoviocytes
|
[3H]-BK binding |
IP accumulation |
IL-6 release |
IL-8 release |
|||||
|---|---|---|---|---|---|---|---|---|
| pKi | n | pKB | n | pIC50 | n | pIC50 | n | |
| MEN16132 | 8.9 ± 0.02 | 3 | 9.9 ± 0.09 | 14 | 8.1 ± 0.33 | 5 | 8.4 ± 0.19 | 5 |
| Icatibant | 8.4 ± 0.20 | 3 | 8.1 ± 0.03 | 14 | 6.6 ± 0.35 | 5 | 6.7 ± 0.25 | 5 |
Experimental conditions for each assay are detailed in Methods. Data are expressed as the mean ± SEM of n independent experiments.
IL, interleukin.
BK activation of phospholipase C (IP accumulation assay) and antagonism by MEN16132 and icatibant
In the IP accumulation assay, BK induced a concentration-dependent response: the observed Emax was about 10-fold over the basal at 10 µM BK concentration, and the EC50 value was 0.45 nM (0.33–0.62, 95% c.l.).
Both MEN16132 (1 nM–1 µM) and icatibant (10 nM–10 µM) induced a concentration-dependent rightward shift of BK concentration-response curves (Figure 2A, B). The analysis of Schild regression indicated a competitive antagonism for both MEN16132 and icatibant (Figure 2C), and the slope values were not statistically different from unity: 1.096 (0.941–1.251, 95% c.l.) for MEN16132 and 1.118 (0.942–1.294, 95% c.l.) for icatibant. The apparent potency values calculated as pKB from single experiments are reported in Table 1, and indicate MEN16132 about 80-fold more potent than icatibant in this assay.
Figure 2.
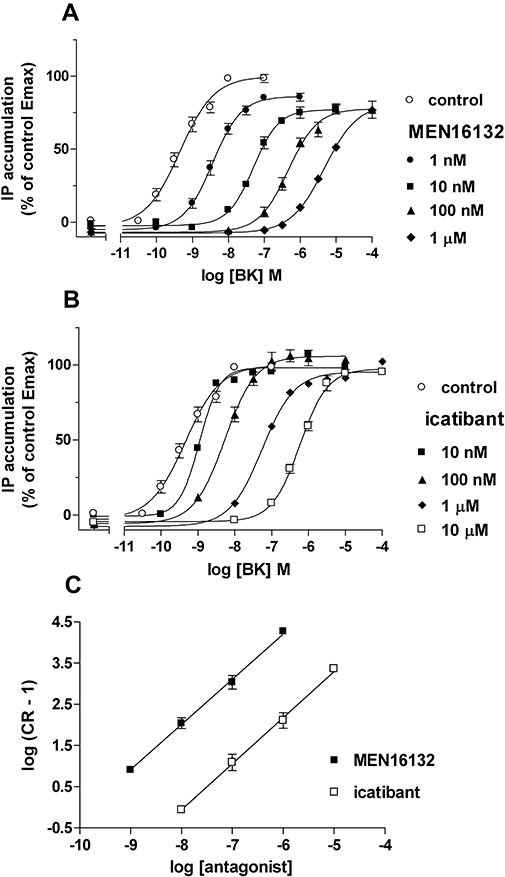
MEN16132 (A) and icatibant (B) antagonist activity towards BK-induced activation of IP production. Antagonists were added at the indicated concentrations 15 min before the agonist incubation (60 min). C: Schild analysis of data presented in panels A and B. Data are expressed as mean ± SEM of three to four independent experiments, each one performed in triplicate. IP, inositol phosphates.
Both antagonists did not modify the basal IP accumulation at any of the tested concentrations.
Long-term incubation of synovial cells with BK increases the release of IL-6 and IL-8
After 24 h of incubation, control synoviocytes produced basal IL-6 or IL-8 levels of 742 ± 68 pg·mL−1 (n= 32) or 1361 ± 97 pg·mL−1 (n= 52) respectively. BK (1 nM–10 µM, 24 h) stimulated the release of IL-6 and IL-8 in a concentration-dependent manner with EC50 values of 216 nM (with 95% confidence limits (c.l.),118–396) for IL-6 (Figure 3A) and 52.9 nM (25.8–108, 95% c.l.) for IL-8 (Figure 3B) respectively. The Emax observed at 10 µM BK concentration was about 3.5-fold over the basal for IL-6 and 2.5-fold over the basal for IL-8. Similar cytokine release into the cell supernatants were obtained by using 0.1 ng·mL−1 of the cytokine TNFα, which under the same experimental conditions stimulated the IL-6 release by 3.6 ± 0.5-fold and the IL-8 release by 3.4 ± 0.3-fold (n= 3) over the basal levels.
Figure 3.
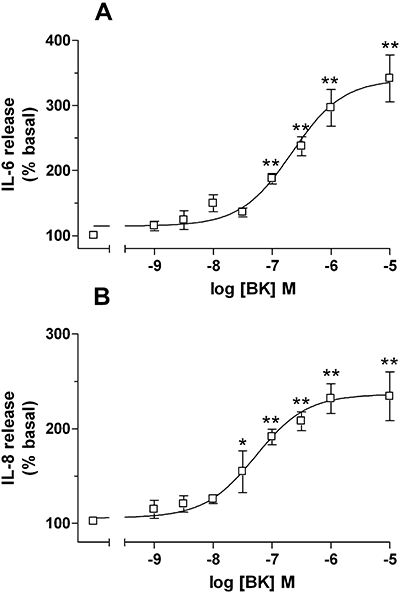
Bradykinin (BK)-induced release of IL-6 (A) and IL-8 (B) in human synoviocytes. Cells were incubated for 24 h with BK (1 nM–10 µM). Data (basal release = 100%) are expressed as the mean ± SEM of seven independent experiments, each one performed in triplicate. *P < 0.05, **P < 0.01 versus basal values, one-way anova followed by Dunnett's post hoc test. IL, interleukin.
MEN16132 and icatibant inhibit IL-6 and IL-8 release induced by BK
The relative potency of the two antagonists was evaluated by their blockade of the increased release of IL-6 and IL-8 induced by BK (1 µM). MEN16132 (0.1 nM–1 µM) and icatibant (1 nM–10 µM) induced a concentration-dependent inhibition of BK-induced IL-6 and IL-8 release (Figure 4A, B). The calculated pIC50 values are reported in Table 1, and indicated that MEN16132 was more potent than icatibant, by 30-fold or 50-fold, in inhibiting BK-induced release of IL-6 or IL-8 respectively.
Figure 4.
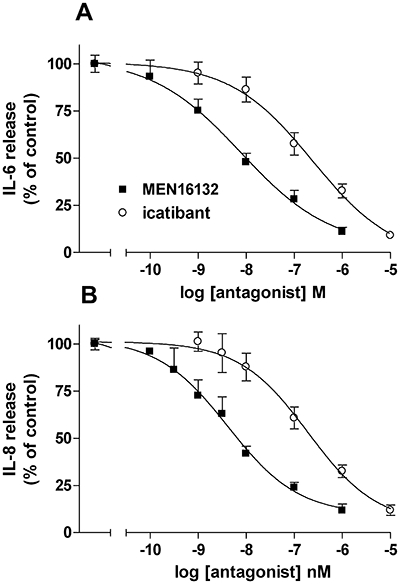
Inhibitory effect of MEN16132 and icatibant on bradykinin (BK)-induced IL-6 (A) or IL-8 (B) release. Human synoviocytes were preincubated for 30 min with the indicated concentrations of antagonists before BK (1 µM, 24 h) stimulation. Data are expressed as % of control response to BK (in the absence of antagonist), and represent the mean ± SEM of five independent experiments, each one performed in triplicate. IL, interleukin.
Both antagonists per se inhibited the basal IL-6 release only at the higher concentration: MEN16132 inhibited basal production of IL-6 by 43 ± 3% at 1 µM and icatibant by 45 ± 5% at 10 µM. On the contrary, basal IL-8 release was not modified by the two antagonists.
Lack of effect by COX and LOX inhibitors on the IL-6 and IL-8 release induced by BK
To evaluate the participation of COX and LOX products in the BK-induced release of IL-6 and IL-8, the non-selective COX inhibitor indomethacin and the non-selective LOX inhibitor NDGA were used. Both inhibitors were pre-incubated with the cells for 60 min before BK stimulation (1 µM, 24 h).
Although no significant differences could be detected through post hoc comparisons, indomethacin slightly inhibited both basal and BK-induced IL-6 release (two-way anova: F indomethacin = 8.62, d.f. 1, 32, P= 0.006), but the COX inhibitor did not prevent the stimulatory effect of BK, compared with controls (Figure 5A) (two-way anova:, F interaction = 0.6838, d.f. 1,32).
Figure 5.
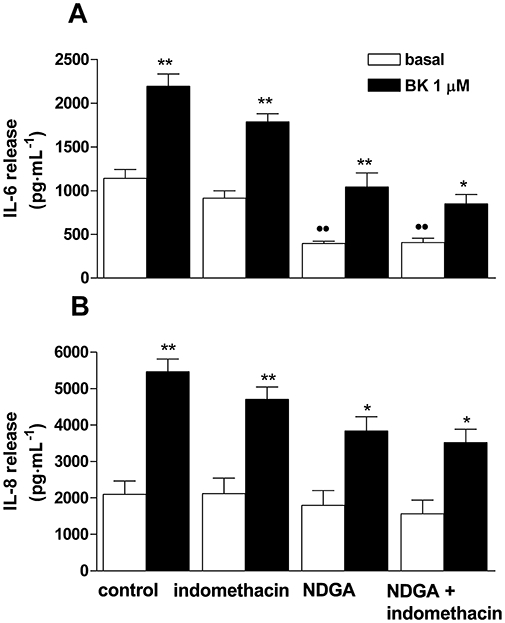
Effect of indomethacin and nordihydroguaiaretic acid (NDGA) on bradykinin (BK)-induced IL-6 (A) or IL-8 (B) release. Human synoviocytes were preincubated for 60 min with indomethacin (10 µM) or NDGA (10 µM), alone or in combination, before BK (1 µM, 24 h) stimulation. Data are expressed in pg·mL−1, and represent the mean ± SEM of three to five independent experiments, each one performed in triplicate. ••P < 0.01 versus control basal; *P < 0.05, **P < 0.01 versus the corresponding basal condition; two-way anova followed by Bonferroni's post hoc test. IL, interleukin.
On the other hand, indomethacin neither affected basal, nor BK-induced IL-8 release, and no antagonism was observed, compared with control experiments (Figure 5B) (two-way anova: F interaction = 1.107, d.f. 1,53).
When cells were treated with NDGA (10 µM) the basal release of IL-6 in the supernatants was significantly reduced (Figure 5A), being 34 ± 2% of the basal control, but no further reduction was observed when NDGA and indomethacin were administered together. BK induced a significant increase of IL-6 release both in cells treated with NDGA alone and in those treated with both inhibitors. As compared with control experiments, the stimulatory effect induced by BK on IL-6 release was not modified by either NDGA alone or both NDGA and indomethacin, as indicated by the non-significant effects on the BK response in both experimental conditions (F interaction = 2.864, d.f. 1,32, F interaction = 8.469, d.f. 1,32, for NDGA and NDGA/indomethacin groups respectively) (Figure 5A).
No differences in the basal release of IL-8 was observed with either the NDGA or the NDGA plus indomethacin treatment (Figure 5B), and BK stimulated IL-8 release in both experimental conditions (NDGA or NDGA+indomethacin) in a manner similar to control experiments (two-way anova: F interaction = 2.781, d.f. 1,45; F interaction = 3.255, d.f. 1,44 for NDGA and NDGA/indomethacin groups respectively) (Figure 5B).
In the same samples, the release of PGE2 was prevented by indomethacin (10 µM) both under basal and BK stimulated conditions: in the supernatants of cells pretreated with indomethacin the measured levels of PGE2 content were 18 ± 2 and 20 ± 2% of the control basal release (n= 5). On the contrary, no detectable levels of leukotrienes were measured in the same samples.
Effect of different inhibitors on the IL-8 release induced by BK
A panel of inhibitors towards different intracellular signalling mechanisms were used to identify the pathways involved in BK induced IL-8 release by synovial cells. All the inhibitors used here were pre-incubated 30 min before BK (1 µM) stimulation, except for dexamethasone which was pre-incubated for 60 min. The results from these inhibitor studies are shown in Table 2. The PLC inhibitor U73122 (3 µM) inhibited the BK induced IL-8 increase by 61%, whereas the inactive isomer U73343 (10 µM) did not affect it. The inhibitors for MAPKs p38 (SB203580, 10 µM), JNK (SP600125, 30 µM) and ERK 1/2 (PD98059, 30 µM) inhibited the control response by 40%, 38% and 33% respectively. Furthermore, the BK-induced IL-8 release was inhibited by the PI3K inhibitor (LY294002, 10 µM) by 43% and by the NF-κB inhibitor (BAY-117085, 5 µM) by 35%. Pretreatment with the glucocorticoid dexamethasone (100 nM) markedly inhibited the BK induced IL-8 release by 77%.
Table 2.
Effect of different inhibitors on the IL-8 release induced by BK in human synoviocytes
| Inhibitor | Time of pre-incubation |
IL-8 release (%) |
|
|---|---|---|---|
| BK (1 µM) | n | ||
| Control | – | 100 ± 7 | 4 |
| U73122 (3 µM) | 30 min | 39 ± 10a | 4 |
| U73343 (10 µM) | 30 min | 97 ± 6 | 4 |
| PD98059 (30 µM) | 30 min | 67 ± 2a | 4 |
| SP600125 (30 µM) | 30 min | 62 ± 2a | 4 |
| SB203580 (10 µM) | 30 min | 60 ± 5a | 4 |
| LY294002 (10 µM) | 30 min | 57 ± 9a | 4 |
| BAY-117085 (5 µM) | 30 min | 65 ± 2a | 4 |
| Dexamethasone (100 nM) | 60 min | 23 ± 2a | 3 |
Cells were preincubated with each inhibitor, at the concentration and for the time reported in the table, and then exposed to BK (1 µM, 24 h).
Data are expressed as the mean ± SEM of n independent experiments, each one performed in triplicate.
P < 0.001, one-way anova followed by Bonferroni's post hoc test.
BK, bradykinin; IL, interleukin.
Neither greater concentrations of single inhibitors nor their combined effect could be investigated because they produced toxic effects on the cells under the present experimental conditions.
Discussion and conclusions
In the present study we show the pharmacological profile of BK and the two antagonist compounds, icatibant and MEN16132, in human synovial cells by using different assays. In addition, the current study highlights for the first time the capability of BK to induce IL-8 production by synoviocytes, and investigates the mechanisms involved.
As mentioned in the Introduction, the expression of BK B2 receptors, detected by means of radioligand binding using tritiated BK, had already been reported in human synovial cells (Bathon et al., 1992a; Uhl et al., 1992). Under the present experimental conditions, we measured an abundant presence of receptors (Bmax 121 550 sites per cell) which matched the results from previous reports (82 500 sites per cell, Uhl et al., 1992). Also, the measured Kd value of BK (Kd 1.1 nM, Ki 0.9 nM) agreed with previous published results (Kd 2.9 nM, Uhl et al., 1992; Kd 2.3 nM, Bathon et al., 1992a). Lastly, data obtained from inhibition curves indicated MEN16132 as potent as BK and slightly (threefold) more potent than icatibant. From these experiments the nM affinity of icatibant (Ki 4.6 nM) is in agreement with that previously reported (IC50 3 nM, Bathon et al., 1992a).
Although icatibant was shown to inhibit BK-induced intracellular calcium release (IC50 10 nM) or PGE2 production (IC50 20 nM) in human synoviocytes (Bathon et al., 1992a; 1994;), a complete characterization of its antagonist properties in this cell system has not been reported thus far. Here, we first describe the quantitative pharmacology of BK in activating PLC, and the competitive antagonist mechanism of action of the two tested antagonists, as indicated by the Schild analysis (slope not different from unity), which implies a concentration-dependent shift of the whole BK concentration-response curve. The antagonist potency values, as well as the competitive antagonism type, of icatibant and MEN16132 detected in the IP accumulation assay (pKB values 8.1 and 10.0, respectively, present results) are close to earlier results obtained under analogous experimental conditions in a recombinant cell system expressing the human B2 receptor (icatibant pKB 8.5, Bellucci et al., 2004; MEN16132 pKB 10.3, Cucchi et al. (2005), or in the smooth muscle contractility of the human detrusor preparation (icatibant pKB 8.4, Meini et al., 2000; MEN16132 pKB 9.9, Cucchi et al., 2005).
Bradykinin is known to induce both IL-6 and IL-8 release in different human cell types, such as smooth muscle (Pang and Knox, 1998; Huang et al., 2003; Zhu et al., 2003), epithelial cells (Koyama et al., 1998; Rodgers et al., 2002), fibroblasts (Hayashi et al., 1998; Hayashi et al., 2000; Koyama et al., 2000) and osteoblasts (Rahman et al., 1992; Kondo and Togari, 2004). Very recently, BK was reported to release IL-6 release from human synoviocytes isolated from patients with rheumatoid arthritis (RA) (Lee et al., 2008). In the present study we show that BK, after a long-term incubation (24 h, as in the papers quoted above) was able to increase the release of IL-6 and IL-8 from human synoviocytes, in a concentration-dependent manner and with comparable potency values. It is worth pointing out that this release produced by BK occurs over the concentration range of endogenous BK levels detected in the synovial fluid of OA and RA patients (ranging between 98 and 427 ng·mL−1, corresponding to 1 to 4 × 10−7 M, Bond et al., 1997). Furthermore, this IL-6 and IL-8 release induced by BK was comparable to that produced by the pleiotropic cytokine TNFα at a concentration (0.1 ng·mL−1) which has been detected in the OA and RA synovial fluid (0.08–0.17 ng·mL−1, Kahle et al., 1992). The present results also showed that both B2 receptor antagonists are capable of preventing the BK-induced release of IL-6 and IL-8, and that MEN16132 was 30- to 50-fold more potent than icatibant.
The reasons for discrepancy between the greater antagonist potency of MEN16132 relative to icatibant in all the assays used here, compared with the much closer potencies when the ligand affinity is measured at the [3H]-BK binding site, have been previously discussed at length (Meini et al., 2004; 2009; Cucchi et al., 2005), and are possibly related to the different modes of interaction, shown by structurally dissimilar antagonist compounds, with the receptor.
The participation of prostanoids in the production of IL-6 and IL-8 induced by BK appears to be dependent on the cell system studied. In human airway smooth muscle cells, the production of IL-8 was totally prevented by the non-selective COX inhibitor indomethacin, and was due to an increase of the inducible COX-2 enzyme caused by BK (Pang and Knox, 1998; Zhu et al., 2003). In type II alveolar cells, indomethacin could reduce the BK induced IL-8 release only by 30% (Rodgers et al., 2002), thus suggesting that different mechanisms were involved. On the other hand, in human airway smooth muscle cells a different mechanism accounted for BK induced release of IL-6 which could be inhibited only in part by treatment with the COX inhibitor (Huang et al., 2003). In the current investigation we present evidence that, in human synoviocytes, the BK-induced release of both IL-6 and IL-8 was not affected at all by indomethacin, although it did prevent the PGE2 production in the same samples, thus excluding prostanoids as mediators of the response to BK in this cell system.
Moreover, because of the role of leukotrienes in inducing chemotactic responses, the effects of the non-selective LOX inhibitor NDGA, alone or together with indomethacin, were evaluated. Our finding that NDGA treatment, both alone or co-administered with indomethacin, reduced the basal release of IL-6 and IL-8 indicated that LOX products may per se contribute to the release of these cytokines. On the other hand, the ability of BK to induce release of cytokines irrespective of the inhibition exerted by NDGA, suggests that even LOX products were not the major mediators of this effect of BK. The fact that we could not detect, under the investigated experimental conditions, any leukotriene release, agrees with previously reported inability of fibroblast-like synovial cells, and fibroblasts in general, to produce leukotrienes (or products of the 5-LOX) but rather 12-hydroxy-eicosatetraenoic acid compounds (or products of the 12-LOX) (Zurier, 2009), which in turn might be involved in the release of cytokines. Clarification of this aspect will necessitate further studies.
The mechanism of BK-induced IL-6 release, via B2 receptors in synovial cells was recently investigated, and shown to be dependent on PLC and also NF-κB signalling, in turn responsible for the increased IL-6 expression (Lee et al., 2008). In the current investigation, we present pharmacological evidence that PLC and NF-κB activation were also involved in the BK-mediated release of IL-8, as suggested by the partial inhibition observed in cells pretreated with the selective PLC inhibitor U73122 (but not its inactive isoform U73343) or the NF-κB inhibitor BAY-117085. The participation of NF-κB in the mechanism leading to release of IL-8 by BK was previously demonstrated in human airway smooth muscle cells, although, contrary to what we found in synoviocytes, the release also involved COX products, as the release was totally inhibited by indomethacin (see above, Zhu et al., 2003). Furthermore, in a recombinant system, NF-κB activation by BK can be mediated through a PLC-PI3K dependent pathway (Xie et al., 2000), which would be compatible with the inhibitory effect of the PI3K inhibitor LY294002 in the present study. In addition, BK B2 receptor activation has been linked to the activation of several kinase cascades, such as the ERK 1/2, p38 and JNK MAPK in the BK-stimulated release of IL-8 from human airway fibroblasts and smooth muscle cells (Hayashi et al., 2000; Huang et al., 2003) or in the synergistic induction of COX-2 by BK and IL-1 in human osteoblasts (Brechter and Lerner, 2007). Our data with pharmacological inhibitors of different intracellular kinases suggest that these MAPKs can all participate, although only partly, in the release of IL-8 induced by BK in synoviocytes. Lastly, consistent with previous reports of human airway smooth muscle cells, we found that dexamethasone pretreatment blocked the IL-8 release induced by BK. This effect has been ascribed, at least in part, to an inhibitory mechanism on different transcription factors such as NF-κB (Pang and Knox, 1998; Zhu et al. 2003).
As a whole, our findings suggest that BK and B2 receptors are involved in the modulation of mechanisms participating in inflammatory synovitis, and that MEN16132 is a high affinity receptor antagonist capable of blocking responses to BK produced in human fibroblast-like synoviocytes.
Acknowledgments
Portions of this work were previously presented at the 2008 OARSI Congress on Osteoarthritis, 18–21 September Rome, Italy. The authors thank Dr Alessandro Lecci for his advice on statistical analysis, and Ms Lisa Mylander for the revision of English language.
Glossary
Abbreviations:
- BK
bradykinin
- COX
cyclooxygenase
- LOX
lipoxygenase
- NDGA
nordihydroguaiaretic acid
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathon JM, Manning DC, Goldman DW, Towns MC, Proud D. Characterization of kinin receptors on human synovial cells and upregulation of receptor number by interleukin-1. J Pharmacol Exp Ther. 1992a;260:384–392. [PubMed] [Google Scholar]
- Bathon JM, Proud D, Mizutani S, Ward PE. Cultured human synovial fibroblasts rapidly metabolize kinins and neuropeptides. J Clin Invest. 1992b;90:981–991. doi: 10.1172/JCI115975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathon JM, Croghan JC, MacGlashan DW, Jr, Proud D. Bradykinin is a potent and relatively selective stimulus for cytosolic calcium elevation in human synovial cells. J Immunol. 1994;153:2600–2608. [PubMed] [Google Scholar]
- Bellucci F, Meini S, Cucchi P, Catalani C, Giuliani S, Zappitelli S, et al. The N-terminal of icatibant and bradykinin interact with the same Asp residues in the human B2 receptor. Eur J Pharmacol. 2004;491:121–125. doi: 10.1016/j.ejphar.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Bellucci F, Cucchi P, Santicioli P, Lazzeri M, Turini D, Meini S. Characterization of kinin receptors in human cultured detrusor smooth muscle cells. Br J Pharmacol. 2007;150:192–199. doi: 10.1038/sj.bjp.0706976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003;33:28–37. doi: 10.1016/s8756-3282(03)00086-3. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Broggini M, Balzarini P, Franchi S, Sacerdote P. Effects of nimesulide on pain and on synovial fluid concentrations of substance P interleukin-6 and interleukin-8 in patients with knee osteoarthritis: comparison with celecoxib. Int J Clin Pract. 2006;61:1270–1277. doi: 10.1111/j.1742-1241.2007.01453.x. [DOI] [PubMed] [Google Scholar]
- Bond AP, Lemon M, Dieppe PA, Bhoola KD. Generation of kinins in synovial fluid from patients with arthropathy. Immunopharmacol. 1997;36:209–216. doi: 10.1016/s0162-3109(97)00023-4. [DOI] [PubMed] [Google Scholar]
- Bonnet CS, Walsh DA. Osteoarthritis, angiogenesis and inflammation. Rheumatology. 2005;44:7–16. doi: 10.1093/rheumatology/keh344. [DOI] [PubMed] [Google Scholar]
- Brechter AB, Lerner UH. Bradykinin potentiates cytokine-induced prostaglandin biosynthesis in osteoblasts by enhanced expression of cyclooxygenase 2, resulting in increased RANKL expression. Arthritis Rheum. 2007;56:910–923. doi: 10.1002/art.22445. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cucchi P, Meini S, Bressan A, Catalani C, Bellucci F, Santicioli P, et al. MEN16132, a novel potent and selective nonpeptide antagonist for the human bradykinin B2 receptor. In vitro pharmacology and molecular characterization. Eur J Pharmacol. 2005;528:7–16. doi: 10.1016/j.ejphar.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Eisen V. Plasma kinins in synovial exudates. Br J Exp Pathol. 1970;51:322–327. [PMC free article] [PubMed] [Google Scholar]
- Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39:237–246. [PubMed] [Google Scholar]
- Hayashi R, Yamashita N, Matsui S, Maruyama M, Sugiyama E, Sugiyama S, et al. Bradykinin stimulates interleukin-8 production by human lung fibroblasts. Immunology. 1998;95:507–511. doi: 10.1046/j.1365-2567.1998.00649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, Yamashita N, Matsui S, Fujita T, Araya J, Sassa K, et al. Bradykinin stimulates IL-6 and IL-8 production by human lung fibroblasts through ERK- and p38 MAPK-dependent mechanisms (2000) Eur Respir J. 2000;16:452–458. doi: 10.1034/j.1399-3003.2000.016003452.x. [DOI] [PubMed] [Google Scholar]
- Hock FJ, Wirth K, Albus U, Linz W, Gerhards HJ, Wiemer G, et al. Hoe 140 a new potent and long acting bradykinin-antagonist: in vitro studies. Br J Pharmacol. 1991;102:769–773. doi: 10.1111/j.1476-5381.1991.tb12248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CD, Tliba O, Panettieri RA, Jr, Amrani Y. Bradykinin induces interleukin-6 production in human airway smooth muscle cells: modulation by Th2 cytokines and dexamethasone. Am J Respir Cell Mol Biol. 2003;28:330–338. doi: 10.1165/rcmb.2002-0040OC. [DOI] [PubMed] [Google Scholar]
- Kahle P, Saal JG, Schaudt K, Zacher J, Fritz P, Pawelec G. Determination of cytokines in synovial fluids: correlation with diagnosis and histomorphological characteristics of synovial tissue. Ann Rheum Dis. 1992;51:731–734. doi: 10.1136/ard.51.6.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin TP. Competitive antagonism. In: Kenakin TP, editor. Pharmacologic Analysis of Drug-Receptor Interaction. 3rd. Philadelphia, PA: Publishers Press; 1997. pp. 331–373. Lippincott-Raven. [Google Scholar]
- Kondo A, Togari A. Activation of osteoblastic functions by a mediator of pain, bradykinin. Biochem Pharmacol. 2004;68:1423–1431. doi: 10.1016/j.bcp.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Koyama S, Sato E, Nomura H, Kubo K, Miura M, Yamashita T, et al. Bradykinin stimulates type II alveolar cells to release neutrophil and monocyte chemotactic activity and inflammatory cytokines. Am J Pathol. 1998;153:1885–1893. doi: 10.1016/S0002-9440(10)65702-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Sato E, Numanami H, Kubo K, Nagai S, Izumi T. Bradykinin stimulates lung fibroblasts to release neutrophil and monocyte chemotactic activity. Am J Respir Cell Mol Biol. 2000;22:75–84. doi: 10.1165/ajrcmb.22.1.3752. [DOI] [PubMed] [Google Scholar]
- Lee CH, Shieh DC, Tzeng CY, Chen CP, Wang SP, Chiu YC, et al. Bradykinin-induced IL-6 expression through bradykinin B2 receptor, phospholipase C, protein kinase Cdelta and NF-kappaB pathway in human synovial fibroblasts. Mol Immunol. 2008;45:3693–3702. doi: 10.1016/j.molimm.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Leeb-Lundberg LM, Marceau F, Muller-Ester W, Pettibone DJ, Zuraw BL. International Union of Pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- Loeuille D, Chary-Valckenaere I, Champigneulle J, Rat AC, Toussaint F, Pinzano-Watrin A, et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005;52:3492–3501. doi: 10.1002/art.21373. [DOI] [PubMed] [Google Scholar]
- Meini S, Maggi CA. Knee osteoarthritis: a role for bradykinin? Inflamm Res. 2008;57:351–361. doi: 10.1007/s00011-007-7204-1. [DOI] [PubMed] [Google Scholar]
- Meini S, Patacchini R, Giuliani S, Lazzeri M, Turini D, Maggi CA, et al. Characterization of bradykinin B2 receptor antagonists in human and rat urinary bladder. Eur J Pharmacol. 2000;388:177–182. doi: 10.1016/s0014-2999(99)00882-1. [DOI] [PubMed] [Google Scholar]
- Meini S, Cucchi P, Bellucci F, Catalani C, Faiella A, Rotondaro L, et al. Site-directed mutagenesis at the human B2 receptor and molecular modelling to define the pharmacophore of non-peptide bradykinin receptor antagonists. Biochem Pharmacol. 2004;67:601–609. doi: 10.1016/j.bcp.2003.09.034. [DOI] [PubMed] [Google Scholar]
- Meini S, Cucchi P, Bellucci F, Catalani C, Giuliani S, Santicioli P, et al. Comparative antagonist pharmacology at the native mouse bradykinin B2 receptor: radioligand binding and smooth muscle contractility studies. Br J Pharmacol. 2007;150:313–320. doi: 10.1038/sj.bjp.0706995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meini S, Cucchi P, Catalani C, Bellucci F, Giuliani S, Santicioli P, et al. Pharmacological characterization of the bradykinin B2 receptor antagonist MEN16132 in rat in vitro bioassays. Eur J Pharmacol. 2009;615:10–16. doi: 10.1016/j.ejphar.2009.04.057. [DOI] [PubMed] [Google Scholar]
- Melmon KL, Webster ME, Goldfinger SE, Seegmiller JE. The presence of a kinin in inflammatory synovial effusion from arthritides of varying etiologies. Arthritis Rheum. 1967;10:13–20. doi: 10.1002/art.1780100103. [DOI] [PubMed] [Google Scholar]
- Newcombe DS, Fahey JV, Ishikawa Y. Hydrocortisone inhibition of the bradykinin activation of human synovial fibroblasts. Prostaglandins. 1977;13:235–244. doi: 10.1016/0090-6980(77)90005-3. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Segami N, Kaneyama K, Suzuki T, Miyamaru M. Relationships between pain-related mediators and both synovitis and joint pain in patients with internal derangements and osteoarthritis of the temporomandibular joint. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:328–332. doi: 10.1067/moe.2002.124106. [DOI] [PubMed] [Google Scholar]
- Pang L, Knox AJ. Bradykinin stimulates IL-8 production in cultured human airway smooth muscle cells: role of cyclooxygenase products. J Immunol. 1998;161:2509–2515. [PubMed] [Google Scholar]
- Pap T, Gay S. Fibroblasts and fibroblast-like synoviocytes. In: Firestein GS, Budd RC, Harris ED Jr, Mclnnes IB, Ruddy S, Sergent JS, editors. Kelley's Textbook of Rheumatology. 8th. Philadelphia, PA: Saunders Elsevier; 2009. pp. 201–214. [Google Scholar]
- Rahman S, Bunning RA, Dobson PRM, Evans DB, Chapman K, Jones TH, et al. Bradykinin stimulates the production of prostaglandin E2 and interleukin-6 in human osteoblast-like cells. Biochim Biophys Acta. 1992;1135:97–102. doi: 10.1016/0167-4889(92)90172-8. [DOI] [PubMed] [Google Scholar]
- Rodgers HC, Pang L, Holland E, Corbett L, Range S, Knox AJ. Bradykinin increases IL-8 generation in airway epithelial cells via COX-2-derived prostanoids. Am J Physiol Lung Cell Mol Physiol. 2002;283 doi: 10.1152/ajplung.00483.2001. L612-L618. [DOI] [PubMed] [Google Scholar]
- Sakao K, Takahashi KA, Mazda O, Arai Y, Tonomura H, Inoue A, et al. Enhanced expression of interleukin-6, matrix metalloproteinase-13, and receptor activator of NF-kappaB ligand in cells derived from osteoarthritic subchondral bone. J Orthop Sci. 2008;13:202–210. doi: 10.1007/s00776-008-1227-5. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Mateus MM, Defresne MP, Crielaard JM, Reginster JY, Henrotin YE. Metabolism of human articular chondrocytes cultured in alginate beads. Longterm effects of interleukin 1beta and nonsteroidal antiinflammatory drugs. J Rheumatol. 2002;29:772–782. [PubMed] [Google Scholar]
- Song IH, Althoff CE, Hermann KG, Scheel AK, Knetsch T, Burmester GR, et al. Contrast-enhanced ultrasound in monitoring the efficacy of a bradykinin receptor-2 antagonist in painful knee osteoarthritis compared to magnetic resonance imaging. Ann Rheum Dis. 2009;68:75–83. doi: 10.1136/ard.2007.080382. [DOI] [PubMed] [Google Scholar]
- Uhl J, Singh S, Brophy L, Faunce D, Sawutz DG. Role of bradykinin in inflammatory arthritis: identification and functional analysis of bradykinin receptors on human synovial fibroblasts. Immunopharmacology. 1992;23:131–138. doi: 10.1016/0162-3109(92)90037-d. [DOI] [PubMed] [Google Scholar]
- Valenti C, Cialdai C, Giuliani S, Lecci A, Tramontana M, Meini S, et al. MEN16132, a novel potent and selective nonpeptide kinin B2 receptor antagonist: in vivo activity on bradykinin-induced bronchoconstriction and nasal mucosa microvascular leakage in anesthetized guinea pigs. J Pharmacol Exp Ther. 2005;315:616–623. doi: 10.1124/jpet.105.088252. [DOI] [PubMed] [Google Scholar]
- Valenti C, Cialdai C, Giuliani S, Tramontana M, Quartara L, Maggi CA. MEN16132, a kinin B2 receptor antagonist, prevents the endogenous bradykinin effects in guinea-pig airways. Eur J Pharmacol. 2008;579:350–356. doi: 10.1016/j.ejphar.2007.10.025. [DOI] [PubMed] [Google Scholar]
- Wang CT, Lin YT, Chiang BL, Lin YH, Hou SM. High molecular weight hyaluronic acid down-regulates the gene expression of osteoarthritis-associated cytokines and enzymes in fibroblast-like synoviocytes from patients with early osteoarthritis. Osteoarthritis Cartilage. 2006;14:1237–1247. doi: 10.1016/j.joca.2006.05.009. [DOI] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci USA. 2000;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Browing DD, Hay N, Mackman N, Ye RD. Activation of NF-κB by bradykinin through a Gαq- and Gβγ-dependent pathway that involves phosphoinositide 3-kinase and Akt. J Biol Chem. 2000;275:24907–24914. doi: 10.1074/jbc.M001051200. [DOI] [PubMed] [Google Scholar]
- Zhu YM, Bradbury DA, Pang L, Knox AJ. Transcriptional regulation of interleukin (IL)-8 by bradykinin in human airway smooth muscle cells involves prostanoid-dependent activation of AP-1 and nuclear factor (NF)-IL-6 and prostanoid-idependent activation of NF-κB. J Biol Chem. 2003;278:29366–29375. doi: 10.1074/jbc.M301785200. [DOI] [PubMed] [Google Scholar]
- Zurier RB. Prostaglandins, leukotrienes, and related compounds. In: Firestein GS, Budd RC, Harris ED Jr, Mclnnes IB, Ruddy S, Sergent JS, editors. Kelley's Textbook of Rheumatology. 8th. Philadelphia, PA: Saunders Elsevier; 2009. pp. 343–377. [Google Scholar]