

Abstract
Photodynamic therapy (PDT) uses a photosensitizer, light and oxygen to produce extensive oxidative damage to organelles housing the photosensitizer. Although PDT is an efficient trigger of apoptosis, it also induces autophagy in many kinds of cells. Autophagy can serve as both a cell survival and a cell death mechanism. Our previous study indicates that autophagy contributes to cell death after PDT, especially in apoptosis-deficient cells. Here, we provide further evidence to support the role of autophagy in cell killing after PDT. Autophagy was blocked by knockdown of one essential factor, LC3 or Atg7, in MCF-7 cells. The cells were exposed to a range of doses of PDT sensitized by the phthalocyanine Pc 4; steps in autophagy were monitored by western blotting for LC3-II and by fluorescence microscopy for the uptake of monodansylcadaverine or for the distribution of transfected GFP-LC3; and overall cell death was monitored by MTT assay and by clonogenic assay. We find that blocking autophagy increased the survival of MCF-7 cells after PDT and increased the shoulder on the dose-response curve. In response to Pc 4-PDT, Atg7-deficient MCF-7 cells remained capable of robust accumulation of LC3-II, but were defective in comparison to Atg7+ cells in the formation of autophagosomes. We conclude that apoptosis-deficient cells rely on autophagy for cell death after Pc 4-PDT and that the strong activation of LC3 maturation in response to PDT could occur even in cells with limited or no Atg7 expression.
Keywords: autophagy, LC3, Atg7, apoptosis, photodynamic therapy, cytotoxicity
Introduction
Macroautophagy (hereafter referred to as autophagy) is a process for lysosomal degradation of organelles and long-lived proteins. During autophagy, cytoplasmic contents are sequestered within double-membrane vacuoles called autophagosomes; then the vacuole membranes fuse with the lysosomal membrane to deliver the contents into the autolysosome, where they are degraded.1-4 Although autophagy was originally described as a survival response to starvation, allowing the recycling of materials, it may also serve as a mechanism of cell death when cells are treated with toxic stimuli, including ionizing radiation,5 rapamycin6 or photodynamic therapy.7-9 Autophagy as a means of induction of cancer cell death has received increasing attention recently, especially in circumstances of apoptosis-resistant cells.10
Photodynamic therapy (PDT) is a potent cancer treatment which employs photosensitizers and visible light to kill cells and ablate tumors.11,12 PDT with a variety of photosensitizers induces apoptosis in many kinds of cancer cells.13 More recently, it was found that PDT induces autophagy in cells representing both lymphoid7 and solid7-9 malignancies as well as in murine embryonic fibroblasts.8 Autophagy was observed in human prostate cancer DU145 cells, which are deficient in the pro-apoptotic factor Bax, in response to PDT sensitized by 9-capronyloxytetrakis (methoxyethyl) porphycene (CPO)7 or the phthalocyanine Pc 4.9 Autophagy was also triggered following Pc 4-PDT in both procaspase-3-deficient and -overexpressing human breast cancer MCF-7 cells, i.e., whether or not they were capable of typical apoptosis.9 The common occurrence of autophagy following PDT has raised the question of its role in the response of cells to PDT.
Although autophagy has been extensively studied, little was known about its molecular mechanism until the discovery of autophagy-related genes (ATG) in yeast.14 To date, 30 ATG genes have been identified. The corresponding gene products comprise the “core” machinery15 that coordinates the specific steps in the autophagic pathway. There are two ubiquitin-like conjugation systems for autophagy; i.e., the ATG12 and ATG8 conjugation systems.16-18 Atg12 is activated by an E1-like enzyme, Atg7,19 and finally conjugated to Atg5 in a reaction similar to ubiquitination. Atg7 can also activate Atg8 and thereby participates in the ATG8 conjugation system. In mammalian cells, Atg7 is essential for the autophagy conjugation system, the formation of autophagosomes, and starvation-induced degradation of proteins and organelles.20
Based on the importance of Atg7 in autophagy, its function has been widely studied in recent years.
Kessel et al.21,22 depleted Atg7 in murine leukemia L1210 cells by shRNA knockdown and noted that the deficient cells were more sensitive than Atg7-replete cells to the lethal effects of a low PDT dose. This suggested that autophagy served a survival function in L1210 cells. Similar results were found for camptothecin (CPT)-treated MCF-7 cells.23 In contrast, other laboratories have reported that Atg7 knockdown protected against cell death.24-26 Similarly, we found that the chemical inhibitors of autophagy, 3-methyladenine (3-MA) and wortmannin, provided greater protection against loss of viability to apoptosis-deficient than to apoptosis-competent MCF-7 cells.9 This result suggested that the cells that were deficient in apoptosis (because of the absence of caspase-3) were more dependent on autophagy for cell death. Although 3-MA and wortmannin are commonly used inhibitors of autophagy, they are general phosphatidylinositol-3-kinase (PI3K) inhibitors and could affect cell death by mechanisms other than direct inhibition of autophagy. In addition, both 3-MA and wortmannin were mildly toxic (producing about 34% and 40% cell killing, respectively, under the conditions of our experiments). Therefore, we have re-visited the role of autophagy in the PDT response of human carcinoma cells by studying cells in which an important autophagy protein was depleted by inhibitory RNA techniques. Here, we report the response of MCF-7 cells to knockdown of Atg 8 (LC3) or Atg7, in order to examine the relationship between autophagy and cell death following PDT of human carcinoma cells.
Results
In order to examine the contribution of autophagy to cell viability in Pc 4-PDT-treated cells, MCF-7v and MCF-7c3 cells were transiently transfected with siRNA against LC3 (the mammalian homologue of the yeast Atg8 protein) to block the autophagy pathway, then treated with PDT. Figure 1A confirms that Pc 4-PDT induces a strong time-dependent increase in the level of LC3-II, an autophagy marker, in the nontransfected cells; in both apoptosis-deficient MCF-7v and apoptosis-competent MCF-7c3, a marked accumulation of LC3-II was found by 22 h post-PDT, as reported previously.9 In contrast, in cells transfected with LC3 siRNA, the PDT-induced accumulation of LC3-II was undetectable at 2 h post-PDT or at the same time after transfection in the controls (i.e., suggesting that LC3 knockdown was nearly complete). At 22 h post-PDT, the PDT-induced increase in LC3-II was markedly reduced in the siRNA-transfected cells in comparison to the nontransfected cells. Under all of these conditions, the level of LC3-I was not much changed, perhaps due to rapid processing of LC3-I into LC3-II, as reported by others.28,29
Figure 1.

The effects of LC3 siRNA transfection on LC3-II levels and PDT-induced cell death. (A) MCF-7v and MCF-7c3 cells were transfected with LC3 siRNA, as described in Materials and Methods. Twenty-four hours later, nontransfected and transfected cells were loaded with 200 nM Pc 4 for 3 h, then irradiated with 200 mJ/cm2 of red light and incubated for an additional 2 h or 22 h. Cells were collected, and protein from whole cell lysates was separated on SDS-PAGE gels, transferred to PVDF membranes, and probed with an anti-LC3 antibody. (B) MTT assay. MCF-7v and MCF-7c3 cells grown in 96-well plates were transfected with LC3 siRNA, as described above. After 24 h, the nontransfected and transfected cells were loaded with the indicated doses of Pc 4 for 4 h and then irradiated with 200 mJ/cm2 red light and post incubated for 24 h before assay. The viability of cells transfected with siRNA alone was 70.6% and 77.2% of the untreated control cells for MCF-7v and MCF-7c3, respectively. Data are the mean ± standard deviation of results from 3 independent experiments (*p < 0.05, t-test).
Figure 1B shows results of an MTT assay for increasing doses of Pc 4-PDT on MCF-7v and MCF-7c3 cells with or without LC3 siRNA transfection. A dose-dependent loss of cell viability was observed in both cell lines; however, LC3 knockdown greatly reduced the effectiveness of PDT against MCF-7v cells but had no significant effect on MCF-7c3 cells. In the MCF-7v cells, inhibition of autophagy led to an increase in the shoulder on the dose-response curve. The results agree with those obtained with the chemical inhibitors9 and suggest that cells that are defective in typical apoptosis die mainly by non-apoptotic mechanism(s) after PDT, which may include autophagy. Blockage of autophagy in cells already defective in apoptosis results in reduced cell killing. In contrast, when apoptosis and autophagy function simultaneously, apoptosis appears to be dominant, and as a result, LC3 knockdown had a little or no effect on MCF-7c3 cells.
Atg7, a ubiquitin-activating enzyme E1-like protein, which mediates a critical step in Atg5-Atg12 and Atg8-phosphatidylethanolamine complex formation, is a key autophagy intermediate. In the absence of Atg7, autophagy is severely depressed. Furthermore, a cell line with Atg7 stably knocked down would allow more detailed studies of the role of autophagy in PDT response. Thus, we obtained MCF-7 cells stably transfected with Atg7 shRNA (Atg7−) and a control line transfected with scrambled RNA (Atg7+) from Dr. A. Kelekar (University of Minnesota). Western blot analysis (Fig. 2A) confirmed that Atg7 was expressed in the control cell line with or without exposure to PDT but was not detectable in the Atg7-knockdown cells in either condition.
Figure 2.

Autophagy in PDT-treated Atg7+ and Atg7 − MCF-7 cells (from A. Kelekar). (A) The cells were untreated or treated with 200 nM Pc 4 and 200 mJ/cm2 of red light and post-incubated for 20 h. Cells were collected and western blot analysis was performed with anti-Atg7 as well as anti-actin (for a loading control) antibodies. (B) Images of MDC-labeled control and PDT-treated cells. Cells were treated with 0, 25 or 150 nM Pc 4 and 200 mJ/cm2 red light and post-incubated for 17 h. At the end of the incubation, cells were stained with MDC for 20 min and examined immediately. (C and D) PDT-induced autophagosome formation in MCF-7/Atg7+ and MCF-7/Atg7− cells. Cells were transiently transfected with GFP-LC3 construct, then PDT-treated with 150 nM Pc 4 and 200 mJ/cm2 red light and post-incubated for 6 or 24 hours. At the end of the incubation, GFp-positive cells were examined and photographed. Arrows in a representative fluorescence micrograph (C) indicate the autolysosomes/autophagosomes in PDT-treated cells. (D) The percentage of GFp-expressing cells with punctate GFP-LC3. Cells treated as described in (C) were visualized by fluorescence microscopy, and the GFP-expressing cells were counted as punctate or diffuse. The results are mean ± standard deviation of data from three independent experiments (*p < 0.01, t-test). (E) Expression of Beclin 1 in MCF-7 cells. Untreated cells of each MCF-7 sub-line were analyzed by western blot with anti-Beclin 1 and anti-actin (loading control) antibodies. Lane 1, MCF-7/Atg7+; lane 2, MCF-7/Atg7−; lane 3, MCF-7c3; lane 4, MCF-7v.
The induction of autophagy by PDT in MCF-7 cells has been identified by electron microscopic observation of the presence of double-membrane autophagosomal structures and by immunoblot observation of the time- and dose-dependent increase in the level of LC3-II.9 Here, the role of Atg7 in the formation of autophagosomes was assessed by monitoring the distribution of the fluorescent dye monodansylcadaverine (MDC). This dye has been used to detect the presence of late-stage autophagosomes and other acidic vesicles.30 When cells are viewed with a fluorescence microscope, autophagosomes stained by MDC appear as distinct dot-like structures distributed within the cytoplasm or localizing in the perinuclear region. Figure 2B shows a dose-dependent increase in the number of MDC-labeled vesicles in PDT-treated MCF-7 (Atg7+) cells compared with the untreated cells (top). The development of MDC positivity in the Atg7+ cells was also time dependent (data not shown). In contrast, PDT did not increase the fluorescence density or the number of MDC-labeled particles in the Atg7− cells (bottom). It appears that MDC staining of autophagosomes is limited to the Atg7+ cells after PDT, providing evidence that PDT-induced autophagy was severely compromised in the Atg7− cells.
Another method to monitor autophagy is to visualize the intracellular localization of GFP-tagged LC3 by fluorescence microscopy.31 In untreated cells, GFP-LC3 is diffusely dispersed in the expressing cells, whereas during autophagy, it relocalizes to autophagosomes and appears as punctate fluorescence. To determine the effect of PDT on the distribution of GFP-LC3, MCF-7/Atg7+ and MCF-7/Atg7− cells were transiently transfected with GFP-LC3, then exposed to PDT (150 nM Pc 4 and 200 mJ/cm2) and further incubated for 6 h or 24 h. As shown in Figure 2C, GFP fluorescence was diffuse in essentially all GFP-expressing cells in the absence of PDT, and PDT induced a higher frequency of punctate GFP-LC3 in MCF-7/Atg7+ cells than in MCF-7/Atg7− cells. To quantify these data, GFP-expressing cells with diffuse or punctate fluorescence were counted. It can be seen from Figure 2D that at both post-PDT times, 40–45% of MCF-7/Atg7+ cells displayed punctate fluorescence. In contrast, only ~8% of MCF-7/Atg7− cells exhibited punctate GFP-LC3. The difference between the response of the two cell lines was highly statistically significant (p = 0.0007 and 0.0003 for 6 h and 24 h post-PDT, respectively). There was no significant difference in the distribution of GFP-LC3 between the two cell lines in the absence of PDT. There was also no significant difference between the data for the two time points (p = 0.06 and p = 0.7 for MCF-7/Atg7+ and MCF-7/Atg7− cells, respectively), which indicates that the extended incubation time did not induce a significant increase in GFP-LC3 positive cells. Based on the results with MDC and with GFP-LC3, it appears that Atg7 is essential for the formation of autophagosomes during PDT-induced autophagy.
Some MCF-7 cultures have been reported to lack expression of Beclin 1.32,33 An absence of Beclin 1 might affect our results by allowing LC3-II formation in the absence of Atg7. Therefore, we examined each of the four MCF-7 sub-lines used in this study for Beclin 1 expression. As shown in Figure 2E, all of them express readily detectable levels of this protein.
As autophagy can be a dual-function process, both protecting against and promoting cell death, its role in MCF-7 cells was examined by the direct measure of the ability of PDT to reduce cell clonogenicity. Exponential cultures of the Atg7+ and Atg7− MCF-7 cells were exposed to increasing doses of Pc 4-PDT, then collected and plated for colony formation. As shown in Figure 3, throughout the range of doses used, the Atg7− cells were more resistant to PDT, and at the lowest doses, the survival curve of the autophagy-defective cells had a markedly greater shoulder than that for the Atg7+ cells. Because the cells were not given G418 (agent used for selection of cells expressing Atg7 shRNA) for 9 days during the incubation for colony formation, it was important to ascertain that the Atg7-knockdown was stable without selection. When the Atg7− cells were grown without G418 for 9 days, no Atg7 was detected by western blot analysis (data not shown), indicating that the differential response between the two cell lines resulted from the differential expression of Atg7.
Figure 3.
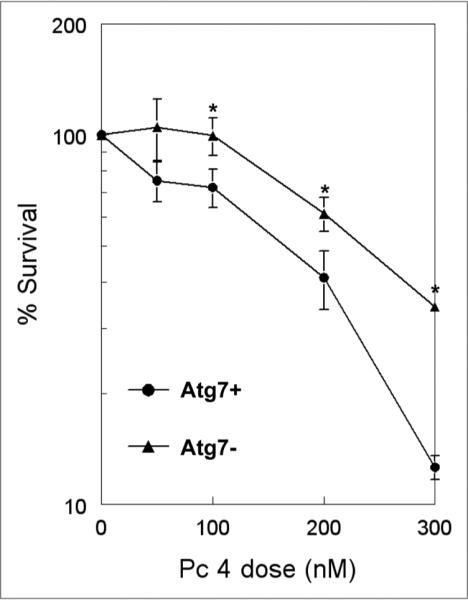
Clonogenic survival of PDT-treated Atg7+ and Atg7− MCF-7 cells. Cells were incubated with various concentrations of Pc 4 for 18 h, as indicated on the abscissa, then irradiated with 200 mJ/cm2 red light, collected and diluted to the appropriate concentrations for plating. Data for PDT-treated cells were normalized to the plating efficiency of the corresponding untreated controls (on average, 49.5% for Atg7+ cells and 38.4% for Atg7− cells). Each datum is the mean ± standard deviation of at least triplicate results from three independent experiments (*p < 0.05, t-test).
Although MCF-7 cells are procaspase-3-deficient, they express other caspases, and both caspase-dependent and caspase-independent apoptotic pathways could be functionally active in these cells under some circumstances.34 To test caspase-dependent apoptosis after PDT, the cleavage of PARP was examined (Fig. 4). As positive controls, some cells were treated with camptothecin (CPT) or staurosporine (STS). For the Atg7+ cells, PARP cleavage was observed in response to STS but not after PDT or CPT. In the Atg7− cells, there was also no PARP cleavage before or after PDT; however, PARP was extensively cleaved upon exposure to CPT (Fig. 4A) or STS (Fig. 4B).
Figure 4.
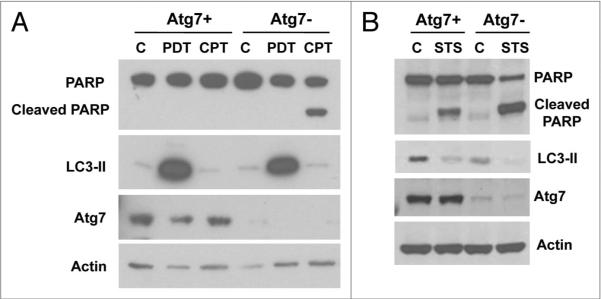
Atg7 knockdown enhanced PARP cleavage in CPT- or STS-treated, but not in PDT-treated, MCF-7 cells. C: Control. MCF-7 cells (Atg7+ and Atg7−) were exposed to 200 nM Pc 4 and 200 mJ/cm2 red light then further incubated for 24 h or treated with 2 µM CPT for 24 h (A). Cells were also incubated in 1 µM sTs for 6 h (B). At the end of the incubation, whole cell lysates were subjected to electrophoresis and analyzed by western blot for the presence of PARP cleavage, and the levels of LC3 and Atg7.
Discussion
The present study shows that human breast cancer MCF-7 cells that are deficient in typical apoptosis in response to PDT become resistant to PDT-induced cell killing when autophagy is also impaired. This was seen for MCF-7v cells when autophagy was inhibited by 3-MA or wortmannin9 or by knockdown of LC3 (Figs. 1 and 2), and for the MCF-7/Atg7− cells in comparison to their Atg7+ counterparts (Fig. 3). In contrast, the PDT response of MCF-7c3 cells, which overexpress procaspase-3 and are apoptosis-competent, was little affected by inhibition of autophagy (reviewed in ref. 9 and Fig. 2). A third pattern of response to PDT during inhibition of autophagy was found by Kessel et al. in murine leukemia L1210 cells;21,22 i.e., sensitization to PDT when autophagy was downregulated by shRNA knockdown of Atg7. Although there are several differences between the conditions of our and Kessel’s experiments (adherent human breast carcinoma vs. murine leukemia cells growing in suspension; different photosensitizers), it is tempting to speculate that one important factor determining the differential responses is the contribution of apoptosis in each cell type. For the leukemia cells, apoptosis is rapidly induced by PDT; for MCF-7 cells, apoptosis is deficient in lines that have not been transfected with procaspase-3, and even in apoptosis-competent MCF-7c3 cells, the rate of apoptosis is not as rapid as in lymphoid-derived cells.35 Thus, it is possible that the speed with which apoptosis proceeds may be a determinant of the role of autophagy in PDT response.
Both protection against cell death and sensitization to other (non-PDT) stressors has been observed when Atg7 is knocked down. A dramatic inhibition of cell death was observed in both Atg7 knockdown HCT116 (TRAIL-sensitive) and U87 (TRAIL-resistant) cells in response to HW1 (human single-chain fragment variable (scFv) antibody against TRAIL receptor 2).24 Similarly, for human osteosarcoma cells (U2OS) treated with avicin D, a plant triterpenoid, cell death was reduced when Atg7 was silenced.26 Yu et al.25 found that reducing expression of Atg7 or Beclin 1 inhibited zVAD-induced death of L929 and U937 cells. In other cases, sensitization of cells has resulted from decreased expression of Atg7. Kessel et al.22 reported that when L1210 leukemia cells were PDT-treated with either of two photosensitizers, CPO or MC, L1210/Atg7− cells were more sensitive than L1210 wild-type cells at a low dose, but the two cell lines exhibited comparable cytotoxicity at a higher PDT dose. When MCF-7 cells were treated with camptothecin (CPT), a DNA-damaging agent, knockdown of Atg7 activated apoptosis, resulting in reduction of cell viability.23 Cell type and stimulus-dependent cell response has also been observed for knockdown of expression of other ATG genes. Mouse embryonic fibroblasts (MEFs) deficient in Atg5 were more sensitive to treatment with STS or tunicamycin but more resistant to treatment with menadione or UVC radiation.36 However, Atg5− HeLa cells were more sensitive to tunicamycin, but not to STS, than were Atg5+ HeLa cells.37 Kim et al.38 found that inhibition of autophagy by knock-down of Atg5 and Beclin 1 results in ionizing radiation resistance in Bax/Bak double knock-out MEFs. Apel et al.39 studied the interference with autophagy by siRNA for several different autophagy proteins in a variety of human cancer cell lines exposed to ionizing radiation. They found that downregulation generally sensitized the cells to X radiation, although there were several combinations of siRNA knockdowns and radiation doses where some cells were protected from cell death. Thus, the role of autophagy, as revealed by the elimination of Atg5 or Atg7 expression, is dependent upon the cellular stress/toxin and the capacity of the cell to carry out apoptosis. In this context, the finding of different roles for autophagy in PDT-treated cells is not surprising. Our results demonstrate that whether or not apoptosis is activated in MCF-7 cells is stimulus-dependent. When MCF-7/Atg7− cells were treated with CPT or STS, more apoptosis, as indicated by PARP cleavage, was induced than in MCF-7/Atg7+ cells, which confirms the report of Abedin et al.23 In contrast, when MCF-7 cells were treated with PDT, or when L929 and U937 cells were treated with an inhibitor of apoptosis, z-VAD,25 autophagy but not apoptosis was a prominent pathway of cell death, and Atg7 knockdown protected against cell death (Fig. 3 and reviewed in ref. 25).
Recently, Nishida et al.40 reported that MEFs lacking Atg5 were capable of carrying out autophagy in response to etoposide or starvation. Furthermore, when both Atg5 and Atg7 were silenced, autophagy remained functional, providing evidence for an Atg5/Atg7-independent pathway of macroautophagy. However, MEFs lacking Atg5 or Atg7 did not generate LC3-II in response to etoposide. It is unlikely that the major autophagy pathway operating in our MCF7 cells in response to PDT is identical to that revealed by Nishida et al.40 because high levels of LC3-II accumulated after PDT irrespective of the presence of Atg7 (Fig. 4). Studies with transfected GFP-LC3 are useful for evaluating the ability of cells to incorporate LC3-II into the maturing autophagosomal membrane; during this process, the pattern of GFP-LC3 fluorescence changes from diffuse to punctate. In response to Pc 4-PDT, MCF-7/Atg7+ cells developed the punctate pattern of GFP fluorescence, whereas the pattern remained mostly diffuse in Atg7− cells, indicative of a defect in the incorporation of the lipidated LC3 into autophagosomes (Fig. 2C and D). These observations suggest that the accumulation of high levels of LC3-II in PDT-treated cells is at least partially a result of PDT-induced damage to a later step in autophagy, preventing LC3-II turnover. Experiments with the protease inhibitors E64d and pepstatin A are consistent with this notion, since they did not further increase the accumulation of LC3-II.9 The use of PDT as the triggering agent has the potential to reveal previously undiscovered or atypical elements of the autophagic mechanism.
Atg7 is considered to be essential in both arms of the pathway for expanding the autophagosomal membrane. As clearly described in Cecconi and Levine,41 in the first arm, Atg7 promotes conjugation of Atg8 (LC3) with phosphatidylethanolamine (PE), generating LC3-II. In the second arm, Atg7 conjugates Atg12 to Atg5, forming a complex that is required to insert LC3-II into the growing autophagosomal membrane.
Our data suggest that elimination of Atg7 from MCF7 cells has greater consequences for the Atg5-Atg12 arm of the pathway than for the Atg8 arm following PDT, since the MCF7/Atg7− cells were not impaired in the formation of LC3-II after PDT (Fig. 4) but were defective in the insertion step (Fig. 2). However, since the knockdown may not be complete, there is the possibility that a small amount of residual Atg7 may be sufficient for LC3-II formation or another unidentified component of the system may be able to substitute for Atg7 during LC3-II formation but not during Atg5-Atg12 conjugation. Because Levine and co-workers did not observe Beclin 1 in their MCF-7 cultures,32,33 we considered the possibility that a lack of Beclin 1 may allow LC3-II formation in the absence of Atg7. However, all of the MCF-7 lines we have studied express Beclin 1 (Fig. 2E), so that its absence cannot be driving the atypical LC3-II formation following Pc 4-PDT.
Another interesting difference between our observations and those of Kessel and Reiners22 relates to the ability of the cells to generate LC3-II in response to PDT when expression of Atg7 is deficient. Whereas L1210/Atg7− cells are clearly defective in LC3-II formation after PDT with CPO or mesochlorin (MC) as photosensitizer, as mentioned above MCF7/Atg7− cells appear to be fully capable of producing high levels of LC3-II in response to Pc 4-PDT. CPO and MC bind mainly to the endoplasmic reticulum (ER) and mitochondria, respectively,22 and Pc 4 localizes to both mitochondria and ER.12 Thus, subcellular localization of the photosensitizer may not be the primary cause of the differential roles of autophagy in PDT response. However, a systematic investigation of how these differences might contribute to the opposing effects of Atg7 knockdown on photocytotoxicity will be explored in future work.
Materials and Methods
Cell culture
Human breast cancer MCF-7 cells transfected with Atg7 shRNA (Atg7−) or a scrambled sequence (Atg7+) were provided by Dr. A. Kelekar, University of Minnesota. The cells were grown in minimal essential medium (MEM) with 10% fetal bovine serum (FBS), 1% nonessential amino acids and 1 mM sodium pyruvate. MCF-7 cells transfected with human procaspase-3 cDNA (MCF-7c3) or empty vector (MCF-7v) were obtained from Dr. C. Froehlich, Northwestern University, and cultured in RPMI-1640 medium containing 10% FBS. All cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 in air.
Photodynamic treatment
The phthalocyanine photosensitizer Pc 4, HOSiPcOSi(CH3)2(CH2)3N(CH3)2, was provided by Dr. Malcolm E. Kenney, Case Western Reserve University Department of Chemistry.27 It was dissolved in dimethylformamide to 0.5 mM and further diluted in complete medium. Cells were loaded with Pc 4 by addition of an aliquot of the stock solution to the culture medium either ~18 h or ~4 h (for MTT assay) before irradiation. The light source was an EFOS LED array (EFOS, Mississauga, ONT, Canada) delivering red light (λmax ≈ 675 nm). All irradiations were performed at room temperature.
siRNA transfection
A pool of four siRNA duplexes targeting human LC3 RNA were purchased from Dharmacon Inc., (L-012846-00). Transfection was performed using Lipofectamine 2000 (Invitrogen, 11668-027) according to the manufacturer’s protocol.
GFP-LC3 transfection and fluorescence
MCF-7 cells were transiently transfected with GFP-LC3 construct (generous gift from Dr. T. Yoshimori, National Institute of Genetics, Japan) using Lipofectamine 2000 (Invitrogen, 11668-027). Twenty-four hours after transfection, cells were PDT-treated, then returned to the incubator for various times up to an additional 24 hours. At the end of the post-incubation, cells were examined by fluorescence microscopy and photographed. For quantitation, the percentage of GFP-LC3-positive cells with punctate fluorescence was determined. For each experimental condition, at least 300 GFP-positive cells per sample were counted from each of three separate experiments. Statistical analysis was performed using Student’s t-test.
MDC labeling
Cells grown in 35-mm Petri dishes were untreated or treated with Pc 4-PDT. At various times thereafter, cells were incubated in medium with 10 µM monodansylcadaverine (MDC, Sigma, 30432) for 20 min at 37°C, then washed with PBS twice. Images were examined immediately by fluorescence microscopy.
Clonogenic assay
Atg7+ and Atg7− MCF-7 cells were collected from the culture monolayer with trypsin immediately after PDT. Aliquots of the cells were plated in triplicate into 6-cm Petri dishes in amounts sufficient to yield 50–100 colonies per dish. After incubation for ~10 days, the cells were stained with 0.1% crystal violet in 20% ethanol, and colonies containing at least 50 cells were counted. The average plating efficiencies of untreated Atg7+ and Atg7− MCF-7 cells were 49.5% (range 38–60%) and 38.4% (range 34–44%), respectively.
Assay of cell viability
Cells were seeded in 96-well plates at 1.5 × 104 cells/well and allowed to attach overnight. The next day, cells were transfected with LC3 siRNA. After 24 h, the medium was removed and replaced with fresh medium with or without various doses of Pc 4. The cells were incubated for ~4 h, then photoirradiated and incubated for an additional 24 h. Cell viability was measured using MTT (Sigma-Aldrich, M-2128), according to the manufacturer’s instructions.
Western blot analysis
Cells were washed twice with cold PBS and lysed in 1X SDS sample buffer (31.25 mM Tris, pH 6.8, 1% SDS, 2.5% mercaptoethanol and 5% glycerol) and sonicated. Equivalent amounts of protein were loaded onto polyacrylamide gels, subjected to electrophoresis, transferred to a PVDF membrane and incubated with anti-LC3 antibody (a kind gift from Dr. T. Yoshimori, National Institute of Genetics, Japan), anti-Atg7 antibody (ProSci Inc., 3617), anti-PARP antibody (BD Pharmingen, 556362), anti-Beclin 1 antibody (BD Transduction Laboratories, 612113), or anti-actin antibody (NeoMarkers, MS-1295). The immune complexes were detected by ECL system (Amersham, RPN2106).
Acknowledgements
The authors are grateful to Dr. A. Kelekar (University of Minnesota) for the MCF7/Atg7+ and MCF7/Atg7− cells, to Dr. C. Froehlich (Northwestern University) for the MCF7v and MCF7c3 cells, and to Dr. T. Yoshimori (National Institute of Genetics, Japan) for the GFP-LC3 cDNA and the anti-LC3 anti-body. This research was supported by R01 CA106491 from the National Cancer Institute, DHHS.
REFERENCES
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12:1542–52. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76. [PMC free article] [PubMed] [Google Scholar]
- Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- Kessel D, Vicente MG, Reiners JJ., Jr. Initiation of apoptosis and autophagy by photodynamic therapy. Lasers Surg Med. 2006;38:482–8. doi: 10.1002/lsm.20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. Faseb J. 2006;20:756–8. doi: 10.1096/fj.05-4305fje. [DOI] [PubMed] [Google Scholar]
- Xue LY, Chiu SM, Azizuddin K, Joseph S, Oleinick NL. The death of human cancer cells following photodynamic therapy: apoptosis competence is necessary for Bcl-2 protection but not for induction of autophagy. Photochem Photobiol. 2007;83:1016–23. doi: 10.1111/j.1751-1097.2007.00159.x. [DOI] [PubMed] [Google Scholar]
- Wang M, Tan W, Zhou J, Leow J, Go M, Lee HS, Casey PJ. A small molecule inhibitor of isoprenyl-cysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J Biol Chem. 2008;283:18678–84. doi: 10.1074/jbc.M801855200. [DOI] [PubMed] [Google Scholar]
- Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, et al. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleinick NL, Morris RL, Belichenko I. The role of apoptosis in response to photodynamic therapy: what, where, why and how. Photochem Photobiol Sci. 2002;1:1–21. doi: 10.1039/b108586g. [DOI] [PubMed] [Google Scholar]
- Oleinick NL, Evans HH. The photobiology of photodynamic therapy: cellular targets and mechanisms. Radiat Res. 1998;150:146–56. [PubMed] [Google Scholar]
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–74. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP and MAP-LC3. J Biol Chem. 2001;276:1701–6. doi: 10.1074/jbc.C000752200. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel D, Arroyo AS. Apoptotic and autophagic responses to Bcl-2 inhibition and photodamage. Photochem Photobiol Sci. 2007;6:1290–5. doi: 10.1039/b707953b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel D, Reiners JJ., Jr. Apoptosis and autophagy after mitochondrial or endoplasmic reticulum photodamage. Photochem Photobiol. 2007;83:1024–8. doi: 10.1111/j.1751-1097.2007.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–10. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- Park KJ, Lee SH, Kim TI, Lee HW, Lee CH, Kim EH, et al. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res. 2007;67:7327–34. doi: 10.1158/0008-5472.CAN-06-4766. [DOI] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- Xu ZX, Liang J, Haridas V, Gaikwad A, Connolly FP, Mills GB, Gutterman JU. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell Death Differ. 2007;14:1948–57. doi: 10.1038/sj.cdd.4402207. [DOI] [PubMed] [Google Scholar]
- Oleinick NL, Antunez AR, Clay ME, Rihter BD, Kenney ME. New phthalocyanine photosensitizers for photodynamic therapy. Photochem Photobiol. 1993;57:242–7. doi: 10.1111/j.1751-1097.1993.tb02282.x. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–9. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 2005;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Simstein R, Burow M, Parker A, Weldon C, Beckman B. Apoptosis, chemoresistance and breast cancer: insights from the MCF-7 cell model system. Exp Biol Med. 2003;228:995–1003. doi: 10.1177/153537020322800903. [DOI] [PubMed] [Google Scholar]
- Ke MS, Xue LY, Feyes DK, Azizuddin K, Baron ED, McCormick TS, et al. Apoptosis mechanisms related to the increased sensitivity of Jurkat T-cells vs. A431 epidermoid cells to photodynamic therapy with the phthalocyanine Pc 4. Photochem Photobiol. 2008;84:407–14. doi: 10.1111/j.1751-1097.2007.00278.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem. 2008;283:4766–77. doi: 10.1074/jbc.M706666200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Mutter RW, Cao C, Albert JM, Freeman M, Hallahan DE, Lu B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J Biol Chem. 2006;281:36883–90. doi: 10.1074/jbc.M607094200. [DOI] [PubMed] [Google Scholar]
- Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell dealth. Dev Cell. 2008;15:344–57. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]