

Abstract
The development of renewable alternatives to diesel and jet fuels is highly desirable for the heavy transportation sector, and would offer benefits over the production and use of short-chain alcohols for personal transportation. Here, we report the development of a metabolically engineered strain of Escherichia coli that overproduces medium-chain length fatty acids via three basic modifications: elimination of β-oxidation, overexpression of the four subunits of acetyl-CoA carboxylase, and expression of a plant acyl–acyl carrier protein (ACP) thioesterase from Umbellularia californica (BTE). The expression level of BTE was optimized by comparing fatty acid production from strains harboring BTE on plasmids with four different copy numbers. Expression of BTE from low copy number plasmids resulted in the highest fatty acid production. Up to a seven-fold increase in total fatty acid production was observed in engineered strains over a negative control strain (lacking β-oxidation), with a composition dominated by C12 and C14 saturated and unsaturated fatty acids. Next, a strategy for producing undecane via a combination of biotechnology and heterogeneous catalysis is demonstrated. Fatty acids were extracted from a culture of an overproducing strain into an alkane phase and fed to a Pd/C plug flow reactor, where the extracted fatty acids were decarboxylated into saturated alkanes. The result is an enriched alkane stream that can be recycled for continuous extractions. Complete conversion of C12 fatty acids extracted from culture to alkanes has been demonstrated yielding a concentration of 0.44 g L−1 (culture volume) undecane.
Keywords: metabolic engineering, Escherichia coli, fatty acid, alkane, biofuel, diesel
Introduction
The production of transportation fuels from renewable sources is necessary to meet continuing demand in the face of dwindling petroleum supplies while also curbing the release of greenhouse gases. Although much focus has been placed on developing biomass-derived gasoline alternatives such as ethanol and other short-chain alcohols (Atsumi et al., 2008; Houghton et al., US Department of Energy SC-0095, 2006), higher energy density distillates such as diesel and jet fuel are needed by the heavy transportation sector owing to weight and range constraints. Diesel and jet fuel currently account for more than half of the world refinery output destined for use in vehicles (US Department of Energy ORNL-6984, ). Furthermore, diesel engines improve fuel efficiency in small passenger vehicles over gasoline engines, and have already been widely adopted in Europe. Medium- and long-chain hydrocarbons can potentially serve as replacements for diesel, rendering them an attractive target for microbial production from lignocellulosic feedstocks. Unlike ethanol, the low water solubility of hydrocarbons would result in low recovery costs and reduced toxicity in microbial fermentations. Hydrocarbons would also be compatible with existing transport and storage infrastructure and vehicle engines.
Microbially synthesized fatty acids are logical precursors to diesel-like hydrocarbons, and offer the flexibility to exploit a variety of biomass-derived carbon sources. Fatty acid biosynthesis (Fig. 1) is initiated by the conversion of acetyl-coenzyme A (CoA) to malonyl-CoA by acetyl-CoA carboxylase (ACC). Fatty acid biosynthesis is highly regulated at this initial rate-limiting step (Davis et al., 2000; Li and Cronan, 1993). From malonyl-CoA, the condensing intermediate malonyl-acyl carrier protein (ACP), and ultimately the initial species that enters the elongation cycle, acetoacetyl-ACP, are generated. The elongation cycle fully reduces the β-ketoacyl-ACP to an acyl-ACP, which is then condensed with malonyl-ACP to lengthen the fatty acid molecule. Long chain length acyl-ACPs are substrates for two acyltransferases, PlsB and PlsC, as part of phospholipid biosynthesis. The accumulation of acyl-ACPs feedback inhibits multiple enzymes in fatty acid biosynthesis, representing one mode of coordination of lipid production to the growth rate (Davis and Cronan, 2001; Heath and Rock, 1996). The most dramatic examples of decoupling fatty acid biosynthesis from normal modes of regulation come from the heterologous expression of plant acyl-ACP thioesterases. Although several have been expressed in Escherichia coli (Ghosh et al., 2007; Jha et al., 2006; Serrano-Vega et al., 2005; Yuan et al., 1995), the FatB-type medium chain acyl-ACP thioesterase from Umbellularia californica demonstrated the largest increase in total fatty acid production, with a chain length distribution heavily skewed toward C12 and C14 species (Voelker and Davies, 1994). One metabolic engineering strategy for overproducing free fatty acids in E. coli utilizing the acyl-ACP thioesterase from Cinnamomum camphorum and a cytosolic form of thioesterase I (TesA) from E. coli (Cho and Cronan, 1993) has been reported (Lu et al., 2008).
Figure 1.

Fatty acid biosynthesis pathway in Escherichia coli and metabolic engineering strategy for overproduction of free fatty acids. Acetyl-CoA is converted to malonyl-CoA by the four subunits of acetyl-CoA carboxylase (AccABCD). Malonyl-CoA is converted to malonyl-ACP (acyl–acyl carrier protein) by malonyl-CoA:ACP transacylase (FabD), and to the acetoacetyl-ACP, the first β-ketoacyl-ACP in the fatty acid elongation cycle, by multiple pathways catalyzed by FabD, β-ketoacyl-ACP synthase III (FabH), and β-ketoacyl-ACP synthase I (FabB). The ketoacyl-ACP is reduced twice and dehydrated once to yield an acyl-ACP in the elongation cycle by β-ketoacyl-ACP reductase (FabG), enoyl-ACP reductase (FabI), and β-hydroxyacyl-ACP dehydratase (FabZ). The acyl-ACP is then condensed with malonyl-ACP by FabB or β-ketoacyl-ACP synthase II (FabF). Glycerol-3-phosphate acyltransferase (PlsB) and 1-acylglycerol-3-phosphate acyltransferase (PlsC) utilize C16 to C18 acyl-ACPs as substrates for phospholipid biosynthesis. Acyl-ACP thioesterases hydrolyze acyl-ACPs to yield free fatty acids. Free fatty acids can be degraded, ultimately to acetyl-CoA, by enzymes of β-oxidation. This process is initiated by acyl-CoA synthetase (FadD), which converts free fatty acids to acyl-CoAs. Bolded arrows represent reactions enhanced by overexpression (ACC) or heterologous expression (BTE) of indicated enzymes. X represents a disruption of the indicated pathway by gene deletion.
In this study, we sought to further understand previously reported instabilities of plasmids expressing the U. californica acyl-ACP thioesterase (Voelker and Davies, 1994) by varying the copy number of a codon-optimized version of the gene (BTE) on plasmids having different origins of replication, in an effort to balance the level of active protein with possible metabolic burden and product toxicity (Jones et al., 2000). Previous studies expressing acyl-ACP thioesterases in E. coli have been conducted at 30°C owing to this putative instability (Feng and Cronan, 2009; Lu et al., 2008). However, no additional studies have been conducted to determine any rationale for this instability. With this information, E. coli was engineered to overproduce free fatty acids via three modifications: (1) expression of a codon-optimized acyl-ACP thioesterase from U. californica (BTE) on a suitable plasmid; (2) deletion of the fadD gene encoding acetyl-CoA synthetase, the first enzyme involved in β-oxidation; and (3) over-expression of ACC.
Fatty acids can be converted to useful liquid fuels by chemical catalytic or enzymatic esterification (Fjerbaek et al., 2008; Kalscheuer et al., 2006; Vasudevan and Briggs, 2008), putative enzymatic decarboxylation (Banerjee et al., 2002), and catalytic decarboxylation (Mäki-Arvela et al., 2007). Other strategies utilizing intermediates in fatty acid biosynthesis to enzymatically produce fatty alcohols and olefins have been reported in the patent literature but have not been reported in peer-reviewed journals (Keasling et al., patent application WO/2007/136762; Friedman and Rude, patent application WO 2008/113041). Here, the overproduced free fatty acids were extracted from culture and catalytically decarboxylated to alkanes. Coupling the microbial fermentation to a scalable existing chemical technology precludes the need to heterologously express a new in vivo pathway to convert the fatty acids to a useful liquid product, which even if properly balanced could reduce the overall yield of fatty acids owing to increased cellular metabolic burdens. Decarboxylation is a preferable conversion because alkanes have more desirable properties, such as higher energy density and lower viscosity, than corresponding esters that would be produced by catalytic esterification of fatty acids. Furthermore, the final medium chain alkane product can be recycled for reuse as an extractant of fatty acids from the culture medium and as the solvent for the catalytic decarboxylation. E. coli has been selected because of its ease of genetic manipulation, well-understood physiology (especially with regards to fatty acid biosynthesis), and rapid growth rate. Because all prokaryotic and eukaryotic organisms possess the ability to produce fatty acids as part of membrane lipid biosynthesis, small modifications to the methods presented should be broadly applicable to other industrial microorganisms.
Materials and Methods
Strain Construction
Bacterial strains, plasmids, and oligonucleotide primers used in this study are listed in Tables I and II. Oligonucleotide primers were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). Chemicals and reagents were purchased from Fisher Scientific (Pittsburgh, PA) unless otherwise specified. E. coli K-12 MG1655 was obtained from the E. coli Genetic Stock Center (New Haven, CT) and was the background strain used in this work. Strain RL01 was constructed by deleting the fadD gene from the MG1655 chromosome by λRed-mediated recombination (Datsenko and Wanner, 2000) using the λRed recombinase encoded on plasmid pKD46. Plasmid pKD13 was used as the template for amplification of the linear cassette using primers 1 and 2. The kan cassette was removed by expressing the FLP recombinase encoded on plasmid pCP20. Strain NRD204 (K-12 MG1655 araBAD::cat; De Lay and Cronan, 2007) was generously donated by Dr. Cronan (University of Illinois at Urbana-Champaign). Strain RL07 was constructed by P1 phage transduction (Thomason et al., 2007) of the araBAD::cat insertion into strain RL01 via a modified protocol utilizing selective plates containing 1.25 mM sodium pyrophosphate as a calcium chelator. The cat cassette was removed using pCP20 producing strain RL08. To minimize the possible presence of multiple insertions by λRed recombination, all recombinant strains were used as donors to transduce back into the parent strain. Gene insertions and deletions were verified by colony polymerase chain reaction (PCR) using primers 3 and 4 for fadD and 5 and 6 for araBAD, and by the absence of growth after plating on M9 agar supplemented with either 0.1% (w/v) sodium oleate (TCI America, Portland, OR) as previously described (Overath et al., 1969), or 0.4% (w/v) L-arabinose (Calbiochem, San Diego, CA) as carbon sources (Supplementary Fig. 1).
Table I.
Bacterial strains and plasmids used in this study.
| Strain/plasmid | Relevant genotype/property | Source or reference |
|---|---|---|
| Strains (Escherichia coli) | ||
| K-12 MG1655 | F− λ− ilvG− rfb-50 rph-1 | CGSC |
| RL01 | K-12 MG1655 fadD::kan | This report |
| RL02 | K-12 MG1655 ΔfadD | This report |
| NRD204 | K-12 MG1655 araBAD::cat | De Lay and Cronan (2007) |
| RL07 | K-12 MG1655 ΔfadD araBAD::cat | This report |
| RL08 | K-12 MG1655 ΔfadD ΔaraBAD | This report |
| DH10B | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ− rpsL nupG | Invitrogen |
| Plasmids | ||
| pKD13 | Template plasmid, R6K gamma origin, AmpR, KanR | Datsenko and Wanner (2000) |
| pKD46 | Carries Red recombinase under ParaB control, R101 origin, repA101(ts), AmpR | Datsenko and Wanner (2000) |
| pCP20 | Carries yeast FLP recombinase under constitutive promoter, pSC101 origin, λcI857+, λpR Repts, AmpR, CmR | Cherepanov and Wackernagel (1995) |
| pBAD33 | PBAD promoter, pACYC origin, CmR | Guzman et al. (1995) |
| pBAD18 | PBAD promoter, pBR322 origin, AmpR | Guzman et al. (1995) |
| pBAD24 | PBAD promoter, pBR322 origin, AmpR | Guzman et al. (1995) |
| pRL1 | pBAD33 carrying accD under PBAD control, CmR | This report |
| pRL2 | pRL1 carrying accDA under PBAD control, CmR | This report |
| pBAD33-ACC | pRL2 carrying accDABC under PBAD control, CmR | This report |
| pBAD18-BTE | pBAD18 carrying BTE under PBAD control, AmpR | This report |
| pBAD18-BTE-H204A | pBAD18 carrying BTE with H204A under PBAD control, AmpR | This report |
| pBT-2 | pBBR1-MCS origin, KanR | Lynch and Gill (2006) |
| pUC19 | pUC origin, lacZα, AmpR | New England Biolabs |
| pBAD34 | PBAD promoter, pUC origin, AmpR | This report |
| pBAD34-BTE | pBAD34 carrying BTE under PBAD control, AmpR | This report |
| pBAD34-BTE-H204A | pBAD34 carrying BTE-H204A under PBAD control, AmpR | This report |
| pBAD35 | pBT-2 containing araC -PBAD-MCS-rrnB fragment from pBAD18, KanR | This report |
| pBAD35-BTE | pBAD35 carrying BTE under PBAD control, KanR | This report |
| pBAD35-BTE-H204A | pBAD35 carrying BTE-H204A under PBAD control, KanR | This report |
Amp, ampicillin; Cm, chloramphenicol; Kan, kanamycin; R, resistance; ts, temperature-sensitive.
Table II.
Oligonucleotide primers used in this study.
| Primer name | Sequence (5′–3′)a |
|---|---|
| 1. fadDKO_fwd | GGTTGCGATGACGACGAACACGCATTTTAGAGGTGAAGAAGTGTAGGCTGGAGCTGCTTC |
| 2. fadDKO_rev | CGCCGGATTAACCGGCGTCTGACGACTGACTTAACGCTCAATTCCGGGGATCCGTCGACC |
| 3. fadDKO_colPCR_fwd | ACGGCATGTATATCATTTGGG |
| 4. fadDKO_colPCR_rev | CTTTAGTGGGCGTCAAAAAAAAC |
| 5. araBADKO_colPCR_fwd | AAGCGGGACCAAAGCCATGAC |
| 6. araBADKO_colPCR_rev | AGGAGACTTCTGTCCCTTGCG |
| 7. accD_fwd | CCCGAGCTCAGGTCCCTAATGAGCTGGATTGAAC |
| 8. accD_rev | CCCCCCGGGTCAGGCCTCAGGTTCCTGATCC |
| 9. accA_fwd | GGGCCCGGGAGGAATACTATGAGTCTGAATTTCCTTG |
| 10. accA_rev | GGGGTCGACCTCGAGTTTACGCGTAACCGTAGCTCATC |
| 11. accBC_fwd | CCCCTCGAGACGGAACCCACTCATGGATATTC |
| 12. accBC_rev | CCCGCATGCTTATTTTTCCTGAAGACCGAGTTTTTTC |
| 13. BTE-H204A_mega_rev | TCTCATCCGCCAAAAC |
| 14. BTE-H204A_mega_mut | TGTTAATCAGGCTGTGAACAACCTGAAATACG |
| 15. BTE-H204A_mega_fwd | TTGGGCTAGCGAATTC |
| 16. pBAD18-to-pBT_fwd | TTATGACAACTTGACGGCTACATC |
| 17. pBAD18-to-pBT_rev | AGAGTTTGTAGAAACGCAAAAAGGC |
| 18. BTE-to-pBAD35_fwd | ACGCTTTTTATCGCAACTCTC |
| 19. BTE-to-pBAD35_rev | GGGGCATGCTTAAACACGAGGTTCGCGC |
| 20. pBAD33ara_fwd | GGGCTCGAGTTATGACAACTTGACGGCTACATC |
| 21. pBAD33ara_rev | GGGAGATCTAGAGTTTGTAGAAACGCAAAAAGGC |
| 22. pUC19ara_fwd | GGGCTCGAGGTGCCTAATGAGTGAGCTAACTC |
| 23. pUC19ara_rev | GGGAGATCTTAGTTAAGCCAGCCCCGACAC |
| 24. qPCR_BTE_fwd | CTGTCTACCATCCCGGAC |
| 25. qPCR_BTE_rev | TCAGTTTTTGCAGTTTCTTGATTTCG |
| 26. qPCR_ompA_fwd | TGTTGAGTACGCGATCACTC |
| 27. qPCR_ompA_rev | GTTGTCCGGACGAGTGC |
Restriction sites are underlined.
Gene Synthesis
The 897-bp portion of the DNA sequence of BTE lacking the 83 amino acids at the N-terminus that appear to be involved in thylakoid targeting (Voelker et al., 1992) and containing the XbaI site formerly used to clone a functional part of the gene in E. coli (Ohlrogge et al., 1995) was codon-optimized for expression in E. coli, with common restriction sites eliminated. An artificial ribosome-binding site (RBS; AGGAGG), spacer sequence, start codon, and bases to create an in-frame sequence were added upstream of the gene fragment. The full sequence (Supplementary Material) was custom-synthesized (Integrated DNA Technologies, Inc.) and was received in plasmid pUC57 (pUC57-BTE).
Plasmid Construction
All cloning was performed in E. coli DH10B cells (Invitrogen, Carlsbad, CA), and all enzymes for cloning were purchased from New England Biolabs (Ipswich, MA). The accD gene encoding the β-subunit of acetyl-CoA carboxyltransferase, the accA gene encoding the α-subunit of acetyl-CoA carboxyltransferase, and the accBC operon encoding biotin carboxyl carrier protein and biotin carboxylase were amplified by PCR from MG1655 genomic DNA with their putative upstream RBS using primers 7 and 8, 9 and 10, and 11 and 12, respectively. These PCR products were sequentially inserted into pBAD33 (Guzman et al., 1995) between the SacI and XmaI sites (pRL1), SalI and XbaI sites (pRL2), and SphI and XhoI sites to create the artificial operon accDABC in plasmid pBAD33-ACC.
The BTE fragment from plasmid pUC57-BTE between the XmaI and HindIII sites was inserted into plasmid pBAD18 (Guzman et al., 1995) to generate plasmid pBAD18-BTE. To create a nonfunctional BTE for use as a negative control, a catalytic histidine at amino acid 204 was identified based on prior alignments of BTE with other plant acyl-ACP thioesterases (Yuan et al., 1996). A two-step megaprimer PCR procedure (Xu et al., 2003) was used to mutagenize the first two nucleotides of the histidine codon at position 204 to create an alanine codon. Primers 13 and 14 were used in the first reaction to generate a 3′ megaprimer from pBAD18-BTE template. This purified megaprimer and primer 15 were used to generate the complete BTE-H204A fragment, which was inserted as described for BTE to form plasmid pBAD18-BTE-H204A.
To generate a low copy vector harboring the PBAD promoter system, the 1697-bp fragment between the start of the araC gene and the end of the rrnB terminator was amplified from pBAD18 (Guzman et al., 1995) with primers 16 and 17. This insert was treated with T4 polynucleotide kinase and blunt ligated into EcoRV-digested plasmid pBT-2 (Lynch and Gill, 2006) to form plasmid pBAD35. A fragment containing the BTE gene was amplified from pBAD18-BTE using primers 18 and 19 and inserted into pBAD35 between the XmaI and SphI sites to generate plasmid pBAD35-BTE. The same procedure was performed using pBAD18-BTE-H204A as a template to form plasmid pBAD35-BTE-H204A.
To generate a high copy vector harboring the PBAD promoter system, the 1693-bp fragment between the start of the araC gene and the end of the rrnB terminator was amplified from pBAD33 with primers 20 and 21. The 2284-bp fragment of pUC19 (New England Biolabs) containing the origin and AmpR marker was amplified with primers 22 and 23. These two fragments were digested with XhoI and BglII and ligated to form plasmid pBAD34. The XmaI/HindIII fragments containing BTE or BTE-H204A from pBAD18-BTE and pBAD18-BTE-H204A were inserted into pBAD34 to form plasmids pBAD34-BTE and pBAD34-BTE-H204A. All plasmid constructs thus described were verified by sequencing.
Cell Transformation, Media, and Growth
Plasmids were electroporated into strain RL08 and selected on Luria–Bertani (LB) agar (Becton Dickinson, Cockeysville, MD) containing 25 μg mL−1 of kanamycin (pBAD35 constructs) or 50 μg mL−1 of ampicillin (pBAD18 constructs) and 34 μg mL−1 of chloramphenicol. All cultures were grown in a 37°C shaker at 250 rpm. Overnight cultures inoculated from single colonies were used to inoculate shake flasks containing LB medium (Becton Dickinson) supplemented with 0.4% glycerol and antibiotics. The cultures were induced at an optical density (OD600) of 0.2 with 0.2% (w/v) of L-arabinose.
Quantitative PCR for Determination of Copy Number
Immediately prior to induction at OD600 0.2, 1 mL of cell culture was collected and centrifuged at 16,000g for 1 min. The cell pellet was resuspended in 100 μL of deionized water. One microliter of resuspended cell pellet was used directly as the template in a quantitative PCR (qPCR) reaction with Maxima™ SYBR green/fluorescein qPCR master mix (Fermentas, Glen Burnie, MD). Primers 24 and 25 were used for BTE amplification, and primers 26 and 27 for chromosomal ompA amplification. SYBR Green fluorescence was monitored with a Bio-Rad CFX96 optical reaction module (Bio-Rad, Hercules, CA). Threshold cycle (Ct) values were calculated by Bio-Rad CFX Manager software.
Fatty Acid Extraction and Methylation
To 2.5-mL samples of cell culture (three replicates for each culture at each sampling time), 5 μL of 10 mg mL−1 heptadecanoic acid (Fluka, Buchs, Switzerland) dissolved in ethanol and 50 μL of 10 mg mL−1 pentadecanoic acid (Acros Organics, Geel, Belgium) dissolved in ethanol were added as internal standards. Next, 100 μL of glacial acetic acid and 5.0 mL of a 1:1 (v/v) chloroform/methanol mixture were added (Bligh and Dyer, 1959). The samples were inverted several times, vortexed vigorously, and centrifuged. The aqueous layer and cell debris were removed by aspiration and the chloroform layer was stored at −80°C until further processing. To methylate the fatty acids, the chloroform layer was thawed and evaporated under a nitrogen stream. Residual water was removed by lyophilization for approximately 1 h. To the dried residue, 0.5 mL of 1.25 M HCl in methanol (Fluka) was added, and the reaction was incubated overnight (14–16 h) at 50°C. The reaction mixtures were quenched by the addition of 5 mL of 100 mg mL−1 aqueous NaHCO3 (Sigma-Aldrich Corp., St. Louis, MO), and fatty acid methyl esters were extracted twice into 0.5 mL of hexane. The hexane layers were collected for analysis.
Gas Chromatography/Mass Spectrometry of Fatty Acid Methyl Esters
Gas chromatography/mass spectrometry (GC/MS) analysis was performed on a model 7890 Agilent GC (Agilent Technologies, Inc., Santa Clara, CA) with a 30 m × 0.25 mm DB-5 capillary column (Agilent Technologies, Inc.) and a model 5975 mass spectrometer. The oven temperature program was 100°C for 2 min, 150°C for 4 min, and a ramp to 250°C at a rate of 4°C min−1. One microliter of sample was injected with a 1:10 split ratio. Peak identification was achieved by comparison with internal standards and to the NIST Mass Spectral Database. Quantification was achieved by comparison of integrated peaks with calibration curves of a fatty acid methyl ester standard (Supelco No. 18918) with added methyl heptadecanoate (Fluka) and methyl pentadecanoate (Fluka). As a result of the high concentration of dodecanoic acid in BTE-expressing cultures, 20-fold dilutions were injected to accurately quantify this species. Concentrations of decenoic, dodecenoic, and tetradecenoic methyl esters were estimated using the sensitivity ratio of hexadecenoic to hexadecanoic methyl esters in the external standard. Calculated sample concentrations were normalized to the recovered concentrations of internal standards and averaged for all replicates.
Decane Extraction of Fatty Acids
After 34 h, 40 mL of decane (Acros Organics) was added to approximately 410 mL of each culture, and the mixtures were placed in a shaker at 250 rpm for 30 min. The resulting emulsions were acidified to facilitate phase separation by the addition of 10 mL of concentrated hydrochloric acid, shaken for 1 min, and centrifuged at 2,500g for 20 min or 16,000g for 10 min. The decane layer was collected for catalytic conversion.
Catalytic Decarboxylation
The extracted fatty acids dissolved in decane were decarboxylated to alkanes over a 1 wt% Pd/C catalyst (Sigma-Aldrich Corp.) in a plug flow reactor. The catalyst (1.1 g) was loaded into a 0.25-in. tubular stainless steel reactor operating in an upflow configuration surrounded by aluminum blocks heated externally by a well-insulated furnace. The catalyst bed was held in place by plugs of quartz wool at the reactor entrance and exit. Prior to reaction, fresh catalyst was reduced in flowing hydrogen (250 cm3 [STP] min−1 at 300°C [5°C min−1 ramp] for 4 h). The temperature was maintained at 300°C and the pressure was maintained at 12 bar for reaction experiments. A liquid solution containing the fatty acids in the decane extraction was introduced (0.05 mL min−1) into the upflow reactor using a high-performance liquid chromatography (HPLC) pump along with a 5% H2 co-feed flow of 250 cm3 (STP) min−1. The effluent liquid was collected at room temperature in a gas–liquid separator (Penberthy, Prophetstown, IL) and drained for GC/MS analysis (Shimadzu GC-2010; Shimadzu, Kyoto, Japan; with an mass MS and DB-5ms column from Alltech Deerfield, IL). Alkane concentrations were quantified by comparison with external standards containing undecane, dodecane, pentadecane, and hexadecane in a decane solvent. The tridecane peak was identified by its mass spectrum (Supplementary Fig. 2) and its concentration was determined using an estimated response factor. A small quantity of undecane (2 μmol mL−1) was present as an impurity in the decane received from the manufacturer. This concentration was subtracted from all undecane concentrations of converted culture extractions to yield the reported concentrations.
Results and Discussion
Initial Strain Construction and Optimization of BTE Copy Number
The development of an initial fatty acid overproducing strain followed the metabolic engineering strategy presented in Figure 1. First, to prevent catabolism of free fatty acids, the gene encoding acyl-CoA synthetase (fadD) was deleted from the chromosome. Second, to prevent consumption of the inducing agent L-arabinose, the araBAD operon was deleted. This operon encodes three genes involved in the initial steps of L-arabinose degradation: L-ribulokinase, L-arabinose isomerase, and L-ribulose-5-phosphate 4-epimerase. The resulting strain, K-12 MG1655 ΔfadD ΔaraBAD, is designated as strain RL08. Finally, to hydrolyze free fatty acids from acyl-ACP, a codon-optimized plant acyl-ACP thioesterase (BTE) from U. californica was cloned into various arabinose-inducible plasmids and transformed into strain RL08. The four selected plasmids were pBAD34 (pUC origin), pBAD18 (pBR322 origin without rop), pBAD33 (pACYC origin), and pBAD35 (pBBR1 origin). These plasmids range from very high reported copy number with the pUC origin (pBAD34; Yanisch-Perron et al., 1985), to medium copy number in pBAD18 (Cronan, 2006; Guzman et al., 1995) to low copy number in pBAD33 (Chang and Cohen, 1978; Guzman et al., 1995). The only reported copy number of the pBBR1 origin in E. coli is 30–40 copies per cell (Antoine and Locht, 1992). An identical set of plasmids expressing a nonfunctional version of BTE with histidine-204 mutagenized to an alanine were also transformed into strain RL08 to serve as negative controls.
To determine the optimal plasmid copy number for expressing BTE, cultures of strain RL08 harboring each plasmid (with both functional and nonfunctional BTE) were grown in shake flasks containing 50 mL of LB medium supplemented with 0.4% (v/v) glycerol. The OD600 was monitored (Fig. 2), and relative copy numbers of each plasmid were determined by qPCR from cell cultures immediately before induction of transcription at OD 0.2, and during early stationary phase after an elapsed time of 7.7 h from inoculation (Table III). Dramatically lower cell densities were observed after approximately 5 h when BTE was expressed on plasmids pBAD34 and pBAD18 (Fig. 2). Nonfunctional BTE-H204A did not exhibit similarly reduced cell densities, strongly suggesting that the decreased cell densities were a result of thioesterase activity.
Figure 2.

Time course of growth of Escherichia coli RL08 cultures harboring plasmids expressing BTE (filled markers) or nonfunctional BTE-H204A (open markers) monitored by optical density (OD600). Cells were grown at 37°C in shake flasks containing Luria–Bertani medium supplemented with 0.4% (v/v) glycerol and appropriate antibiotics for each vector. Cells were induced at an OD600 of 0.2 by the addition of a final concentration of 0.2% (w/v) L-arabinose.
Table III.
Calculated copy numbers of BTE per copy of chromosomal gene ompA from cultures of strain RL08 harboring BTE on the plasmids shown.
| Plasmid | Copy number (per ompA)
|
|
|---|---|---|
| Exponential | Stationary | |
| pBAD34-BTE | 100 ± 9 | 1200 ± 240 |
| pBAD34-BTE-H204A | 96 ± 15 | 840 ± 170 |
| pBAD18-BTE | 24 ± 3 | 110 ± 10 |
| pBAD18-BTE-H204A | 24 ± 2 | 70 ± 8 |
| pBAD33-BTE | 5 ± 1 | 19 ± 2 |
| pBAD33-BTE-H204A | 7 ± 1 | 13 ± 3 |
| pBAD35-BTE | 3 ± 0 | 5 ± 0 |
| pBAD35-BTE-H204A | 4 ± 0 | 2 ± 0 |
Culture samples were harvested during early exponential phase (OD600 of approximately 0.2) immediately prior to induction and during early stationary phase (7.7 h after inoculation). Errors are propagated standard deviations about the mean of three replicate samples.
Relative copy numbers trended as expected (Table III), with pBAD34 exhibiting the highest number of copies per copy of the selected chromosomal internal standard, ompA, during preinduced exponential growth and early stationary phase. The poorly characterized pBBR1 origin (present in pBAD35) is shown here to be present at low copy numbers (~5), similar to that of the pACYC origin during exponential growth. As expected, the pBAD18 construct was shown to have an intermediate copy number. BTE copy numbers relative to ompA increased in early stationary phase for all vectors except pBAD35, which showed no change. This is likely because of the presence of multiple replication forks on the chromosome during rapid growth (Nordström and Dasgupta, 2006), or the onset of nutrient limitations in the medium that stop replication from oriC while replication continues from many plasmid origins (Friehs, 2004). The plasmids expressing BTE-H204A all displayed lower copy numbers during stationary phase than the corresponding plasmids expressing BTE, possibly as a result of increased cell lysis in these induced cultures. Variation in the lysis of whole cell templates has been previously implicated as a source of error in determining plasmid copy numbers by qPCR (Providenti et al., 2006). Consistent with this hypothesis, no discrepancy in copy number was observed between plasmids harboring BTE and BTE-H204A during exponential growth prior to induction.
To determine whether copy number correlated with fatty acid production, total fatty acids were extracted and derivatized for analysis by GC/MS after 23 h (Fig. 3). In strains expressing functional BTE, the predominant fatty acid chain length shifted dramatically from C16 to C12, with C12 fatty acids accounting for up to 75% of the total fatty acid composition. For the culture expressing pBAD33-BTE, approximately 15% of C12 and 59% of C14 fatty acids were unsaturated. Lower but significant levels of hydroxylated C12, an intermediate in the fatty acid elongation cycle, were also observed but not quantified in functional BTE-expressing strains (Supplementary Fig. 3). Of the functional BTE-expressing plasmids, the lowest titer (0.25 ± 0.01 g L−1) was observed from the highest copy number plasmid, pBAD34-BTE. The two plasmids with the lowest overall copy number in both exponential and early stationary phase, pBAD33-BTE and pBAD35-BTE, accumulated the highest titers of fatty acids (0.54 ± 0.00 and 0.48 ± 0.00 g L–1, respectively), with medium copy number plasmid pBAD18-BTE accumulating a slightly lower quantity (0.40 ± 0.03 g L−1). Strains harboring a nonfunctional BTE gene on the various plasmids accumulated similar quantities of predominantly C16 fatty acids, as expected.
Figure 3.
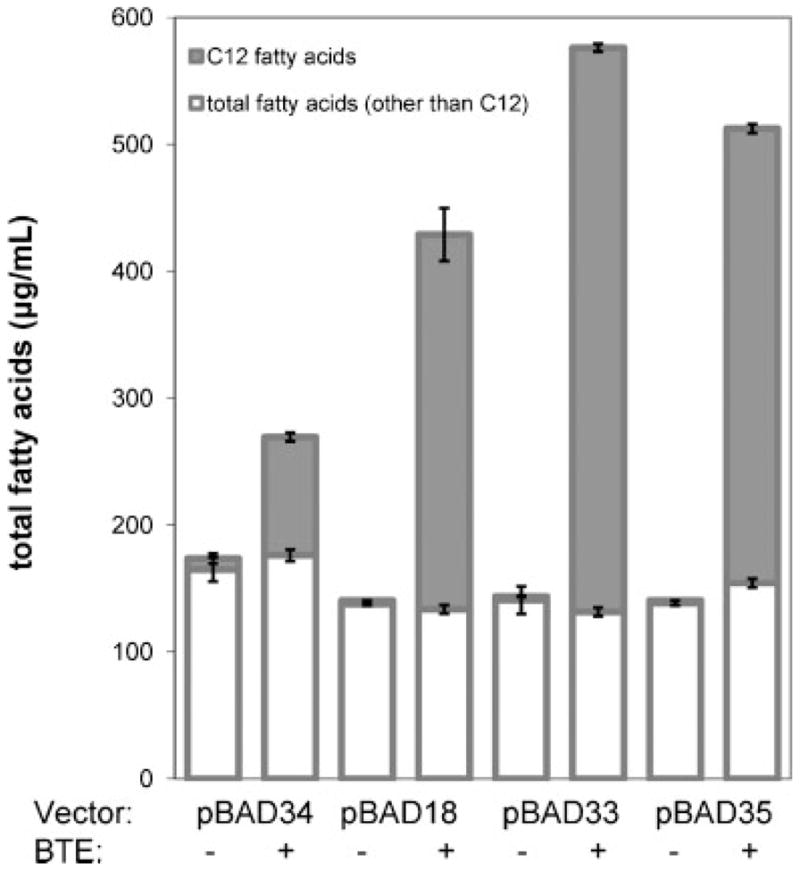
Total fatty acid titers (μg mL−1 culture medium, open bars) and C12 chain length fatty acid titers (saturated and estimated unsaturated, filled bars) extracted from cultures shown in Figure 2 at approximately 23 h postinoculation. Error bars represent standard deviations about the mean of two or three replicate samples for either C12 fatty acids (lower bars) or total fatty acids including C12 (upper bars).
It can therefore be concluded that expression of BTE on a low copy number plasmid results in optimal fatty acid production. One possible explanation is that higher quantities of functional thioesterase are produced from the high copy number plasmids, resulting in an initially rapid rate of accumulation of medium-chain free fatty acids. As there is no known protein-mediated export mechanism for free fatty acids, they likely pass across the inner membrane via a transmembrane flip (Black and DiRusso, 2003). Dodecanoic acid, the dominant BTE product, has a much higher water solubility than longer chain fatty acids (Vorum et al., 1992) and can possibly cross the outer membrane through porins (Hearn et al., 2009). A very rapid accumulation of C12 and C14 free fatty acids in the inner membrane may disrupt the membrane integrity and result in cell lysis. A second possible explanation is that higher quantities of functional thioesterase significantly deplete C12 and C14 acyl-ACPs destined for membrane phospholipid incorporation, resulting in a reduced number of viable cells.
Co-Overexpression of Acetyl-CoA Carboxylase
The conversion of acetyl-CoA to malonyl-CoA by ACC has been identified as a rate-limiting step in fatty acid biosynthesis (Davis et al., 2000). To see if higher production could be achieved over expression of BTE alone, the four subunits of ACC were cloned as an artificial operon (accDABC) on a low copy arabinose-inducible plasmid to yield pBAD33-ACC. To co-express BTE, pBAD35-BTE was selected owing to its compatibility with pBAD33-ACC and its high level of fatty acid overproduction. Four cultures of E. coli strain RL08 harboring combinations of either pBAD33 or pBAD33-ACC, and pBAD35-BTE or pBAD35-BTE-H204A, were grown in shake flasks in 500 mL of LB medium supplemented with 0.4% (v/v) glycerol as a carbon source. Fatty acids were extracted and derivatized for analysis by GC/MS at several times (Fig. 4; Supplementary Fig. 4).
Figure 4.

Growth and fatty acid production of strain RL08 harboring combinations of plasmids pBAD33 or pBAD33-ACC, and pBAD35-BTE-H204A or pBAD35-BTE. Cells were grown at 37°C in shake flasks containing Luria–Bertani medium supplemented with 0.4% (v/v) glycerol, 50 μg mL−1 of ampicillin, and 34 μg mL−1 of chloramphenicol. A: OD600 as a function of time from inoculation; (B) total (filled bars) and C12 chain length (saturated and estimated unsaturated, open bars) of fatty acid titers (μg mL−1 culture medium) for selected times during cell growth as indicated (6, 10, 18, 29, and 34 h). Error bars represent standard deviations about the mean for three replicate samples for either C12 fatty acids (upper bars) or total fatty acids including C12 (lower bars).
Elevated levels of fatty acids were detected at the onset of stationary phase in cultures expressing BTE, with maximum accumulation observed at approximately 29 h postinoculation. At this time, the strain expressing both BTE and overexpressing ACC exhibited a small increase in fatty acid titer (0.81 ± 0.02 g L−1) relative to a strain expressing BTE alone (0.70 ± 0.01 g L−1). Preliminary microarray data from our lab (not shown) suggests that expression of BTE alone dramatically increases gene expression of the four subunits of ACC, possibly by an uncharacterized transcriptional activation mechanism, which may explain the modest effect. This result is in agreement with a previous observation by Ohlrogge et al. (1995) wherein expression of BTE in E. coli increases the levels of biotin carboxyl carrier protein (AccB). The expression levels of AccA, AccC, and AccD were not quantified in this prior work. Additionally, heterologous expression of BTE in the seeds of the rapeseed plant, Brassica napus was shown to increase expression levels of its native biotin carboxyl carrier protein and biotin carboxylase (Eccleston and Ohlrogge, 1998). Both plants and bacteria have similar multisubunit acetyl-CoA carboxylases (Cronan and Waldrop, 2002). The strain expressing only ACC does not overproduce fatty acids, as previously observed owing to feedback inhibition of ACC by accumulated acyl-ACPs (Davis and Cronan, 2001).
Extraction and Conversion of Fatty Acids to Alkanes
To demonstrate a complete process for fuel production, fatty acids were extracted from approximately 400 mL of the overproducing culture (the remaining volume from an original culture volume of 500 mL) overexpressing both ACC and BTE at 34 h, with a total measured saturated and unsaturated C12 fatty acid titer of 0.36 ±0.02 g L−1. Decane was selected to facilitate analysis of the dominant undecane product, owing to the presence of significant undecane impurities in other commercially available alkanes of higher molecular weight, such as tridecane. Although decane is mildly toxic to microbes (Sardessai and Bhosle, 2002), larger alkanes such as dodecane are essentially nontoxic and their use to extract metabolites during cell growth has been previously demonstrated (Janikowski et al., 2002; Newman et al., 2006). The emulsion resulting from decane addition was acidified and centrifuged to facilitate phase separation, and the decane layer was collected. Approximately 60% of the decane added could be collected as a de-emulsified liquid layer. Fatty acids in the decane extractions were decarboxylated at 100% conversion in the presence of hydrogen over a 1 wt% Pd/C catalyst in a plug flow reactor operating at 300°C and 12 bar. The catalytic decarboxylation of fatty acids over Pd/C catalysts has previously been demonstrated using a semi-batch reactor operating at 300°C (Mäki-Arvela et al., 2007). Under these conditions unsaturated fatty acids are fully hydrogenated, which is desirable for the stability of the product during storage. In the collected alkane product, 0.44 ± 0.3 g L−1 (culture volume) of undecane was obtained (Fig. 5), representing a complete recovery and conversion of C12 fatty acids from the culture medium. Smaller amounts of tridecane and pentadecane were also present, as expected from the fatty acid composition (Supplementary Table I).
Figure 5.
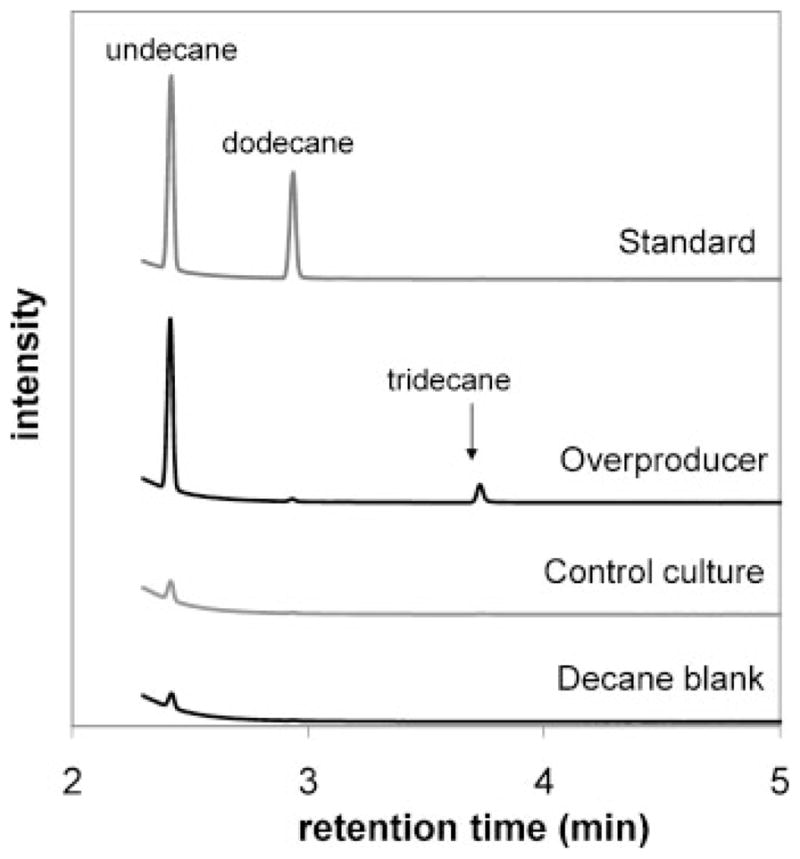
Chromatograms of collected decane layers from a 34-h fatty acid overproducing culture (RL08/pBAD33-ACC/pBAD35-BTE, second from top trace) and a negative control culture (RL08/pBAD33/pBAD35-BTE-H204A, second from bottom trace) following decarboxylation at 300°C in a plug flow reactor containing 1% (w/w) Pd/C catalyst in the presence of hydrogen. A standard containing undecane and dodecane in a decane solvent (top trace) and a blank decane sample (bottom trace) are shown for comparison.
Ultimately, an industrial process can be envisioned in which a desired hydrocarbon product is used to both extract fatty acids from a fermentor and act as the solvent for the decarboxylation reaction. A product stream could be continuously or semi-continuously collected that matches the fatty acid production rate, with the remainder of the alkane phase recycled as an extractant in a two-phase partitioning bioreactor.
Conclusion
A strain of E. coli that exhibits an approximately sevenfold increase in fatty acid production (predominantly C12 fatty acids) over the baseline strain (RL08) was metabolically engineered. A key aspect of the strategy was utilizing a low copy number vector for expression of BTE. The successful conversion of overproduced fatty acids to a useful enriched liquid alkane stream was demonstrated by a novel process that couples microbial production of free fatty acids to a catalytic reaction step. Further genetic and process improvements are currently underway to increase fatty acid yields and alkane recovery.
Supplementary Material
Acknowledgments
This work was funded in part by the DOE Great Lakes Bioenergy Research Center (GLBRC; DOE Office of Science BER DE-FC02-07ER64494) and startup funds from the University of Wisconsin-Madison Graduate School. R.M.L. was supported as a trainee in the Chemistry-Biology Interface Training Program (NIH). The authors are grateful to Wesley Marner, Sydnor Withers, Alan Higbee, Spencer Hoover, and Anna Mielke for their contributions.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Antoine R, Locht C. Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from Gram-positive organisms. Mol Microbiol. 1992;6:1785–1799. doi: 10.1111/j.1365-2958.1992.tb01351.x. [DOI] [PubMed] [Google Scholar]
- Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451:86–89. doi: 10.1038/nature06450. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Sharma R, Chisti Y, Banerjee U. Botryococcus braunii: A renewable source of hydrocarbons and other chemicals. Crit Rev Microbiol. 2002;22:245–279. doi: 10.1080/07388550290789513. [DOI] [PubMed] [Google Scholar]
- Black PN, DiRusso CC. Transmembrane movement of exogenous long-chain fatty acids: Proteins, enzymes, and vectorial esterification. Microbiol Mol Biol Rev. 2003;67:454–472. doi: 10.1128/MMBR.67.3.454-472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Chang ACY, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Cho H, Cronan JE., Jr Escherichia coli thioesterase I, molecular cloning and sequencing of the structural gene and identification as a periplasmic enzyme. J Biol Chem. 1993;268:9238–9245. [PubMed] [Google Scholar]
- Cronan JE. A family of arabinose-inducible Escherichia coli expression vectors having pBR322 copy control. Plasmid. 2006;55:152–157. doi: 10.1016/j.plasmid.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Cronan JE, Jr, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MS, Solbiati J, Cronan JE., Jr Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2000;275:28593–28598. doi: 10.1074/jbc.M004756200. [DOI] [PubMed] [Google Scholar]
- Davis MS, Cronan JE., Jr Inhibition of Escherichia coli acetyl coenzyme A carboxylase by acyl–acyl carrier protein. J Bacteriol. 2001;183:1499–1503. doi: 10.1128/JB.183.4.1499-1503.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay NR, Cronan JE. In vivo functional analysis of the type II acyl carrier proteins of fatty acid biosynthesis. J Biol Chem. 2007;282:20319–20328. doi: 10.1074/jbc.M703789200. [DOI] [PubMed] [Google Scholar]
- Eccleston VS, Ohlrogge JB. Expression of lauroyl-acyl carrier protein thioesterase in Brassica napus seeds induces pathways for both fatty acid oxidation and biosynthesis and implies a set point for triacylglycerol accumulation. Plant Cell. 1998;10:613–621. doi: 10.1105/tpc.10.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Cronan JE. Escherichia coli unsaturated fatty acid synthesis: Complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J Biol Chem. 2009;284:29526–29535. doi: 10.1074/jbc.M109.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjerbaek L, Christensen KV, Norddahl B. A review of the current state of biodiesel production using enzymatic transesterification. Biotechnol Bioeng. 2008;102:1298–1315. doi: 10.1002/bit.22256. [DOI] [PubMed] [Google Scholar]
- Friehs K. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol. 2004;86:47–82. doi: 10.1007/b12440. [DOI] [PubMed] [Google Scholar]
- Ghosh SK, Bhattacharjee A, Jha JK, Mondal AK, Maiti MK, Basu A, Ghosh D, Ghosh S, Sen SK. Characterization and cloning of a stearoyl/oleoyl specific fatty acyl–acyl carrier protein thioesterase from the seeds of Madhuca longifolia (latifolia) Plant Physiol Biochem. 2007;45:887–897. doi: 10.1016/j.plaphy.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn EM, Patel DR, Lepore BW, Indic M, van den Berg B. Transmembrane passage of hydrophobic compounds through a protein channel wall. Nature. 2009;458:367–370. doi: 10.1038/nature07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath RJ, Rock CO. Regulation of fatty acid elongation and intitiation by acyl–acyl carrier protein in Escherichia coli. J Biol Chem. 1996;271:1833–1836. doi: 10.1074/jbc.271.4.1833. [DOI] [PubMed] [Google Scholar]
- Janikowski TB, Velicogna D, Punt M, Daugulis A. Use of a two-phase partitioning bioreactor for degrading polycyclic aromatic hydrocarbons by a Sphingomonas sp. Appl Microbiol Biotechnol. 2002;59:368–376. doi: 10.1007/s00253-002-1011-y. [DOI] [PubMed] [Google Scholar]
- Jha JK, Maiti MK, Bhattacharjee A, Basu A, Sen PC, Sen SK. Cloning and functional expression of an acyl-ACP thioesterase FatB type from Diploknema (Madhuca) butyracea seeds in Escherichia coli. Plant Physiol Biochem. 2006;44:645–655. doi: 10.1016/j.plaphy.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Jones KL, Kim SW, Keasling JD. Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab Eng. 2000;2:328–338. doi: 10.1006/mben.2000.0161. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R, Stölting T, Steinbüchel A. Microdiesel: Escherichia coli engineered for fuel production. Microbiology. 2006;152:2529–2536. doi: 10.1099/mic.0.29028-0. [DOI] [PubMed] [Google Scholar]
- Li SJ, Cronan JE., Jr Growth rate regulation of Escherichia coli acetyl coenzyme A carboxylase, which catalyzes the first committed step of lipid biosynthesis. J Bacteriol. 1993;175:332–340. doi: 10.1128/jb.175.2.332-340.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Vora H, Khosla C. Overproduction of free fatty acids in E. coli: Implications for biodiesel production. Metab Eng. 2008;10:333–339. doi: 10.1016/j.ymben.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Lynch MD, Gill RT. Broad host range vectors for stable genomic library construction. Biotechnol Bioeng. 2006;94:151–158. doi: 10.1002/bit.20836. [DOI] [PubMed] [Google Scholar]
- Mäki-Arvela P, Kubickova I, Snåre M, Eränen K, Murzin DYu. Catalytic deoxygenation of fatty acids and their derivatives. Energy Fuels. 2007;21:30–41. [Google Scholar]
- Newman JD, Marshall J, Chang M, Nowroozi F, Paradise E, Pitera D, Newman KL, Keasling JD. High-level production of amorpha-4,11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli. Biotechnol Bioeng. 2006;95:684–691. doi: 10.1002/bit.21017. [DOI] [PubMed] [Google Scholar]
- Nordström K, Dasgupta S. Copy-number control of the Escherichia coli chromosome: A plasmidologist’s view. EMBO Rep. 2006;7:484–489. doi: 10.1038/sj.embor.7400681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlrogge J, Savage L, Jaworski J, Voelker T, Post-Beittenmiller D. Alteration of acyl–acyl carrier protein pools and acetyl-CoA carboxylase expression in Escherichia coli by a plant medium chain acyl–acyl carrier protein thioesterase. Arch Biochem Biophys. 1995;317:185–190. doi: 10.1006/abbi.1995.1152. [DOI] [PubMed] [Google Scholar]
- Overath P, Pauli G, Schairer HU. Fatty acid degradation in Escherichia coli. An inducible acyl-CoA synthetase, the mapping of old-mutations, and the isolation of regulatory mutants. Eur J Biochem. 1969;7:559–574. [PubMed] [Google Scholar]
- Providenti MA, O’Brien JM, Ewing RJ, Paterson ES, Smith ML. The copy-number of plasmids and other genetic elements can be determined by SYBR-Green-based quantitative real-time PCR. J Microbiol Methods. 2006;65:476–487. doi: 10.1016/j.mimet.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Sardessai Y, Bhosle S. Tolerance of bacteria to organic solvents. Res Microbiol. 2002;153:263–268. doi: 10.1016/s0923-2508(02)01319-0. [DOI] [PubMed] [Google Scholar]
- Serrano-Vega MJ, Garcés R, Martínez-Force E. Cloning, characterization and structural model of a FatA-type thioesterase from sunflower seeds (Helianthus annuus L.) Planta. 2005;221:868–880. doi: 10.1007/s00425-005-1502-z. [DOI] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol. 2007;1:17. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- Vasudevan PT, Briggs M. Biodiesel production—Current state of the art and challenges. J Ind Microbiol Biotechnol. 2008;35:421–430. doi: 10.1007/s10295-008-0312-2. [DOI] [PubMed] [Google Scholar]
- Voelker TA, Worrell AC, Anderson L, Bleibaum J, Fan C, Hawkins DJ, Radke SE, Davies HM. Fatty acid biosynthesis redirected to medium chains in transgenic oilseed plants. Science. 1992;257:72–74. doi: 10.1126/science.1621095. [DOI] [PubMed] [Google Scholar]
- Voelker TA, Davies HM. Alteration of the specificity and regulation of fatty acid synthesis of Escherichia coli by expression of a plant medium-chain acyl–acyl carrier protein thioesterase. J Bacteriol. 1994;176:7320–7327. doi: 10.1128/jb.176.23.7320-7327.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorum H, Brodersen R, Kragh-Hansen U, Pedersen AO. Solubility of long-chain fatty acids in phosphate buffer at pH 7.4. Biochim Biophys Acta. 1992;1126:135–142. doi: 10.1016/0005-2760(92)90283-2. [DOI] [PubMed] [Google Scholar]
- Xu Z, Colosimo A, Gruenert DC. Site-directed mutagenesis using the megaprimer method. Methods Mol Bio. 2003;235:203–207. doi: 10.1385/1-59259-409-3:203. [DOI] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Yuan L, Voelker TA, Hawkins DJ. Modification of the substrate specificity of an acyl–acyl carrier protein thioesterase by protein engineering. Proc Natl Acad Sci USA. 1995;92:10639–10643. doi: 10.1073/pnas.92.23.10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Nelson BA, Caryl G. The catalytic cysteine and histidine in the plant acyl–acyl carrier protein thioesterases. J Biol Chem. 1996;271:3417–3419. doi: 10.1074/jbc.271.7.3417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.