

Abstract
Recent studies have identified FKBP51 (FK506-binding protein 51) as a sensitive biomarker of corticosteroid responsiveness in vivo. In this work, we have elucidated the molecular mechanisms underlying the induction of FKBP51 by the glucocorticoid receptor (GR) in human A549 lung cancer cells showing robust accumulation of FKBP51 mRNA in response to dexamethasone exposure. Our quantitative chromatin immunoprecipitation scans and enhancer activity analyses indicate that activation of the FKBP51 locus by glucocorticoids in vivo is triggered by the loading of GR to enhancers at about 34 kb 5′ and about 87 kb 3′ of the transcription start site. Interestingly, the region encompassing these enhancers is bordered by CCCTC-binding factor- and cohesin-binding sites. Dexamethasone treatment also decreased the histone density at several regions of the gene, which was paralleled with the occupancy of SWI/SNF chromatin remodeling complexes within the locus. Moreover, silencing of BRM subunit of the SWI/SNF complex blunted the glucocorticoid induction of the locus. The proximal promoter region along with the major intronic enhancer at approximately 87 kb, at which the GR binding peaked, had elevated levels of histone 3 acetylation and H3K4 trimethylation, whereas H3K36 trimethylation more generally marked the gene body and reflected the occupancy of RNA polymerase II. The occurrence of these active chromatin marks within the FKBP51 locus before glucocorticoid exposure suggests that it is poised for transcription in A549 cells. Taken together, these results indicate that the holo-GR is capable of activating transcription and evoking changes in chromatin structure through distant-acting enhancers.
Chromatin immunoprecipitation and enhancer activity assays show that holo-glucocorticoid receptor activates pre-programmed FKBP51 locus through enhancers that reside far from the transcription start site.
Glucocorticoid receptor (GR) is a member of the nuclear receptor superfamily that regulates vital metabolic and developmental processes (1). Glucocorticoids are among the most widely prescribed pharmaceuticals worldwide and key drugs for treating diseases ranging from hematological cancers, such as leukemia, to inflammatory states, including asthma (2, 3, 4). The GR is a ligand-regulated transcription factor that in the absence of glucocorticoids resides in the cytoplasm associated with a heat shock protein 90 chaperone complex that keeps the receptor inactive. After binding of the ligand, the complex dissociates from GR and the receptor is transported to the nucleus where it activates transcription by associating with high affinity to short DNA sequences, glucocorticoid response elements (GREs), in its target genes (1). GR also inhibits transcription, but the inhibition appears to rely more frequently on the repression (antagonism) of other transcription factors, such as activator protein 1 or nuclear factor-κB, than on direct interaction with DNA target sites (5).
In addition to the basal transcription machinery and RNA polymerase II, activation of genes and transcriptional programs by nuclear receptors requires interactions with coregulator proteins that often operate as subunits in chromatin-modifying complexes (6, 7, 8). Nuclear receptors may recruit the adenosine triphosphatase subunits of SWI/SNF chromatin-remodeling complex, with the alternative recruitment of the BRM or the BRG1 being dependent on the target gene or the tissue (8, 9, 10, 11). The SWI/SNF chromatin-remodeling complexes are thought to alter histone-DNA interactions by sliding or transferring nucleosomes in an ATP-dependent manner. They can thus regulate the accessibility of transcription factor binding sites. Many of the coregulator proteins harbor or recruit enzymatic activities, such as acetyltransferase and methyltransferase activity, which covalently modify specific amino acids in histone tails and thereby modify chromatin structure (6, 12). Histone acetylation is almost invariably linked to activation of transcription. Acetylated core histones are enriched around transcription start sites (TSSs) and proximal promoter regions of active genes. The effects of histone methylations on the gene activity, in turn, depend on the context, i.e. the identity of the modified residue and the gene region (13, 14). For example, trimethylation of histone 3 (H3) at K27, which is enriched across inactive genes, constitutive heterochromatin and in repetitive DNA, is a common repressive mark, whereas di- and trimethylation of H3 at K4 and K36 generally represent active, euchromatin, marks (15, 16). Association of H3K9 trimethylation with gene activity may be more complex, because it is negative on the promoter regions, but may be positive on the coding regions (14).
GR target genes have recently been identified on genome-wide levels (10, 17, 18, 19). On an individual locus and chromatin level, however, the GR function has been addressed in a detailed fashion only on a few targets, such as glucocorticoid-induced leucine zipper (GILZ), tyrosine aminotransferase (TAT), and mouse mammary tumor virus long terminal repeat (MMTV LTR) (Refs. 20 and 21 and references therein). However, these model genes harboring GREs in their proximal or near-proximal promoter regions may not be the best representative GR targets, because recent larger scale screens for GR- and other steroid receptor-binding sites have revealed that most of the steroid-regulated genes lack response elements in their promoters, but binding sites more often reside more than 10 kb from promoters, frequently downstream of them (18, 22, 23, 24). Therefore, long-range activation of transcription is emerging as the major mechanism by which the steroid receptors exert their function on their targets. However, there is still scarce information on the molecular mechanisms of the longer-range regulation by the GR on a single-target locus level (25).
Recent studies have identified FK506-binding protein 51 (FKBP51) as a sensitive biomarker of corticosteroid responsiveness in vivo (26, 27, 28), and it was identified as the most highly glucocorticoid-induced gene in a recent genome-wide profiling of human lung biopsies (26). FKBP51 belongs to the immunophilin protein family and is a component of the heat shock protein 90 chaperone complex (29). Overexpression of FKBP51 may limit GR signaling, and the FKBP51 is therefore a potential mediator of corticosteroid resistance (30, 31, 32). Interestingly, FKBP51 has also been found recently to be a negative regulator of the Akt pathway and suggested to affect the response of cancer cells to chemotherapy (33). Moreover, polymorphisms in the FKBP51 have been associated with differences in GR sensitivity and glucocorticoid signaling in stress-related psychiatric disorders (34).
Due to the high glucocorticoid sensitivity of the FKBP51 and scarce information on the action of GR on the target chromatin level, we have elucidated the locus-wide mechanisms of FKBP51 activation by glucocorticoids in human A549 lung cancer cells that display characteristics of alveolar epithelial cells (35). Our results reveal that the holo-GR exerts long-range regulation of the FKBP51 transcription through distal sites that are located both upstream and far downstream (∼100 kb) from the TSS of the gene. Interestingly, analyses of the histone marks within the gene imply that the locus chromatin was poised for transcription in the A549 cells. We also addressed the occurrence of boundary element/insulator-binding protein CTCF (CCCTC-binding factor), which is implicated in the global organization of chromatin architecture (36), within the FKBP51 and its intergenic regions. Because the FKBP51 locus is similarly induced by androgens (37), we also compared the binding of androgen receptor and CTCF to the same chromosome 6 region in VCaP prostate cancer cells.
Results
Glucocorticoid-induced accumulation of FKBP51 mRNA in A549 cells
According to the University of California Santa Cruz Genome Browser (http://genome.ucsc.edu/), the FKBP51 locus harbors, in addition to the previously investigated transcript (variant 1), a longer transcript (variant 2) of which TSS is located at approximately 40 kb upstream from that of the variant 1 (cf. Fig. 2A). The regulation of FKBP51 by glucocorticoids in human A549 cells was first investigated by isolating cellular RNA at different time points after adding an optimal concentration of glucocorticoid (100 nm dex; Fig. 1A) and performing RT-quantitative PCR (RT-QPCR) analyses using primers specific for variants 1 and 2. As shown in Fig. 1B, FKBP51 mRNA variant 1 was rapidly and strongly induced after glucocorticoid exposure, with the induction being more than or equal to 10-fold at 3 h and reaching maximum (≥40-fold) at 12 h, whereas the accumulation of FKBP51 mRNA variant 2 under these conditions was negligible. GILZ showed more than or equal to 10-fold glucocorticoid induction at 3 h, but its overall induction saturated already at that time point. In comparison with the FKBP51 or the GILZ, another GR target S100P (17) showed clearly delayed and weaker response to dexamethasone (dex).
Fig. 2.

Function of the putative FKBP51 GRE-containing regions as enhancers. A, Schematic locations of the GREs and regions analyzed in reporter and ChIP assays. Bars depict the positions of exons, and arrows indicate the TSSs of the different FKBP51 transcript variants, with the bolded arrow indicating the major transcript start site. Roman numerals depict the in silico-identified GREs, and Arabic numerals indicate the regions amplified for enhancer analyses and ChIP assays. B, Sequences of the potential GREs in FKBP51 gene. Top, Position weight matrix used for identification of putative GREs. The frequency of nucleotides at each of the 15 positions in the GRE in the matrix is shown. Lower, List of the in silico-identified GREs, their localization in the FKBP51 gene, and their sequences. C and D, Transcriptional activity of the GRE-containing 300-bp FKBP51 fragments was assessed by reporter gene assays. A549 (C) and COS-1 (D) cells were transiently transfected with TATA-LUC constructs driven by indicated regions of FKBP51 or empty pTATA-LUC (TATA) as described in Materials and Methods. For COS-1 (D) analyses, pSG5-hGR (0.2 μg/well) was cotransfected with the reporter constructs. Cotransfection of pCMVβ and β-galactosidase activity was used for normalization of transfection efficiency. The cells were treated with vehicle (ethanol, EtOH) or 100 nm dex for 16 h before the cells were harvested for reporter analyses. Results are shown as relative LUC activities and fold inductions of glucocorticoid-treated samples in relation to the activity of ethanol-treated samples are shown above the columns. Columns represent the mean ± sd of three independent experiments.
Fig. 1.
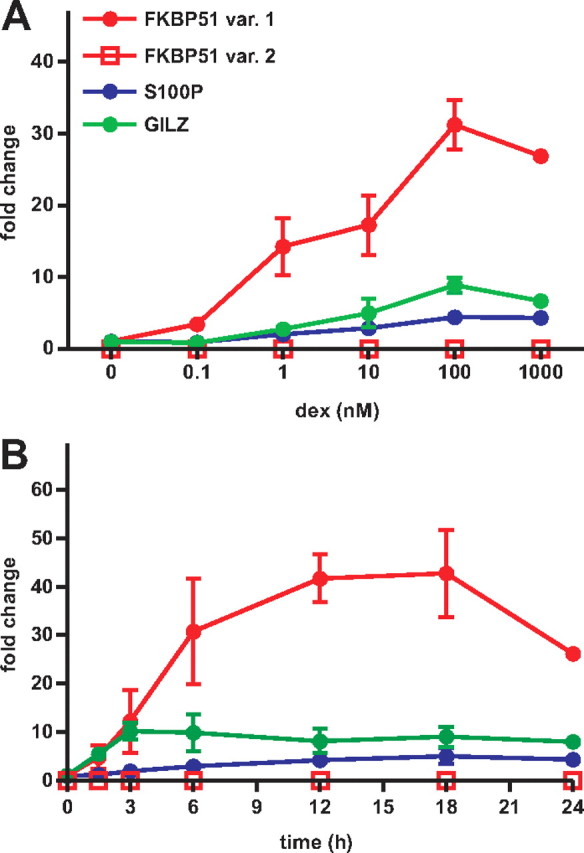
Rapid and robust activation of FKBP51 expression by dex in A549 cells. A, Cells were treated with indicated concentrations of synthetic glucocorticoid (dex) for 12 h, and the amounts of FKBP51 variant 1 (var. 1) and variant 2 (var. 2), S100P, and GILZ mRNAs were analyzed by RT-QPCR and normalized as described in Materials and Methods. B, A549 cells were treated with 100 nm dex for indicated times, and the analysis of mRNAs was performed as above. Columns represent the mean ± sd of three independent experiments.
Function of the putative FKBP51 GRE-containing regions as glucocorticoid-regulated enhancers
The robust induction of FKBP51 in response to glucocorticoids implied that there may be more than one GRE-containing region mediating the effect of holo-GR on the gene transcription. Because the FKBP51 expression is similarly up-regulated by glucocorticoids as androgens and the GR and the androgen receptor (AR) recognize a common consensus sequence, we considered the previously CONSITE program-predicted androgen response elements (AREs) from −20 kb 5′ of the FKBP51 TSS (numbering corresponding to the TSS of the major FKBP51 transcript variant 1) to 20 kb 3′ of its last exon as putative GREs in this study (37, 38) (Fig. 2B). Also, because the FKBP51-neighboring C6orf81 locus turned out to be activated by androgens (cf. Fig. 7, D and E), we extended our in silico searches further upstream from the FKBP51 to cover the intergenic region between these two loci. This process resulted in identification of four additional putative ARE/GREs, GRE-V, GRE-IV, GRE-III, and GRE-II, on the intergenic region between the FKBP51 and the C6orf81, more than 30 kb 5′ of the TSS of the major FKBP51 transcript variant 1. Altogether, the genomic region of chromosome 6 from 35,521,362 to 35,704,858 spanning approximately 180 kb was predicted to contain 17 putative high-affinity binding sites for GR (Fig. 2B).
Fig. 7.

Differential activation of C6orf81 in A549 and VCaP cells by glucocorticoid and androgen, respectively. A, Schematic map of the FKBP51 and its neighboring genes C6orf81 and TULP1 in chromosome 6. Angled arrows indicate the TSSs of different genes, bolded regions show the schematic locations of the genes, and dashed line depicts extra 5′-region of the FKBP51 transcript variant 2. A549 cells and VCaP cells were treated with increasing concentrations of dex (B) and R1881 (D), respectively, for 12 h or with 100 nm dex (C) or 10 nm R1881 (E) for indicated times, and mRNAs were analyzed by RT-QPCR analysis as described in Fig. 1. Columns represent the mean ± sd of three independent experiments. F, Loading of AR to the FKBP51 locus in VCaP cells. Cells were treated with vehicle (EtOH) or 10 nm R1881 for 2 h. ChIP assays were performed using anti-AR antibody, and the precipitated DNAs were used as templates in QPCR. Chr., Chromosome.
Next we tested whether the in silico-predicted GRE-containing regions are capable of conferring glucocorticoid regulation to a reporter gene in A549 cells. Approximately 300-nt regions centering the predicted GREs (cf. Fig. 2A) were cloned in front of a TATA box-containing luciferase plasmid and transfected to the A549 cells treated with or without glucocorticoid. As shown in Fig. 2C, region 10 encompassing GREIX, GREX, and GREXI (with ∼30-fold induction), region 9 containing GREVII and GREVIII (with ∼120-fold induction), and region −3 harboring GRE-III and GRE-II (with ∼6-fold induction) were the only regions conferring marked glucocorticoid activation to the reporter gene in the A549 cells. These three FKBP51 regions were capable of mediating marked glucocorticoid induction of transcription also in COS-1 cells cotransfected with a GR expression vector, but the fold inductions, with the exception of region −3, were weaker in COS-1 than in A549 cells (Fig. 2D). In both cell lines, however, region 9 showed the strongest glucocorticoid induction. Taken together, these results indicate that the region at approximately 34 kb 5′ of the TSS and the regions at about 87 and 100 kb 3′ of the TSS of FKBP51 show characteristics of GR-regulated enhancers. All these three putative enhancer regions encompass more than one potential GRE.
Glucocorticoid-triggered binding of the GR to the distal regions in FKBP51 chromatin
We next investigated the binding of GR to the FKBP51 locus in vivo by performing chromatin immunoprecipitations (ChIPs) in A549 cells with an anti-GR antibody followed by quantitative real-time PCR analysis. In addition to the regions harboring the in silico-predicted GREs, these assays included four regions without predicted GREs, one on the both side of the TSS (regions −1 and 1), one approximately 46 kb downstream of the TSS (region 6) and one 3′ of the gene (region 12). Altogether, 17 regions of FKBP51 were scanned for GR binding (cf. Fig. 2A). A region located in the middle of non-glucocorticoid-responsive DSCAM in chromosome 22 served as a negative control. A549 cells were exposed to dex for various times (0–120 min at 20-min intervals) before ChIPs. Loading of GR was already clearly detectable on region −3 upstream and region 9 downstream from the FKBP51 TSS 20 min after the dex exposure (Fig. 3A). At later time points (≥80 min), regions 5, 8, and 10 also showed more than 10-fold glucocorticoid-stimulated occupancy of the receptor. However, region 9 was the major holo-GR- occupying chromatin region at all time points, with the induction by dex peaking at the 40-min time point (∼45-fold induction). A closer look at the kinetics of the receptor loading during the first 120 min revealed two waves of GR binding to regions −3 and 9, whereas only one delayed wave took place at regions 8 and 10 (Fig. 3B). The loading of holo-GR to regions 5 and 8 was more prominent than expected on the basis of their weak or absent enhancer function, whereas the occupancy of the receptor on chromatin regions −3, 9, and 10 correlated well with their isolated enhancer activity both in A549 cells and also in COS-1 cells. Notably, the FKBP51 region 9 at approximately 87 kb 3′ of the TSS displayed both the strongest enhancer activity and the highest GR chromatin occupancy in response to glucocorticoid exposure in A549 cells.
Fig. 3.
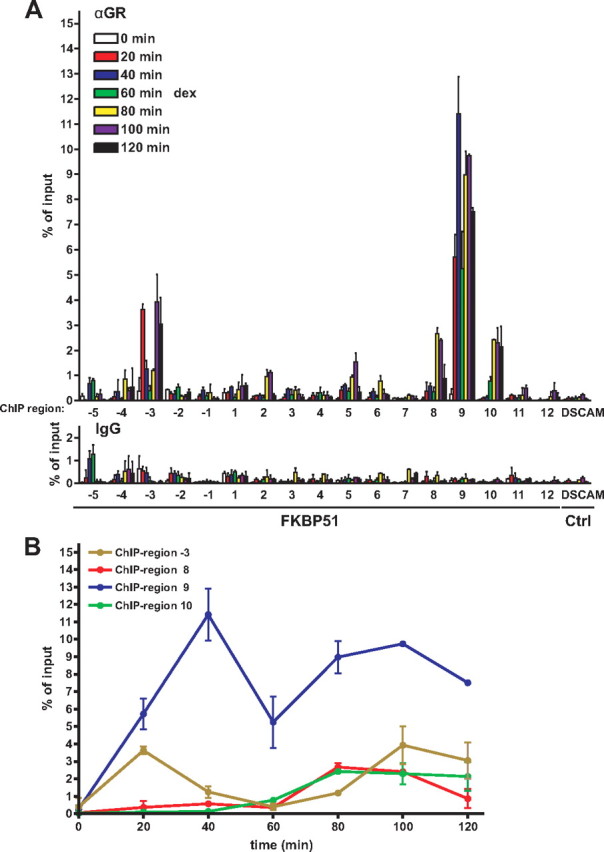
Temporo-spatial loading of GR to the FKBP51 locus in response to glucocorticoid exposure in A549 cells. A, Cells were treated with 100 nm dex for indicated times, and ChIPs with anti-GR antibody followed by real-time PCR were carried out as described in Materials and Methods to investigate the occupancy of GR within 17 different regions spanning approximately 160 kb of chromosome 6 that covers the FKBP51 locus plus its surroundings (cf. Fig. 2A). A region in the middle of DSCAM in chromosome 22 served as a nontarget control. ChIP assays with normal IgG monitored nonspecific binding. Results are shown as percentages of the input samples. Columns represent the mean ± sd of three experiments. B, Binding of GR to the FKBP51 regions −3, 8, 9, and 10 plotted against time after addition of the hormone. Ctrl., Control.
RU486 inhibits the binding of holo-GR onto the FKBP51 locus
We also tested the effect of the antagonist RU486 (mifepristone) on the FKBP51 transcription and whether RU486-occupied GR binds to the locus. Although the antagonist exhibited some partial agonistic activity on the FKBP51 transcription, yielding a small increase in the amount of the FKBP51 mRNA, an excess of RU486 efficiently inhibited (∼84%) the agonistic activity of dex (Fig. 4A). Similar to the effect of RU486 on the accumulation of FKBP51 mRNA, it inhibited the loading of agonist-occupied GR to the FKBP51 locus and was (on its own) inefficient in promoting the interaction of GR with the chromatin (Fig. 4B). These results strongly suggest that one major mechanism by which the RU486 can block the GR activity is by blunting the receptor’s capability to interact with its binding sites in chromatin.
Fig. 4.

Effect of RU486 on the FKBP51 expression and the GR loading to the locus chromatin. A, Accumulation of FKBP51 mRNA in response to exposure of A549 cells to dex (100 nm) or RU486 (1000 nm) alone or in combination relative to the mRNA level with vehicle (EtOH) for 12 h. RNAs were analyzed by RT-QPCR analysis as described in Fig. 1. B, Loading of GR to the FKBP51 locus in response to agonist or antagonist exposure alone or in combination for 2 h in A549 cells. ChIP scans were performed using anti-GR antibody followed by quantitative PCR analysis. Results are shown as percentage of the input samples and represent the mean ± sd of three experiments.
Histone H3 modifications and the occupancy of RNA polymerase II within the FKBP51 locus
Because the above assays suggest that the holo-GR is capable of exerting long-range regulation of transcription, we also monitored the occupancy of RNA polymerase II (PolII) within the FKBP51 locus in A549 cells cultured with or without dex for 2 h. As shown in Fig. 5B, glucocorticoid clearly increased the occupancy of PolII throughout the locus as assessed by ChIP using an antibody against the largest subunit of the PolII. PolII occupancy peaked around the TSS and at the 3′-end of the gene. Because the PolII occupancy does not reflect that of the holo-GR in the locus, this result suggests that chromatin looping is involved in the glucocorticoid-induced transcription of the FKBP51 locus.
Fig. 5.
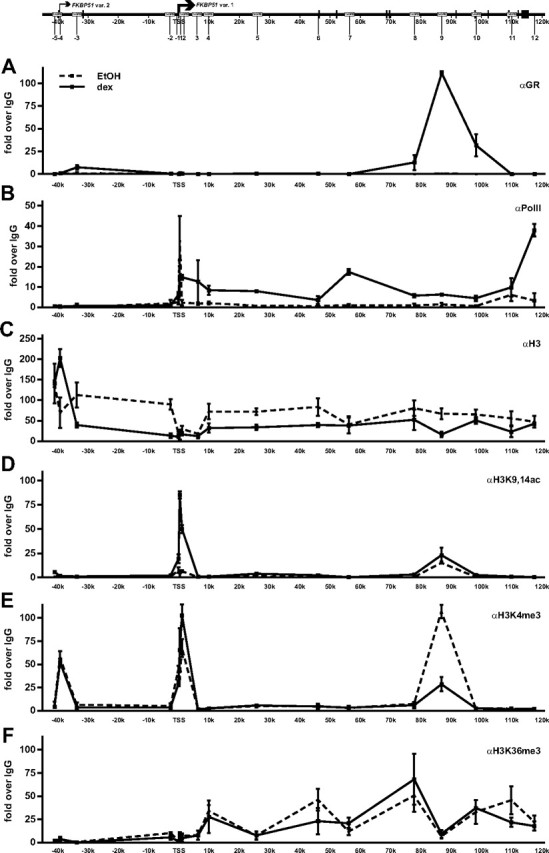
Occupancy of RNA PolII and occurrence of H3 modifications within the FKBP51 locus in A549 cells in the presence and absence of glucocorticoid. Cells were treated with vehicle (EtOH) or 100 nm dex for 2 h, and ChIP assays were performed using antibodies against GR (A), PolII (B), H3 (C), H3K9,14ac (D), H3K4me3 (E), or H3K36me3 (F). ChIP samples were used as templates in qPCR. Results are shown as fold over the normal rabbit IgG-precipitated samples and represent the mean ± sd of three experiments.
We next investigated the occurrence of some histone modifications that have been associated with active chromatin states. To that end, we scanned the levels of H3, which is acetylated at K9 and K14 (H3K9,14ac), tri-methylated at K4 (H3K4me3), or K36 (H3K36me3) with specific antibodies in ChIP analyses before and after glucocorticoid treatment of the A549 cells. Interestingly, the levels of both H3K9,14ac and H3K4me3 around and immediately 3′ of the FKBP51 TSS, as well as on the major distal GR-binding region at approximately 87 kb from the TSS, were elevated already before dex treatment (Fig. 5, D and E). H3K4me3 was also enriched at the predicted promoter of FKBP51 transcript variant 2 (at ∼40 kb upstream from the TSS of the major FKBP51 transcript variant 1), but there was no enrichment of H3K9,14ac or signs of PolII occupancy, strongly suggesting that the variant 2 promoter is not active in this cell type. The glucocorticoid increased the level of H3K9,14ac around the TSS, but it had only a minor effect on the H3K4me3 status around the same region, actually decreasing the latter modification at about 87 kb 3′ of the TSS. H3K36me3 marks were enriched more downstream from the TSS, and they corresponded with the occupancy of PolII throughout the body of the gene (Fig. 5F). Interestingly, the glucocorticoid treatment also reduced the H3 density throughout the FKBP51 locus, except for the region around the TSS that showed lower than average H3 occupancy before glucocorticoid induction (Fig. 5C). Taken together, the occurrence of the histone marks associated with gene activation together with signs of nucleosome depletion around the TSS in the FKBP51 locus before glucocorticoid exposure suggest preprogramming of the locus chromatin for transcription in the A549 cells.
Involvement of the BRM- and the BRG1-containing chromatin-remodeling complexes in the regulation of the FKBP51 locus
The above results, together with recent literature, suggest that chromatin-remodeling complexes of the SWI/SNF type may cooperate with GR in the regulation of the locus (11). To address this issue, we performed ChIP assays to investigate the potential presence of BRM- and BRG1-containing complexes within the locus. Interestingly, signs of BRG1 in the locus were seen before dex treatment, and the hormone treatment enhanced its presence particularly on the region approximately 6 kb 3′ of the TSS (Fig. 6, A and B). Moreover, the hormone induced recruitment of BRM to the region around the TSS. Interestingly, these were the locus regions that also showed the lowest occupancy of H3 occupancy before dex exposure.
Fig. 6.

Evidence for the involvement of SWI/SNF type chromatin-remodeling complexes in the regulation of the FKBP51 locus. The effect of glucocorticoid on the occupancy of BRM- or BRG1-containing complexes within the FKBP51 locus was analyzed in A549 cells treated with vehicle or dex as in Fig. 4 by ChIP assays using antibodies against BRM (A) or BRG1 (B) followed by QPCR analysis. Results are shown as fold over the normal rabbit IgG-precipitated samples and represent the mean ± sd of three experiments. C, Depletion of BRM from A549 cells transfected with BRM-specific siRNA as assessed by immunoblotting. The level of glyceraldehyde-3-phosphate dehydrogenase monitored the loading of protein in the samples. D, The effect of BRM siRNA on the glucocorticoid induction of FKBP51 mRNA as analyzed by RT-QPCR analysis. ***, P < 0.001 compared with the cells transfected with unspecific target siRNA (siSCR) and treated with dex. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
We next tested whether the BRM has any functional effect on the FKBP51 transcription by depleting the protein from A549 cells using RNA interference (RNAi) and measuring the effect of the depletion on glucocorticoid-induced accumulation of FKBP51 mRNA. The efficiency of the BRM small interfering RNA (siRNA) was confirmed by immunoblotting (Fig. 6C). As shown in Fig. 6D, the BRM RNAi significantly (P < 0.001) inhibited the glucocorticoid-induced FKBP51 transcription, resulting in more than or equal to 50% decrease in the amount of FKBP51 mRNA in A549 cells. This result suggests that BRM is among the important factors mediating the remodeling of the FKBP51 chromatin upon activation by the GR.
Effect of glucocorticoid on the expression of FKBP51-neighboring genes
The upstream- and downstream-neighboring genes of FKBP51 are C6orf81 and TULP1, respectively, with these three loci residing within the approximately 250-kb region in chromosome 6 (chr6:35716685-35465651, Fig. 7A). Because the GR has the capability to regulate transcription from very long distances and the intergenic GR-regulated enhancer (chromatin region −3) is located only about 14 kb from the C6orf81 TSS and the linear distance from the major FKBP51 intronic enhancer (chromatin region 9) to the TULP1 TSS is about the same as that to the FKBP51 TSS (∼90 kb) (Fig. 7A), we next tested whether the GR is also able to activate these FKBP51-neighboring genes. However, the expression of C6orf81 or that of TULP1 showed no significant response to dex treatment in A549 cells (Fig. 7, B and C). Because the regulatory characteristics of FKBP51 by the androgen receptor in VCaP prostate cancer cells are similar to those by the GR in A549 cells (37), it was also of interest to check whether the AR is similarly inactive on these FKBP51-neighboring genes. Interestingly, androgen exposure of VCaP cells resulted in a significant induction of C6orf81 mRNA (∼20-fold at 6 h), whereas TULP1 showed no obvious androgen induction (Fig. 7, D and E).
One explanation for the above difference between the activation of C6orf81 by AR and GR could be that the AR might load to region −3 more avidly than the GR. We therefore analyzed the AR loading to the region −3 that remained outside the chr6:35676714-35521362 region scanned in our previous study (37). In comparison with the holo-GR in A549 cells, the occupancy of holo-AR on region −3 in relation to region −9 was markedly higher in VCaP cells, and there were also signs of AR occupancy on the more upstream region −5 (cf. Figs. 7F and 5A).
Occupancy of CCCTC-binding factor within the FKBP51 locus and its surroundings
Insulators are DNA elements with enhancer-blocking and/or chromatin barrier/boundary functions (36). They are recognized by CCCTC-binding factor (CTCF) that also appears to assist in long-range interactions of chromatin domains (36). Even though recent genome-wide ChIP data indicate a significant overlap in the CTCF-binding patterns between different cell types (36, 39, 40), we considered the possibility that the CTCF occupancy in the chromosome 6 region harboring C6orf81, FKBP51, and TULP1 might differ between the A549 and the VCaP cells. We used the genome-wide CTCF-binding information from the CTCF-binding site database for choosing the regions to be ChIP scanned with anti-CTCF antibody (41, 42, 43). Of the chromatin regions scanned, the region approximately 50 kb 5′ of the FKBP51 TSS located very close to the C6orf81 TSS showed the strongest enrichment of CTCF in both A549 and VCaP cells (Fig. 8, B and D). In both cell lines, also the regions at approximately 100 kb and about 160 kb 3′ of the FKBP51 TSS showed CTCF signals that were at least more than 10 times above the control IgG levels, whereas the other putative insulator sites did not show clear signs of CTCF occupancy. Because cohesin was recently shown to cooperate with the CTCF and occupy the same sites (44), we also analyzed whether the cohesin co-occupies the above putative insulator sites by using an antibody against the cohesin subunit RAD21 in ChIP scans. Interestingly, the same three regions showing the highest occupancy of CTCF also revealed a significant enrichment of the cohesin subunit RAD21 in A549 cells (Fig. 8C). These data suggest that difference in the steroid induction of the FKBP51-neighboring C6orf81 between VCaP and A549 cells is more likely to be due to differential usage of a steroid-regulated enhancer(s) than due to differences in the CTCF-linked insulating mechanisms.
Fig. 8.

Occupancy of the CTCF within the FKBP51 locus and its surroundings in A549 and VCaP cells. A, Schematic map of the FKBP51 and its neighboring genes C6orf81 and TULP1 in chromosome 6 with the circles showing the locations of the major GR-binding sites (identified in the preceding experiments). The locations of putative CTCF-binding regions Ins1-Ins9 (depicted as vertical lines) screened by ChIP assays are based on the information in the CTCF-binding site database (41 42 43 ). The other screened regions served as controls. A549 cells and VCaP cells treated with and without cognate agonist for 2 h were analyzed by ChIP with anti-CTCF antibody (B and D) or with anti-RAD21 antibody (C) followed by qPCR analysis. Results are shown as fold over the normal rabbit IgG-precipitated samples and represent the mean ± sd of three experiments. var., Variant; Ins, insulator.
Discussion
It is now evident that the majority of steroid receptor-binding sites are located at considerable distances from target promoters, not concentrated in their proximity (18, 22, 23, 24). Studies addressing the regulatory mechanisms of GR targets in chromatin conformation have been thus far limited, however, to a relatively small number of target loci harboring GREs in their proximal promoter regions. We chose to focus on the GR-dependent chromatin mechanisms underlying the activation of FKBP51, because it is a very sensitive biomarker of corticosteroid responsiveness in vivo (26, 27, 28), and according to previous reporter gene studies, the proximal promoter of FKBP51 does not harbor GREs, but the elements mediating the glucocorticoid activation apparently reside very distal from the promoter (45, 46). In A549 lung cancer cells that display characteristics of alveolar epithelial cells, the FKBP51 was clearly induced more robustly by dex than, for example, the GILZ that contains three closely spaced GREs within its proximal promoter (<2 kb from its TSS). Our evidence strongly supports the notion that the GR exerts long-range regulation of FKBP51 transcription via very distal intronic and intergenic sites. According to our quantitative ChIP and enhancer assays, there are at least three major distal gene regions that significantly bind GR and also function as strong GR-regulated enhancers in isolation. These three sites appear to harbor at least two potential GREs within a given 300-nt region conferring glucocorticoid regulation to a reporter gene. Interestingly, the kinetics of GR loading to the FKBP51 chromatin displayed wave-like characteristics. Upon glucocorticoid stimulation, the GR was first loaded to the intergenic and the intronic enhancer at approximately 34 kb 5′ and about 87 kb 3′ of the TSS, respectively. In the second wave of GR binding, also the other intronic enhancer at about 100 kb as well as the intronic regions at approximately 78 and 26 kb showed delayed occupancy. However, the GR bound to the latter two regions to a lesser extent than to the major intronic enhancer at approximately 87 kb 3′ of the TSS. The region overlaps with the intronic region (pIE2) previously shown by Hubler and Scammel (45) to confer glucocorticoid and progestin responsiveness to a heterologous reporter. It was also identified as a GR-occupied gene region in a recent ChIP microarray-based survey of 100-kb regions surrounding more than 500 glucocorticoid-responsive genes (18). Interestingly, antagonist RU486, in turn, was inefficient in inducing binding of GR to these chromatin regions, and it inhibited the interaction of agonist-loaded receptor with the FKBP51 chromatin.
It is not clear how transcription factors find their specific target sequences that are spread around genome. Cell-imaging studies indicate that most of the transcription factors, including GR, are highly mobile within the nucleus, exhibiting very dynamic and transient binding to the chromatin (47, 48). Recent models suggest that transcription factors may find their specific sites by random three-dimensional scanning of the genome, which may also involve more local sliding and/or hopping along the chromatin fiber (47). Most of the binding events seem to be highly transient, as they take place at nontarget sites, but prolonged binding is thought to occur at specific binding sites. Should the interactions between transcription factors and chromatin be rapid and transient, how are the long-range interactions between a distal enhancer-bound factor, such as GR, and the general transcription machinery at the core promoter established? It is likely that, in addition to the mediator complexes (49), there are mechanisms that aid these interactions. One possibility is that preassembled higher-order structures of chromatin, possibly preformed loops, stabilize or establish boundaries for these long-range interactions. In view of the higher-order chromatin structures, recent accumulating data support key roles for the insulator/boundary element-binding protein CTCF and cohesin (36, 44). Interestingly, our ChIP assays from the A549 cells indicate a significant enrichment of both the CTCF and the cohesin subunit RAD21 at regions approximately 48 kb 5′ and about 105 kb 3′ of the FKBP51 TSS, which border the region encompassing the GR-regulated enhancers. Our results showing a highly similar pattern of CTCF occupancy within the same chromosome region in androgen-regulated VCaP prostate cancer cells are in accordance with the recent data indicating a significant overlap of CTCF binding across diverse cell types (36, 39, 40). As proposed in our schematic model (Fig. 9), the CTCF bordering the GR-regulated enhancers may provide stability for the long-range interactions and be involved in chromatin loops that enable the interactions between GR and the general transcription machinery. Testing the existence of such loops and potentially complex interactions between several GR-binding regions spanning approximately 150-kb of chromatin is a daunting task that is complicated by the challenges associated with design of a proper set of controls needed to distinguish true signals from background caused by random collisions in chromosome conformation capture analyses (50).
Fig. 9.

A hypothetical model of how CTCF/cohesin complexes that border the region encompassing the major GR-binding sites in the FKBP51 locus might contribute to the long-range interactions and loop formation before (on the left) and after glucocorticoid exposure (on the right). CoA, Coactivator.
The presence of active histone marks within the FKBP51 locus before glucocorticoid exposure in A549 cells suggest that it was kept poised for transcription. Interestingly, also the major intronic GR-binding region showed elevated levels of active chromatin marks, H3K4me3 and H3K9,14ac. In addition to the major FKBP51 transcript (variant 1), the University of California Santa Cruz Genome Browser indicates that the locus harbors information for a longer transcript (variant 2) of which TSS is located at approximately 40 kb upstream from that of the major variant 1 (Fig. 2A). However, our data strongly suggest that the promoter of the variant 2 is largely inactive in the A549 cells. This conclusion is based on RT-QPCR analyses with transcript-specific primers, which is supported by ChIP scans of PolII and histone marks associated with active chromatin and transcription. Although the H3K4me3 mark was elevated around the TSSs of both variants, the histone acetylation mark H3K9,14ac, which is a more consistent mark of active gene regions (14), was enriched at the TSS of variant 1 but not at the TSS of variant 2. Moreover, the level of H3K36me3 and the occupancy of PolII were both elevated merely along the body of variant 1. The effect of glucocorticoid on the above mentioned histone marks was relatively modest, but the glucocorticoid additionally brought about a clear decrease in the amount of H3 at most scanned regions of the locus, except for the region at and immediately 3′ of the major TSS, which before glucocorticoid exposure showed lower than the average levels of H3. In keeping with the notion that GR binding induces changes in the nucleosome density or position, ChIP assays also showed evidence for the recruitment of both BRG1- and BRM-containing SWI/SNF chromatin-remodeling complexes to the FKBP51 locus. BRG1 and BRM are highly related, alternative adenosine triphosphatase subunits of SWI/SNF complexes, and especially the BRG1 has been shown previously to play an important role in the regulation of accessibility of GR-binding sites and glucocorticoid induction of a large number of genes (10, 11). As noted in a recent study (51), depletion of the BRM alone by RNAi resulted in a marked attenuation of the glucocorticoid-induced expression of FKBP51 also in this study, suggesting that the occupancy of BRM in the locus is functionally relevant. Induction of the FKBP51 locus by the AR shares several regulatory characteristics with that by the GR (37). This is not unexpected, because these related nuclear receptors can with a very similar affinity recognize DNA elements that are composed of two hexameric half-sites of 5′-AGAACA-3′-type, arranged as inverted, but typically imperfect (5′-RGnACA-3′), repeats separated by 3-bp spacers (52, 53, 54). The same FKBP51 regions that contained at least two potential AREs/GREs were capable of acting as AR- and GR-regulated enhancers in prostate and lung cancer cells. However, the relative strengths of the FKBP51 enhancer regions were different in VCaP and A549 cells. For example, the intronic enhancer at approximately 87 kb (region 9) conferred the strongest glucocorticoid induction in A549 cells, whereas the intronic region at about 100 kb (region 10) and the intergenic one at approximately 34 kb 5′ from the TSS (region −3) were stronger than region 9 in conferring androgen induction in VCaP cells (Ref. 37 and our unpublished data). Moreover, the loading kinetics of the GR onto various FKBP51 chromatin regions differed from that of the AR. The wave-like loading that was seen with the GR was less evident with the AR and the occupancy of the GR on the FKBP51 locus regions, except for the region at about 87 kb, appeared to be more transient than that of the AR. Notably, the intergenic region between the C6orf81 and the FKBP51 showed prolonged binding with the AR, which may explain, to some extent, the induction of the C6orf81 by androgens in VCaP cells, but not by glucocorticoids in A549 cells. Thus, even though the AR and the GR can activate the same genes and recognize the same DNA elements, they seem to utilize their binding sites in chromatin conformation in a somewhat different fashion. Because we were also comparing two distinct cell types, not only two different steroid receptors, it is likely that also disparities in the cellular milieu contributed to the above differences.
Recent genome-wide surveys and our current chromatin-binding and functional data emphasize the importance of distant-acting enhancers in steroid receptor biology. Thus, the focus also in the field of steroid-regulated gene transcription appears to be shifting from the linear genome to the genome in the third dimension and three-dimensional models of gene transcription. It is also of note that an interphase nucleus is a highly compartmentalized organelle that contains substructures, such as chromosome territories and nuclear bodies (55). Therefore, for better understanding of mechanisms regulating the long-range regulation of transcription, information from cell imaging studies also need to be combined with the ChIP data. With the advent of chromatin interaction analysis using paired-end tag sequencing genome topography maps of long-range chromatin interactions may eventually become available (50). However, demonstration of a chromatin interaction per se does not prove that the interaction is functionally important. Ultimately, disruption of an enhancer in the context of whole genome is needed to confirm its physiological significance for the regulation of a given locus in vivo.
Materials and Methods
Cell culture
A549 and VCaP cells were from American Type Culture Collection (Manassas, VA). A549 cells were maintained in F-12K medium containing 10% US defined fetal bovine serum (FBS) (HyClone Laboratories, Logan, UT), 25 U/ml penicillin, and 25 μg/ml streptomycin in a 5% CO2 atmosphere at 37 C. VCaP cells were maintained in DMEM containing FBS, penicillin, and streptomycin as above. COS-1 cells were maintained in DMEM containing 10% FBS, penicillin (6.25 U/ml), and streptomycin (6.25 μg/ml).
DNA constructs
Selected regions of FKBP51 were PCR cloned from human genomic DNA using Phusion DNA polymerase (Finnzymes, Espoo, Finland) and subcloned into the TATA box-containing pGL3-basic (Promega Corp., Madison, WI). Primers used were the same as listed for ChIP except for 10 the forward primer was 5′-TCTTGCCTCCAACACTGCTG-3′ (Supplemental Table 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). All sequences were verified by DNA sequencing using the ALFexpress system. pCMVβ encoding β-galactosidase was from CLONTECH (Mountain View, CA).
Antibodies
Primary antibodies purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) were as follows: GR (sc-1003), PolII (sc-899), BRG1 (sc-10768), TRAP220 (sc-8998), GAPDH (sc-25778), and normal rabbit IgG (sc-2027); antibodies from Abcam (Cambridge, UK) were: H3 (ab1791), H3K4me3 (ab8580), H3K36me3 (ab9050), RAD21 (ab992), and CTCF (ab70303); antibodies from UpState Biotechnology, Inc. (Lake Placid, NY) were: H3K9,14ac (06-599); and from BD Biosciences (San Jose, CA): BRM (610389).
Isolation of RNA and RT-quantitative PCR (RT-QPCR)
A549 cells were seeded onto six-well plates (3 × 105 cells per well) and grown for 36 h in transfection medium (F-12K medium containing 2.5% charcoal-stripped FBS) devoid of steroids. Subsequently, cells were treated with or without dex (Sigma-Aldrich, St. Louis, MO) or RU38486 (RU486) as indicated. VCaP cells were treated and processed as described (37). Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA) and converted to cDNA using Transcriptor First Strand cDNA synthesis Kit (Roche Diagnostics GmbH, Mannheim, Germany) following manufacturer’s instructions. cDNA was used as a template in RT-QPCR, which was carried out using Mx3000P Real-Time PCR System (Stratagene, La Jolla, CA), FastStart SYBR Green Master (Roche Diagnostics GmbH), and specific primers for FKBP51, S100P, GILZ, C6orf81, and TULP1 (Supplemental Table 1). Analyzed GAPDH mRNA levels were used to normalize the amounts of total RNA between the samples. Fold changes were calculated using formula 2−(ΔΔCt), were ΔΔCt is ΔCt(dex) − ΔCt(EtOH), ΔCt is Ct(gene X) − Ct(GAPDH), and Ct is the cycle at which the threshold in crossed.
Reporter gene assay
A549 and COS-1 cells were seeded onto 12-well plates (1.2 × 105 and 1.4 × 105 cells per well, respectively) and grown for 4 h in transfection medium. The cells were transiently transfected with TATA-LUC constructs driven by different regions of FKBP51 (0.46 μg TATA-LUC construct and 0.02 μg pCMVβ/well, and for COS-1 cells, additionally 0.02 μg pSG5-hGR) using TransIT-LT1 (Mirus Bio Corp., Madison, WI) transfection reagent. One day after transfection, either vehicle (ethanol) or 100 nm dex was added for the next 16 h. The cells were lysed in Reporter Lysis Buffer (Promega Corp.), and LUC activity was measured with Luminoskan Ascent(Thermo Electron, Helsinki, Finland) luminometer and β- galactosidase activity as described (56).
ChIP
A549 cells were seeded at approximately 70% confluence onto 75-cm2 bottles and allowed to grow in steroid-depleted transfection medium for 48 h before ChIP. VCaP cells were treated and processed as in Ref. 37 . The experiments were performed essentially as previously described (57), except for short-term assays (20- to 120-min time points), where the cells were treated with 2.5 μm α-amanitin (Sigma-Aldrich, St Louis, MO) for 2 h to further synchronize the cell population (58) before addition of glucocorticoid. α-Amanitin was removed by washing twice with PBS, and dex was added to the medium for indicated times. Specific primers for different regions are listed in Supplemental Table 1. Quantitative PCR analyses were carried out with FastStart SYBR Green Master (Roche) and Mx3000P Real-Time PCR System. Results were calculated using the formula E−(ΔCt) × 10, where E (efficiency of target amplification) is coefficient of DNA amplification by one PCR cycle for a particular primer pair and ΔCt is Ct(ChIP-template) − Ct(Input). In Figs. 5, 6, and 8, results were presented as fold over the value of normal rabbit IgG-precipitated samples.
Immunoblotting
Cell monolayers were washed with ice-cold TBS and harvested in TBS. Cell pellets were suspended in SDS-PAGE sample buffer and lysed by sonication (2 × 10 sec). Samples were heated for 5 min at 95 C and separated on 7.5% SDS-PAGE gels. Proteins were transferred onto nitrocellulose membranes and visualized by indicated antibody and horseradish peroxidase-conjugated antirabbit antibody using enhanced chemiluminescence detection reagents according to the manufacturer’s instructions (Pierce Chemical Co., Rockford, IL).
RNA interference
For silencing of BRM (59), QIAGEN (Valencia, CA) custom siRNAs and negative control siRNA (scramble, SCR) were used (Supplemental Table 1). For siRNA transfections, A549 cells were seeded onto six-well plates (3 × 105 cells per well) and grown for 4 h in transfection medium. The cells were transfected with siRNAs (40 nm final concentration) using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. After 72 h, the cells were treated with vehicle (ethanol) or 100 nm dex for the next 12 h. Finally, either RNA or protein was extracted as described above.
Acknowledgments
We thank Mrs. Merja Räsänen for excellent technical assistance.
NURSA Molecule Pages:
Coregulators: BRG1 | BRM;
Ligands: Dexamethasone | R1881 | RU486;
Nuclear Receptors: GR.
Footnotes
This work was supported by Academy of Finland, Finnish Cancer Organizations, Sigrid Jusélius Foundation, Emil Aaltonen Foundation, and Finnish Cultural Foundation North Savo Regional Fund.
Disclosure Summary: None of the authors have anything to disclose.
First Published Online January 21, 2010
Abbreviations: AR, Androgen receptor; ARE, androgen response element; ChIP, chromatin immunoprecipitation; CTCF, CCCTC-binding factor; dex, dexamethasone; FBS, fetal bovine serum; FKBP51, FK506-binding protein 51; GILZ, glucocorticoid-induced leucine zipper; GR, glucocorticoid receptor; GRE, glucocorticoid response element; H3, histone 3; PolII, RNA polymerase II; RNAi, RNA interference; RT-QPCR, RT-quantitative PCR; siRNA, small interfering RNA; TSS, transcription start site.
References
- 1.Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB2007. Glucocorticoid receptor physiology. Rev Endocr Metab Disord 8:321–330 [DOI] [PubMed] [Google Scholar]
- 2.Gross KL, Lu NZ, Cidlowski JA2009. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol 300:7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Bosscher K, Haegeman G2009. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 23:281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frankfurt O, Rosen ST2004. Mechanisms of glucocorticoid- induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol 16:553–563 [DOI] [PubMed] [Google Scholar]
- 5.Smoak KA, Cidlowski JA2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125:697–706 [DOI] [PubMed] [Google Scholar]
- 6.Rosenfeld MG, Lunyak VV, Glass CK2006. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1428 [DOI] [PubMed] [Google Scholar]
- 7.Perissi V, Rosenfeld MG2005. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6:542–554 [DOI] [PubMed] [Google Scholar]
- 8.Johnson TA, Elbi C, Parekh BS, Hager GL, John S2008. Chromatin remodeling complexes interact dynamically with a glucocorticoid receptor-regulated promoter. Mol Biol Cell 19:3308–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trotter KW, Archer TK2008. The BRG1 transcriptional coregulator. Nucl Recept Signal 6:e004 [DOI] [PMC free article] [PubMed]
- 10.John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, Voss TC, Davis SR, Meltzer PS, Stamatoyannopoulos JA, Hager GL2008. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell 29:611–624 [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Kinyamu HK, Archer TK2006. Changes in attitude, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Mol Endocrinol 20:1–13 [DOI] [PubMed] [Google Scholar]
- 12.Wolf IM, Heitzer MD, Grubisha M, DeFranco DB2008. Coactivators and nuclear receptor transactivation. J Cell Biochem 104: 1580–1586 [DOI] [PubMed]
- 13.Li B, Carey M, Workman JL2007. The role of chromatin during transcription. Cell 128:707–719 [DOI] [PubMed] [Google Scholar]
- 14.Kouzarides T2007. Chromatin modifications and their function. Cell 128:693–705 [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40:897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson AG, Bilenky M, Tam A, Zhao Y, Zeng T, Thiessen N, Cezard T, Fejes AP, Wederell ED, Cullum R, Euskirchen G, Krzywinski M, Birol I, Snyder M, Hoodless PA, Hirst M, Marra MA, Jones SJ2008. Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding. Genome Res 18:1906–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA 101:15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR2007. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 3:e94 [DOI] [PMC free article] [PubMed]
- 19.So AY, Cooper SB, Feldman BJ, Manuchehri M, Yamamoto KR2008. Conservation analysis predicts in vivo occupancy of glucocorticoid receptor-binding sequences at glucocorticoid-induced genes. Proc Natl Acad Sci USA 105:5745–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blind RD, Garabedian MJ2008. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol 109:150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Astrand C, Belikov S, Wrange O2009. Histone acetylation characterizes chromatin presetting by NF1 and Oct1 and enhances glucocorticoid receptor binding to the MMTV promoter. Exp Cell Res 315:2604–2615 [DOI] [PubMed] [Google Scholar]
- 22.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38:1289–1297 [DOI] [PubMed] [Google Scholar]
- 23.Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M2007. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG2009. ChIP-Seq of ERα and RNA polymerase II defines genes differentially responding to ligands. EMBO J 28:1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hakim O, John S, Ling JQ, Biddie SC, Hoffman AR, Hager GL2009. Glucocorticoid receptor activation of the Ciz1-Lcn2 locus by long range interactions. J Biol Chem 284:6048–6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV2007. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA 104:15858–15863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermeer H, Hendriks-Stegeman BI, van der Burg B, van Buul-Offers SC, Jansen M2003. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: a potential marker for glucocorticoid sensitivity, potency, and bioavailability. J Clin Endocrinol Metab 88:277–284 [DOI] [PubMed] [Google Scholar]
- 28.Vermeer H, Hendriks-Stegeman BI, van Suylekom D, Rijkers GT, van Buul-Offers SC, Jansen M2004. An in vitro bioassay to determine individual sensitivity to glucocorticoids: induction of FKBP51 mRNA in peripheral blood mononuclear cells. Mol Cell Endocrinol 218:49–55 [DOI] [PubMed] [Google Scholar]
- 29.Pratt WB, Toft DO1997. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18:306–360 [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Clark AF, Yorio T2008. FK506-binding protein 51 regulates nuclear transport of the glucocorticoid receptor β and glucocorticoid responsiveness. Invest Ophthalmol Vis Sci 49:1037–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denny WB, Valentine DL, Reynolds PD, Smith DF, Scammell JG2000. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology 141:4107–4113 [DOI] [PubMed] [Google Scholar]
- 32.Reynolds PD, Ruan Y, Smith DF, Scammell JG1999. Glucocorticoid resistance in the squirrel monkey is associated with overexpression of the immunophilin FKBP51. J Clin Endocrinol Metab 84:663–669 [DOI] [PubMed] [Google Scholar]
- 33.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L2009. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Binder EB2009. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34S1:S186–S195 [DOI] [PubMed]
- 35.Ballard PL, Mason RJ, Douglas WH1978. Glucocorticoid binding by isolated lung cells. Endocrinology 102:1570–1575 [DOI] [PubMed] [Google Scholar]
- 36.Phillips JE, Corces VG2009. CTCF: master weaver of the genome. Cell 137:1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makkonen H, Kauhanen M, Paakinaho V, Jääskeläinen T, Palvimo JJ2009. Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Res 37:4135–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandelin A, Wasserman WW, Lenhard B2004. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res 32:W249–W252 [DOI] [PMC free article] [PubMed]
- 39.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K2009. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res 19:24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837 [DOI] [PubMed] [Google Scholar]
- 42.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bao L, Zhou M, Cui Y2008. CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res 36:D83–D87 [DOI] [PMC free article] [PubMed]
- 44.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801 [DOI] [PubMed] [Google Scholar]
- 45.Hubler TR, Scammell JG2004. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 9:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magee JA, Chang LW, Stormo GD, Milbrandt J2006. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 147:590–598 [DOI] [PubMed] [Google Scholar]
- 47.Hager GL, McNally JG, Misteli T2009. Transcription dynamics. Mol Cell 35:741–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Misteli T2001. Protein dynamics: implications for nuclear architecture and gene expression. Science 291:843–847 [DOI] [PubMed] [Google Scholar]
- 49.Chen W, Roeder RG2007. The Mediator subunit MED1/TRAP220 is required for optimal glucocorticoid receptor-mediated transcription activation. Nucleic Acids Res 35:6161–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fullwood MJ, Ruan Y2009. ChIP-based methods for the identification of long-range chromatin interactions. J Cell Biochem 107:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324:407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.La Baer J, Yamamoto KR1994. Analysis of the DNA-binding affinity, sequence specificity and context dependence of the glucocorticoid receptor zinc finger region. J Mol Biol 239:664–688 [DOI] [PubMed] [Google Scholar]
- 53.Roche PJ, Hoare SA, Parker MG1992. A consensus DNA-binding site for the androgen receptor. Mol Endocrinol 6:2229–2235 [DOI] [PubMed] [Google Scholar]
- 54.Claessens F, Verrijdt G, Schoenmakers E, Haelens A, Peeters B, Verhoeven G, Rombauts W2001. Selective DNA binding by the androgen receptor as a mechanism for hormone-specific gene regulation. J Steroid Biochem Mol Biol 76:23–30 [DOI] [PubMed] [Google Scholar]
- 55.Kumaran RI, Thakar R, Spector DL2008. Chromatin dynamics and gene positioning. Cell 132:929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palvimo JJ, Kallio PJ, Ikonen T, Mehto M, Jänne OA1993. Dominant negative regulation of trans-activation by the rat androgen receptor: roles of the N-terminal domain and heterodimer formation. Mol Endocrinol 7:1399–1407 [DOI] [PubMed] [Google Scholar]
- 57.Makkonen H, Jääskeläinen T, Pitkänen-Arsiola T, Rytinki M, Waltering KK, Mättö M, Visakorpi T, Palvimo JJ2008. Identification of ETS-like transcription factor 4 as a novel androgen receptor target in prostate cancer cells. Oncogene 27:4865–4876 [DOI] [PubMed] [Google Scholar]
- 58.Métivier R, Penot G, Hübner MR, Reid G, Brand H, Kos M, Gannon F2003. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115:751–763 [DOI] [PubMed] [Google Scholar]
- 59.Wang S, Zhang B, Faller DV2004. BRG1/BRM and prohibitin are required for growth suppression by estrogen antagonists. EMBO J 23:2293–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]