

Abstract
One-hundred years ago, Starling articulated the interdependence of renal control of circulating blood volume and effective cardiac performance. During the past 25 years, the molecular mechanisms responsible for the interdependence of blood pressure (BP), extracellular fluid volume (ECFV), the renin-angiotensin system (RAS), and sympathetic nervous system (SNS) have begun to be revealed. These variables all converge on regulation of renal proximal tubule (PT) sodium transport. The PT reabsorbs two-thirds of the filtered Na+ and volume at baseline. This fraction is decreased when BP or perfusion pressure is increased, during a high-salt diet (elevated ECFV), and during inhibition of the production of ANG II; conversely, this fraction is increased by ANG II, SNS activation, and a low-salt diet. These variables all regulate the distribution of the Na+/H+ exchanger isoform 3 (NHE3) and the Na+-phosphate cotransporter (NaPi2), along the apical microvilli of the PT. Natriuretic stimuli provoke the dynamic redistribution of these transporters along with associated regulators, molecular motors, and cytoskeleton-associated proteins to the base of the microvilli. The lipid raft-associated NHE3 remains at the base, and the nonraft-associated NaPi2 is endocytosed, culminating in decreased Na+ transport and increased PT flow rate. Antinatriuretic stimuli return the same transporters and regulators to the body of the microvilli associated with an increase in transport activity and decrease in PT flow rate. In summary, ECFV and BP homeostasis are, at least in part, maintained by continuous and acute redistribution of transporter complexes up and down the PT microvilli, which affect regulation of PT sodium reabsorption in response to fluctuations in ECFV, BP, SNS, and RAS.
Keywords: natriuresis, angiotensin II, angiotensin I-converting enzyme inhibitors
2009 marked the centennial of the publication of Ernest H. Starling's monograph The Fluids of the Body (38). In this set of eight lectures, Starling gives his personal perspectives on the properties of protoplasm, fluid intake, intestinal absorption, renal output, and fluid balance,
“…believing a presentation from the standpoint of one worker is more likely to excite interest, whether of sympathy or dissent. In this way my readers may be stimulated to search out for themselves, in the wards or in the laboratory, the answers to some of the riddles with which they are here confronted.”
Indeed, this monograph has stimulated many, including myself, to pursue answers for the riddles it posed, guided by one of Starling's central contributions: his articulation of the interdependence of renal control of circulating blood volume and effective cardiac performance.
“The function of the kidney is to keep the composition of the circulating fluid constant, and we can therefore alter the urine in any direction according to the nature of the changes we bring about in the composition of the body (38).”
To accomplish this function, the kidneys filter the entire plasma volume every 30 min and reabsorb two-thirds of the filtrate in the proximal tubule (PT). Fine control of salt, water, and blood pressure homeostasis has been traditionally attributed to regulation of transport in the distal nephron and collecting ducts. However, it is now apparent that sodium transport in the PT is acutely and chronically regulated by 1) blood pressure (BP), 2) extracellular fluid volume (ECFV), 3) the renin-angiotensin system (RAS) and 4) the sympathetic nervous system (SNS), as well as 5) an intrarenal dopamine natriuretic system. In fact, there is now evidence that regulation of PT Na+ transport is a key effector of feedback loops that connect ECFV, BP, and regulation of the RAS and SNS (Fig. 1). My colleagues and I have focused on the mechanisms regulating PT sodium transport in response to changes in these variables with the aim of answering some of the riddles Starling articulated 100 years ago. Taking the lead from Professor Starling, I will also provide “a presentation from the standpoint of one worker,” to provide an overview of how to dissect molecular mechanisms of transport regulation in whole animal models, specifically, how changes in BP, SNS activity, ECFV, and the RAS alter PT sodium transporter distribution and transport.
Fig. 1.
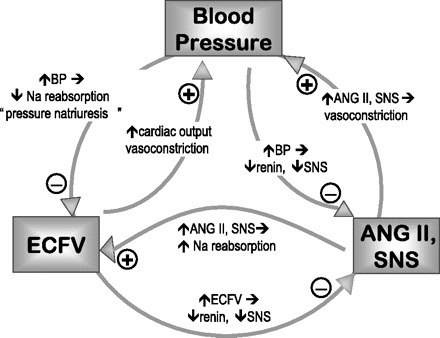
Feedback relationship between blood pressure (BP), ANG II, and sympathetic nervous system (SNS), and extracellular fluid volume (ECFV).
In 1975, I was introduced to the riddles of body fluid balance as a graduate student of S. K. Hong on an international project called Hana Kai II, which investigated physiological responses of human divers to prolonged exposure to a hyperbaric environment. When the habitat was pressurized to 18.6 ATA, urine output of the divers promptly increased from 2 to 2.6 l/day. Careful analysis of many different parameters led the team to conclude that this was not a pressure-diuresis per se, but a diuresis provoked by a decrease in insensible water loss in the hyperbaric environment, which suppressed antidiuretic hormone. This early research experience provided me with not only a template for how to integrate multiple variables to understand ECFV homeostasis, but also insight on the importance of assembling expert collaborators to address a question from multiple vantage points, and it provided me with my first publication (17).
I next joined I. S. Edelman's laboratory. Decades before, Edelman had developed isotope dilution methods that permitted quantitation of body fluid compartment composition in health and disease (6). By the time I arrived, the laboratory was aiming to understand how hormones regulated salt and water homeostasis in these compartments and how salt transport regulated metabolism. It was an era (at University of California, San Francisco, and then Columbia University) when investigators invented molecular biology “from scratch,” sharing methods and approaches to clone and isolate transporters and receptors. In Edelman's group, these cDNAs and antibodies were a means to an end—tools to investigate regulation in intact cells and animals (15, 30). This experience convinced me of the feasibility and utility of addressing homeostasis and disease at multiple levels: from the whole animal to the molecule.
In the 1980s, as an assistant professor at the University of Southern California, I was invited to serve on two dissertation committees. Chung-Lin Chou in Donald Marsh's laboratory was examining the role of the PT in the pressure diuresis response and discovered, using video-densitometric analysis of tubular flow, that raising blood pressure rapidly increased end PT flow rate by 50% (Fig. 2A). They concluded that PT sodium transport was inhibited during acute hypertension and that this response contributed to autoregulation of glomerular filtration rate and renal blood flow by increasing delivery to the macula densa, as well as contributed to pressure diuresis (9, 10). Around the same time, Charles Hensley in Austin Mircheff's laboratory was addressing how parathyroid hormone (PTH) inhibited bicarbonate reabsorption in the PT. When Hensley incubated isolated PTs with PTH and then subjected them to analytical subcellular fractionation, he discovered that Na+/H+ exchanger activity, measured in vesicles from the gradient fractions, redistributed from low-density fractions enriched in brush-border markers to a population at higher density (13, 14). These studies were among the first providing evidence for acute subcellular redistribution, i.e., “trafficking,” as a mechanism for regulating renal sodium reabsorption, although the lack of molecular probes prevented the direct visualization of trafficking. The independent findings from the Marsh and Mircheff laboratories provided the impetus to investigate the hypothesis that the pressure diuresis response was mediated by the rapid trafficking of PT sodium transporters out of the plasma membrane, a mechanism that would decrease sodium reabsorption.
Fig. 2.
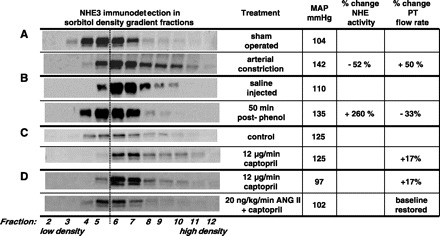
Distinct patterns of sodium hydrogen exchanger (NHE3) trafficking in response to different stimuli. Density distribution pattern of NHE3 before and after a treatment was assessed by fractionating renal cortex on sorbitol density gradients. A constant volume of each of the 12 fractions collected was analyzed by immunoblot with anti-NHE3. A: when mean arterial pressure (MAP) is elevated by arterial constriction, NHE3 moves out of low-density microvilli-enriched membranes to higher-density membranes found at the base of the microvilli. This redistribution is associated with a decrease in proximal tubule (PT) Na+ reabsorption (48) and a decrease in Na+/H+-exchanger activity (57), and an increase in end PT flow rate (10). B: when MAP is elevated by acute phenol injury, which stimulates sympathetic outflow to the kidneys, NHE3 redistributes from higher-density membranes found at the base of the microvilli into the low-density microvilli membranes. This redistribution is associated with an increase in NHE activity and decrease in PT flow rate (23). C: when the angiotensin-converting enzyme inhibitor (ACEI) captopril is infused at a dose that does not change MAP, renal blood flow (RBF), or glomerular filtration rate (GFR), NHE3 moves out of low-density microvilli-enriched membranes to higher-density membranes found at the base of the microvilli. This retraction is associated with an increase in PT flow rate (21). D: when ANG II is added to the captopril infusate at a dose that does not change MAP, RBF, or GFR, NHE3 distribution and PT flow rate return to the baseline distribution (34). (Note: minor variations in the absolute density distributions from one study to another are secondary to person-to-person variation in making and collecting gradients). A dashed line between fractions 5 and 6 is provided as a visual aid to facilitate detecting the redistribution patterns.
Regulation of PT Salt Transport—Anatomy and Transporters
“…the tubules as a whole are endowed with the power of absorbing both water and dissolved substances from the fluid of their lumen. Whether this absorptive power is limited to the cells of Henle's loop or occurs coincidentally with secretion in the cells of the convoluted tubules, as might be imagined from the close analogy between the structure of these cells and that of the intestinal epithelium, we have not sufficient evidence to decide (38).”
The PT has an optimal design for reabsorbing the bulk of the glomerular filtrate. The apical pole has tall brush border formed by densely packed microvilli around an actin filament core, a specialization estimated to increase the apical surface area more than 30-fold (43). Transporters in the apical microvilli include the Na+/H+ exchangers (NHE) and anion transporters to reabsorb most of the sodium chloride and bicarbonate, the Na+-phosphate cotransporter 2 (NaPi2) to reabsorb phosphate, Na+-glucose and Na+-amino acid transporters to reabsorb substrates, and water channels (AQP1) to reabsorb fluid volume. To determine the molecular basis of sodium transport regulation in the PT, we first determined that NHE3 mRNA was expressed in four-fold excess to the other NHE isoforms in the PTs (3), and we identified collaborators that had begun to generate anti-NHE3 antibodies that could be used to examine trafficking at the molecular level. Since the apical sodium entry routes are fairly specific for different regions of the nephron, we concluded that it was not necessary to isolate PTs from the kidney prior to analyzing transporter regulation; NHE3 and NaPi2 detected in renal cortical homogenates could be assumed to be from the PT. The basolateral membranes contain Na+-K+-ATPase, which pumps sodium out of the cell, creating the driving force for sodium entry via the NHE3 and NaPi2. These basolateral membranes are highly infolded and juxtaposed to mitochondria to effectively increase the surface area and energy supply necessary to drive reabsorption of two-thirds of the glomerular filtrate. Unlike the apical transporters, Na+-K+-ATPase is expressed in every cell of the kidney. Our studies in dissected PTs determined that sodium pump abundance and activity are lower than that measured in thick ascending loop of Henle and higher than that in the collecting tubule and collecting ducts (31). However, since the PTs predominate in the cortex, we assumed that significant regulation of PT sodium pump distribution or activity in response to raising blood pressure could be detected in the renal cortex.
To estimate the effects of acute hypertension (or other regulators) on PT sodium transport, we implemented the noninvasive endogenous lithium clearance (CLi+) protocol. Leyssac and Christensen (25) had shown that Li+ (in low concentrations) was handled like Na+ in the PT, but not further down the nephron; thus, a change in CLi+ is a good estimate of the change in volume flow leaving the PT, secondary to inhibition of proximal transport if glomerular filtration rate (GFR) is unchanged.
Sodium transport can be adjusted by changing either apical Na+ entry, basolateral Na+ exit, or both. The literature on natriuretic hormones' actions in the PT (i.e., PTH or dopamine) concurs that transport inhibition is effected via parallel independent inhibition of Na+/H+ exchangers and Na+-K+-ATPase activity (discussed in Ref. 59). This type of regulation allows for a close regulation of intracellular Na+ and K+ levels, as well as cell volume. To illustrate this point, an increase in apical Na+ entry in macula densa cells, which have low sodium pump activity, is not accompanied by a parallel change in basolateral Na+ exit, and the cells swell as part of the tubuloglomerular feedback response (33). Thus, our initial hypothesis was that acute hypertension would provoke parallel decreases in PT Na+ entry and Na+ exit to decrease sodium and volume reabsorption without a change in PT cell volume.
Raising Blood Pressure Inhibits PT Sodium Transport
“…alterations in the blood supply to the kidney, determined by changes on the arterial side, have pronounced effects on the amount of urine formed (38).”
In a collaboration involving five laboratories from four institutions, we ascertained that raising arterial pressure (∼50 mmHg, by constricting various arteries) increased urine output (V) and endogenous CLi+ almost immediately (59). Kidneys were removed 5 min after constricting arteries; the cortex was homogenized and fractionated on sorbitol density gradients, as described by Hensley et al. (13). This study revealed that apical NHE3 was redistributed to higher-density membranes enriched in the intracellular membrane markers (Fig. 2A). Coincidentally, Na+-K+-ATPase total activity decreased, and a distribution shifted to higher-density fractions. Inhibiting PT Na+ reabsorption with a carbonic anhydrase inhibitor similarly increased V and CLi+ but did not alter NHE3 distribution or Na+-K+-ATPase activity, implying that decreasing apical Na+ uptake does not initiate the responses. In the next study, we demonstrated that NHE3 and sodium pumps returned to their control density distributions within minutes after blood pressure was returned to baseline (58). From the results of these studies, we hypothesized that the decrease in PT salt and volume reabsorption during acute hypertension was mediated by both endocytic removal of apical Na+/H+ exchangers and basolateral Na+ pumps, as well as a decrease in total Na+ pump activity. Subsequently, Yip et al. (56) used anti-NHE3 antibodies with confocal microscopy to visualize the pressure-induced redistribution of NHE3 and concluded the transporters moved out of the brush border to the vicinity of the base of microvilli. Yip et al. (57) developed a ratiometric method to measure intracellular pH in PT cells on the surface of the kidney and used it to verify that acute changes in arterial pressure reversibly inhibit apical Na+/H+ exchanger activity (Fig. 2A).
Using the subcellular fractionation methods, we analyzed the natriuretic/diuretic actions of PTH infused in vivo (60), specifically, the effect of PTH 1–34, which couples to adenylate cyclase (AC), phospholipase C (PLC), and phospholipase A2 (PLA2), vs. the PTH 3–34 analog, which couples to PLC and PLA2 but not AC. Interestingly, PTH 1–34 provoked changes in V, CLi+, NHE3, and NaPi2 distribution, and Na+-K+-ATPase activity and distribution were very similar to those measured during acute hypertension. A key observation was that PTH 3–34 inhibited Na+-K+-ATPase activity but failed to increase V or CLi+ or provoke redistribution of NHE3 or NaPi2. The PTH 3–34 results were very informative because they demonstrated that 1) a dissociation occurs between regulation of apical and basolateral sodium transporters, 2) the change in apical transporters appears to be rate limiting for changing PT Na+ reabsorption, and 3) a significant inhibition of basolateral Na+-K+-ATPase activity is not sufficient to depress PT sodium reabsorption. For these reasons, our future studies of PT salt transport focused on the regulation of the rate-limiting apical entry step.
We used a number of different approaches to further investigate the molecular basis for the hypertension-induced inhibition of sodium transport. After 5–30 min of acute hypertension, Yang et al. (45) prepared membrane vesicles from density gradient samples for transport assays and found that Na+/H+ exchanger activity shifted to higher-density membranes along with NHE3 protein without any change in activity/transporter. In the same study, immunofluorescence analysis of NHE3, like those of Yip et al. (56) demonstrated NHE3 moving out of the microvilli but could not determine whether the NHE3 was actually internalized. A cautionary note: even in untreated kidneys, perfusion fixation at elevated pressures causes NHE3 to move to the base of the microvilli, mimicking the effect of hypertension (34). When horseradish peroxidase (HRP) was infused to label the subapical endosomes during the BP elevation, confocal microscopy revealed that NaPi2 was endocytosed to these HRP-containing endosomes, while NHE3 was not (Fig. 3) (48). To determine the precise destination of NHE3 and NaPi2 after high BP or PTH treatment, we were fortunate to engage the expertise of Arvid Maunsbach in electron microscopy (EM) as applied to the PT (27, 28, 48). Immunogold labeling at the EM level revealed that after acute high BP or PTH treatment, NHE3 was concentrated in a distinct domain near the base of the MV, above the coated pits, while NaPi2 moved to endosomes. These results suggested that NHE3 and NaPi2 moved rapidly within the plane of the microvillar membranes to the base during high BP, suggesting that a molecular motor was involved. Biemesderfer et al. (5) had reported the detection of the atypical actin-based motor myosin VI only at the base of the microvilli. When we were careful to avoid elevating perfusion pressures during fixation, we detected myosin VI over the whole length of the microvilli at baseline BP and found that myosin VI moved to the base of the microvilli during acute hypertension, evident by both light and electron microscopy (49). These findings provided the first evidence for a regulated function of myosin VI in the kidney and supported the idea that myosin VI is the molecular motor responsible for moving NHE3 and NaPi2 out of the microvilli during acute hypertension. This hypothesis was supported by the recent study of Blaine et al. (7, 29), who used total internal reflection fluorescence microscopy to demonstrate that myosin VI is necessary for removing NaPi2a from microvilli of cultured LLC-PK1 cells treated with PTH (29).
Fig. 3.
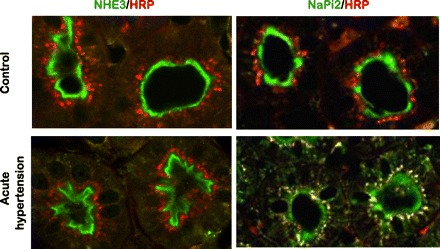
Differential redistribution of NHE3 and NaPi2 during acute hypertension. In this study (48), the endocytic compartment of the PT was labeled by intravenous injection of horseradish peroxidase (HRP), and rats were sham operated (control), or blood pressure was increased for 20 min (acute hypertension). Kidneys were fixed in situ, sectioned, and double labeled with either polyclonal anti-NHE3 or anti-NaPi2 (both green), and with monoclonal anti-HRP (red). Left: NHE3 is retracted from the body to the base of the microvilli during acute hypertension, with no evidence that NHE3 moves into endocytic tracer HRP-labeled compartment. Right: NaPi2 is retracted from the body of the microvilli and is subsequently colocalized with the endocytic tracer HRP (yellow indicates colocalization).
The distinct routes of redistribution of NHE3 vs. NaPi2 in response to high BP or PTH treatment encouraged Riquier et al. (35) to ask whether these transporters were localized to distinct membrane domains (lipid raft vs. nonraft) in the PT microvilli. Using detergent extraction and Optiprep flotation gradients to separate the lighter detergent-resistant lipid rafts from more dense nonrafts, we discovered that >80% of the NHE3, its regulator dipeptidyl peptidase IV (DPPIV) and myosin VI are in the lipid raft-enriched membranes, while ∼70% of the NaPi2, its regulator NHERF-1, ezrin, and the clathrin adaptor AP2 were in nonraft fractions (Fig. 4). The partitioning of NHE3 in rafts and NaPi2 in nonrafts was not changed by raising blood pressure. The distinct domain properties could help to explain why these transporters exhibited distinct trafficking patterns during elevated pressures: NHE3 remains in raft domains settling to the base of the microvilli, while NaPi2 proceed to the nonraft domains in the clathrin-coated pits, where they are freely endocytosed.
Fig. 4.
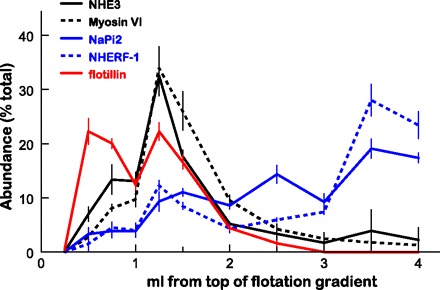
Flotation gradient profiles of NHE3, NaPi2, and associated proteins. Renal cortex total membranes treated with Triton X-100 were fractionated on OptiPrep gradients (35). The top 1.5 ml was collected as six 250-μl samples to delineate the lipid raft domain, and the remainder of the gradient was collected as five 500-μl aliquots. A constant volume of each fraction from untreated Sprague-Dawley rats was assayed by immunoblot. Profiles for NHE3 (solid black), myosin VI (dashed black), NaPi2 (solid blue) NHERF-1 (dashed blue), and the lipid raft marker flotillin (solid red) are compared. Data are expressed as the percentage of the total signal in all 11 fractions (means ± SE).
The signals connecting an increase in arterial pressure to a decrease in sodium transport have been the subject of a number of investigations from a number of laboratories, including our own, and parts of the puzzle are beginning to be identified. Overall, it appears that raising BP acutely stimulates nonautoregulated preglomerular vasculature to release mediator(s) that can inhibit sodium transport in nearby PTs (12). The mediators may include cytochrome P-450 metabolites, nitric oxide, and factors suppressing the RAS. 20-Hydroxyeicosatetraenoic acid (20-HETE), a metabolite(s) of arachidonic acid produced by cytochrome P-450, was one of the first candidates we tested because it is a natriuretic and diuretic (39) and potentiates tubuloglomerular feedback and autoregulation of renal vascular tone (64). Inhibiting cytochrome P-450 metabolism with cobalt chloride significantly blunted the overall diuretic response to acute hypertension and the change in CLi+. At the molecular level, cobalt chloride prevented the redistribution of NHE3 and inhibition of Na+-K+-ATPase activity that accompanied the decrease in PT Na+ reabsorption (61). The Roman laboratory subsequently used a far more specific inhibitor to demonstrate that preventing elevation of renal 20-HETE reduced the pressure-natriuresis response by 40–50% (11, 44). Carey and colleagues (1) used interstitial infusion of inhibitors to suggest that an increase in renal interstitial cGMP is important to the natriuretic response to elevated BP, a result supporting the hypothesis that an elevation in intrarenal nitric oxide (NO) is important to produce the pressure-natriuresis response (12). We postulated that a rise in BP would suppress the RAS, which would suppress PT Na+ reabsorption. Leong et al. (22, 24) clamped ANG II at a subpressor level before raising blood pressure and found that this clamp protocol blunted the pressure natriuretic and diuretic responses about 50% and also blunted the retraction of PT NHE3 from the microvilli (22). Putting together these recent studies on the pressure natriuresis response, we propose that the initial and rapid natriuretic response is driven by rapid generation and action of 20-HETE in the PT and that the repression of the RAS and fall in ANG II, which lags behind the initial response, is important for sustaining the response by reducing sodium transport in ANG II-sensitive regions all along the nephron, including the distal convoluted tubule (20).
“The mechanisms, which determine the adaptation of the organism to changes in the total volume of its fluid content, must come into play with every rise or fall in the general blood pressure (38).”
Starling recognized the linkage between body fluid volume and blood pressure, and we now understand that a transient increase in volume and blood pressure can stimulate a transient increase in urine output to restore homeostasis. At this point, students may ask why the pressure natriuresis does not correct chronic hypertension. The simple answer is that pressure natriuresis is not responsible for maintaining baseline BP; rather, it is designed to maintain fluid and electrolyte homeostasis. For example, an aldosterone-producing tumor that increases sodium reabsorption in the cortical collecting duct (CCD) in an unregulated fashion increases extracellular fluid volume and drives an increase in arterial pressure. The elevated BP will trigger a persistent decrease in sodium reabsorption in the PT, discussed above, as well as in the distal tubule (20, 62) compensating, at least in part, for the increase in CCD Na+ reabsorption. The error signal driving this pressure natriuresis response is elevated blood pressure. That is, chronic hypertension is a homeostatic stimulus that signals the kidneys to excrete more salt to reestablish salt and water balance at the expense of elevated BP. One prediction of this line of logic is that the molecular markers of pressure natriuresis will be evident in the spontaneously hypertensive rat (SHR) model. A number of studies indicate this to be the case. Both the McDonough and Yip laboratories have demonstrated that NHE3 and NaPi2 distributions are normal in very young SHR, before hypertension develops and that by 12 wk of age, NHE3 is partially retracted from the body to the base of the microvilli, and NaPi2 is partially endocytosed to subapical vesicles, analogous to the pattern observed in response to acute hypertension in the Sprague-Dawley rats (26, 46, 56). When comparing molecular differences between hypertensive and control models, it is not always obvious which differences contribute to hypertension and which compensate for hypertension. We postulate that the decrease in PT Na+ and volume reabsorption compensates for Na+ transport elevated elsewhere along the nephron. Reports of decreased PT fluid reabsorption (32) in SHR vs. Wistar-Kyoto (WKY) and increased a fractional excretion of phosphate in SHR vs. WKY (16) provide evidence for physiological consequence of the NHE3 and NaPi2 redistribution in adult SHR. This chronic redistribution of sodium transporters likely contributes, at least in part, to the resistance to natriuretic hormones that is evident in the SHR. Jose and colleagues (40) conclude, in their recent review of the role of the PT in hypertension that, “The increase in renal proximal tubule ion transport in hypertension is due to increased actions by prohypertensive factors that are unopposed by antihypertensive factors.” Elevated blood pressure itself must be considered an antihypertensive factor that decreases sodium transporter activity. Additionally, if NHE3 and NaPi2 are primarily downregulated by hypertension, then the potential to respond to natriuretic hormones acting in the PT will be blunted.
Renal Injury-Sympathetic Nervous System Activation Stimulates PT Sodium Transport
“The blood pressure in man is continually varying. The normal systolic pressure in the brachial artery, when man is at rest, is about 110 mmHg. The slightest excitement or concentration of attention may increase this pressure by 20 or 30 mm (38).”
While Starling recognized that excitement could raise blood pressure, he did not explore the role of the nervous system on the kidneys directly. Campese (8) developed a rat model in which an acute renal injury precipitates hypertension. Specifically, in anesthetized normotensive Sprague-Dawley rats, a single injection of 50 μl 10% phenol into the lower pole of one renal cortex causes blood pressure to rise about 30 mmHg and to remain elevated for at least 5 wk, when the injury is reduced to a microscopic scar (52, 55). The same group went on to demonstrate that the hypertension was associated with a rapid release of norepinephrine from the posterior pituitary and an increase in renal sympathetic nerve activity and that renal denervation or removal of the injected kidney reverses or prevents the hypertension defining this renal injury as a model of neurogenic hypertension (52, 53). The effects of phenol on blood pressure and SNS activity were abolished when tempol or superoxide dismutase-polyethylene glycol was infused in the lateral ventricle before phenol injection (54), indicating that central SNS activation in this model is mediated by increased reactive oxygen species in the brain nuclei involved in the noradrenergic control of blood pressure.
We examined whether the effects of phenol injury-induced hypertension would mimic the effects of vascular constriction hypertension on PT sodium reabsorption and transporter distribution (47). While phenol injection increased blood pressure similarly to that previously reported for vascular constriction (∼30 mmHg), it provoked a very different chain of events consistent with increased, rather than decreased, PT sodium transport. When renal cortex was analyzed on density gradients, we observed a doubling of NHE3 and NaPi2 abundance in apical microvilli-enriched low-density membranes and a corresponding decrease in their abundance in higher-density membranes (Fig. 2B). This redistribution was prevented by renal denervation prior to phenol injection. This redistribution could not be detected by confocal microscopy, which is not surprising given the distribution of NHE3 throughout the microvilli at baseline. We collaborated with the Yip laboratory to determine the impact of phenol injury on PT function (23). The physiological results supported the molecular findings: in the absence of a change in GFR, phenol injury increased PT Na+/H+ exchanger activity 2.6-fold and decreased PT flow rate 33%, while saline injection had no effect on Na+/H+ exchanger activity or PT flow (Fig. 2B). As expected from this set of results, there was only a slight and insignificant pressure diuresis response in this model of neurogenic hypertension. We concluded that the increase and persistence of the hypertension was, at least in part, facilitated by the increase in PT sodium transport, which prevented the pressure diuresis response localized to this region.
Five weeks after the single injection of phenol, blood pressure remained elevated at ∼150 mmHg, and NHE3 and NaPi2 distributions remained shifted to the low-density microvillar membrane-enriched fractions. However, total NHE3 abundance decreased 44% after 5 wk, providing evidence for an “escape” mechanism counteracting the effect of NHE3 redistribution to the microvilli (51).
Angiotensin-Converting Enzyme Inhibitors Decrease PT Sodium Transport
“It has been suggested that the effects of certain diuretics on the kidney … may be largely conditioned, not so much by their influence on the glomerular circulation, as by a paralytic effect on the absorptive functions of the tubules (38).”
Angiotensin-converting enzyme (ACE) catalyzes the conversion of ANG I to ANG II and, as such, is a key rate-limiting component of the RAS. Although ACE inhibitors (ACEI) are among the most popular drugs prescribed to lower blood pressure and slow the progression of heart disease, there was no consensus about how ACEI altered renal sodium transport. We addressed this question by merging physiology and proteomic approaches. Leong et al. (21) identified a dose of the ACEI captopril that, when infused, did not change BP or GFR, but did significantly increase urine output. Our collaborator D. K. P. Yip demonstrated that this dose rapidly increased PT flow rate, evidence that inhibiting ACE inhibits PT Na+ reabsorption. Both subcellular fractionation (Fig. 2C) and microscopy demonstrated that captopril provoked a rapid retraction of NHE3 and myosin VI to the higher-density membranes found at the base of the microvilli. Stimulated by the complete removal of NHE3 out of fraction 4 of the sorbitol gradient (Fig. 2C), we aimed to determine what other PT microvillar proteins redistributed out of the apical membrane-enriched fraction 4 to obtain a more complete picture of the effects of ACEI. We simply resolved this fraction from captopril-infused and saline-infused rats by one-dimensional SDS-PAGE, stained the gel, and visually identified eight bands that were significantly lower in the ACEI-treated set. These bands were identified by matrix-assisted laser desorption/ionization-time of flight microscopy by J. Klein (University of Louisville, Louisville, Kentucky), and their distributions on sorbitol gradients were analyzed by immunoblot. The following PT-resident proteins exhibited redistribution out of fraction 4 to heavier-density fractions: megalin, myosin II-A, clathrin, aminopeptidase N, DPPIV, ezrin, moesin, H+-ATPase β2 subunit. Using a candidate approach, we also identified redistribution of NHERF-1 (21). These findings provide in vivo evidence for acute and coordinate redistribution of NHE3 along with four proteins previously proposed to interact with and/or regulate NHE3:NHERF-1, DPPIV, ezrin, and megalin. The results reveal a very dynamic microvilli, in which many proteins, including transporters, enzymes, and cytoskeleton-associated proteins redistribute along with NHE3 in response to changes in ANG II levels, as well as in response to changes in blood pressure and SNS activity (Fig. 5). In a subsequent study using the same protocol, we showed that captopril also provokes a rapid internalization of distal tubule NCC (37), indicating that the overall diuresis provoked by ACEI is due to depression of Na+ transport at multiple sites along the nephron.
Fig. 5.
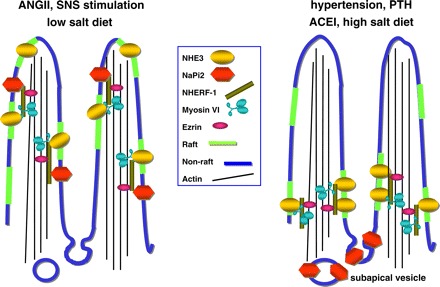
Schematic representing response of renal PT sodium transporters NaPi2 and Na+/H+ exchanger 3 (NHE3) to natriuretic and antinatriuretic stimuli. With the antinatriuretic stimuli, ANG II, SNS simulation/phenol injury and low-salt diet, both NaPi2 and NHE3 are located in the body of the microvilli: NHE3, DPPIV, and myosin VI are enriched in lipid rafts (green), while NaPi2 is enriched in nonlipid rafts (blue) along with NHE regulatory factor 1 (NHERF-1) and ezrin (35). Both NaPi2 and NHE3 have been reported to be associated with the PDZ adaptor protein NHERF-1 and are presumably tethered to the cytoskeleton via ezrin (42). How myosin VI is tethered to the transporters and the actin core remains to be determined. With natriuretic stimuli, such as elevated arterial or perfusion pressure, ACEI, high-salt diet or PTH, NHE3, and NaPi2 are translocated, within the plane of the membrane, out of the body of the microvilli. During PTH treatment, NHERF-1 is phosphorylated and dissociates from NaPi2 (41), presumably allowing the cotransporter to move into the intermicrovillar cleft, where it is endocytosed to subapical dense apical tubules and endosomes (48). In contrast, NHE3 remains in a domain at the base of the microvilli along with myosin VI, both localized to lipid rafts (48, 49).
From these results, can we predict the effects of chronic ACEI treatment on sodium transporters in hypertensive animals? In SHR, we previously determined that NHE3 and NaPi2 were shifted to the base of the microvilli, likely a compensatory response to the elevated blood pressure (26, 56). We saw in the acute captopril study that ACEI also shifted transporters to the base of the microvilli. To determine the combined effects of long-term ACEI and blood pressure lowering, Yang et al. (46) treated adult SHRs with enalapril in their drinking water for 4 wk. Blood pressure was reduced from ∼150 to 100 mmHg. NHE3, already partially retracted in the untreated SHR, fully retracted to the base of the microvilli (with no change in total abundance), and NaPi2 partially retracted to subapical endosomes, and its abundance was reduced. The transporter regulators myosin VI, DPPIV, and NHERF1 were also further redistributed to higher-density membranes, and the abundance of myosin VI was reduced 50%. The loop of Henle Na-K-2Cl cotransporter was also significantly redistributed to higher-density membranes enriched in endosomal markers. This pattern of changes is consistent with the “paralytic effect” predicted by Starling: ACEI likely reduces sodium transport all along the ANG II-sensitive regions of the nephron due to retraction of sodium transporters from the apical membranes of the PT, loop of Henle, and DCT, reducing ECFV and blood pressure. Patients often take ACEI in combination with loop diuretics or thiazide diuretics that act in the DCT. The effect of ACEI would counteract or blunt the development of diuretic resistance by preventing the activation of transport in the regions that are not diuretic targets. These studies provide underpinnings for a molecular explanation for the observation that combining ACEI with diuretics is an effective strategy to reduce renal sodium reabsorption and control blood pressure.
ANG II Increases PT Sodium Transport
“The whole aim of all the mechanisms set into action by change in the volume of the circulating fluid is the maintenance of arterial pressure. Loss of blood is followed by constriction of the arterioles. The flow through all the capillaries, except through those of the brain, is diminished, the pressure in these vessels is also diminished. The arterial side of the circulation is kept filled, so far as possible, at the expense of the venous side. The fall of pressure in the capillaries of the portal system and liver cause a diminution in the flow of lymph from the thoracic duct. In the same way the urinary flow may be diminished or abolished (38).”
We now appreciate that the production of ANG II is key to “all the mechanisms set into action” in the face of decreased ECFV. ANG II constricts the vasculature and stimulates the production of aldosterone and thirst and directly stimulates sodium transport. ANG II's effects on the PT in vivo have been difficult to isolate from the hormone's effect on vascular contractility, RBF, GFR, as well as from the intrarenal production of ANG II. To determine the direct effects of ANG II in the PT, we applied an “ANG II clamp” protocol. First, local and systemic RAS were inhibited with ACEI infused at a level (described in the previous section) that did not alter BP, RBF, or GFR. ACEI treatment, theoretically, also inhibits intrarenal production of ANG II in the PT (63). Second, ANG II was infused along with the captopril at a dose established to be nonpressor (24). Finally, by comparing rats infused with captopril vs. ANG II plus captopril for 20 min, Riquier-Brison et al. (34) were able to isolate the effects of ANG II on PT transport and transporters. The observation that a ANG II clamp decreased PT flow rate compared with an infusion with captopril alone [measured by Yip (21)] provided evidence for an antinatriuretic action of ANG II in the PT (Fig. 2D). After captopril, as discussed above, NHE3 was localized to the base of the microvilli and NaPi2 to subapical cytoplasmic vesicles; after 20-min infusion of ANG II (20 ng·kg−1·min−1), both NHE3 and NaPi2 redistributed into the microvilli (assayed by confocal microscopy and subcellular fractionation): NHE3 from the base of the microvilli and NaPi2 from intracellular pools (Fig. 2D). In addition, other proteins that moved out of the body of the microvilli during captopril treatment (myosin VI, DPPIV, NHERF-1, ezrin, megalin, vacuolar H+-ATPase, aminopeptidase N, and clathrin) returned to the microvilli during the nonpressor ANG II infusion. These results indicate that the acute translocation of this set of transporters, enzymes, and cytoskeleton-associated proteins along the PT microvilli is one of the “mechanisms set into action by change in the volume of the circulating fluid,” specifically, an ANG II-driven antinatriuretic response mediated by a rapid increase in PT salt and water reabsorption that helps to correct and maintain ECFV homeostasis.
If ANG II were to be infused at a dose sufficient to elevate blood pressure, then two counteracting influences would be at play in the PT: hypertension would favor natriuresis, and ANG II would favor antinatriuresis. In collaboration with Susan Gurley and Thomas Coffman (Duke University, Durham, NC), we examined regulation of sodium transporter abundance in wild-type mice infused with 1 μg ANG II·kg−1·min−1 for 2 wk, which increased BP ∼30 mmHg (S. Gurley, unpublished data). We discovered that total abundance of DCT NCC was doubled, likely contributing to the increase in BP, while PT NHE3 abundance was significantly decreased 30%. Whether the NHE3 was located in the body or base of the microvilli remains to be determined.
High-Salt Diet Inhibits PT Sodium Transport
“When sodium chloride is injected into the blood vessels, it escapes with such ease into the tissues that the total hydraemic plethora produced is less than with injection of glucose or sodium sulphate. Moreover, this salt appears to exercise little or no specific influence on the kidney vessels or cells, so that the diuresis comes to an end as soon as the haemoglobin content of the blood, and therefore, its volume, has been restored to normal. On the other hand, the greater molecular concentration of the tissues, caused by the escape into them of the salt, will produce thirst and increased intake of fluid until their concentration is reduced to normal. The absence of the diuretic effect implies that there is no driving force pumping the excess of sodium chloride out of the blood, and therefore, out of the tissues. These indeed seem to possess but little sensitiveness towards the presence of the salt, which forms the most abundant constituent of their normal medium.”
“It is a familiar circumstance that the ingestion of an excessive quantity of salt provokes thirst rather than diuresis. If this excessive ingestion were continued or became chronic, there would be a tendency for the amount of this salt in the body to continually increase, the salt being associated with sufficient water to maintain the molecular concentration of the body fluids at their normal height (38).”
Starling recognized that a high salt intake provokes thirst, which, providing free access to water, increases ECFV and prevents a change in the ECFV salt concentration. However, Starling did not apparently recognize that renal sodium reabsorption would have to be depressed to match sodium intake. M. Sandberg and L. Yang were interested in investigating why a high-salt diet (i.e., elevated ECFV) provoked hypertension in salt-sensitive animals. They first sought to determine the basis for the natriuresis accompanying high-salt intake in normal animals. They applied a systems approach to determine how raising dietary sodium from 0.4 to 4% affected the abundance and subcellular distribution of all of the major transporters along the nephron in Sprague-Dawley rats. After just 18 h, a high-salt diet increased urinary salt excretion 10-fold, and urine volume three-fold, evidence of an active thirst response, but did not change plasma [Na+], as observed by Starling (36, 50). Sandberg, collaborating with Maunsbach, verified that there was a 50% decrease in NCC total abundance, as well as a significant redistribution of the cotransporter from the apical membrane to subapical cytoplasmic vesicles, providing the first evidence for NCC trafficking in vivo (36). The pool size of the other sodium transporters remained unchanged. However, the distribution of the transporters (NHE3, NaPi2, NKCC, NCC, ENaC, Na+-K+-ATPase) on sorbitol gradients shifted to higher-density fractions enriched in membrane markers of intracellular vesicles; similar shifts in distribution of NHE3-associated proteins (DPPIV and myosin VI) were also evident. We concluded that the renal homeostatic response to a high-salt diet involved regulation along the entire nephron; high salt intake reduces Na+ reabsorption by decreasing the abundance of Na+ transporters and their associated regulators in active domains of the plasma membrane primarily by redistribution rather than change in total pool size (only NCC abundance is decreased). Redistribution from one location to another, rather than degradation and synthesis, can provide a mechanism for quickly and continuously adapting to high- or low-sodium intake situations without the need to synthesize or degrade transporters. Reducing the number of sodium transporters in active apical domains would also, theoretically, offset glomerulotubular balance and allow the excretion of a sodium load during high-salt diet. These studies provide a baseline blueprint for future investigations into homeostatic mechanisms in salt-sensitive hypertension.
Overview and Conclusions
“The kidney is a dual organ, and while one part of it acts, so to speak, passively in response to force impressed upon it from without, another part, endowed with sensibility, reacts to the external forces in a direction, which may be opposed to these forces, but is in all cases the appropriate one for the welfare of the whole organism (38).”
During the past 25 years, the molecular mechanisms responsible for the “sensibility” of the kidney have begun to be better understood. This sensibility depends on feedback loops (Fig. 1) that link blood pressure, extracellular fluid volume, the RAS, and SNS. Our studies, and those of other laboratories, revealed the role of the PT in these feedback loops (Fig. 5). Natriuretic stimuli, such as hypertension (acute or chronic), high-salt diet (elevated ECFV), and inhibition of ANG II production depress PT sodium reabsorption; conversely, antinatriuretic stimuli such as ANG II, SNS activation, and low-salt diet (decreased ECFV) increase PT sodium reabsorption (Fig. 4). These variables all regulate the distribution of NHE3 and NaPi2 along the apical microvilli of the PT (Fig. 2).
At the molecular level, our studies show that natriuretic stimuli provoke the dynamic redistribution of a transporter complex that consists of NHE3 and NaPi2 along with their associated regulators (DPPIV, NHERF-1), molecular motors (myosin VI, myosin II-A), and cytoskeleton-associated proteins (ezrin, moesin) to the base of the microvilli. It remains to be determined which members of this complex can be coimmunoprecipitated. The components can be fractionated by preparation of raft vs. nonraft domains, indicating that transporter complex is composed of both lipid raft and nonraft domains (Fig. 3). During natriuretic stimulation, the lipid raft-associated NHE3 remains at the base, and the nonraft-associated NaPi2 is endocytosed. Importantly, the transporters' domain does not change (between raft and nonraft) during this redistribution along the microvilli, which may be important for the differential destinations. While it is clear that NaPi2 transport activity will be decreased by its endocytosis, we have not yet identified the molecular basis for the decrease in NHE3 transport activity that has been measured in intact kidney during hypertension (57). The NHE3 remains in the apical membrane where it could theoretically transport Na+ from lumen to ICF. Assayed in vitro, the Na+/H+ exchanger activity/NHE3 abundance is not different in membrane vesicles prepared from low- vs. high-density gradient fractions. Kocinsky and Aronson have reported that NHE3 is phosphorylated during PTH treatment as it moves to the base of the microvilli; however, they failed to observe a change in NHE activity/NHE3 abundance associated with this phosphorylation (18, 19). One explanation is that NHE activity is regulated by protein-protein associations that are lost with current subcellular fractionation methods.
Antinatriuretic stimuli return the same transporter complex to the body of the microvilli associated with an increase in transport activity and decrease in PT flow rate. That NHE3 and NaPi2 can glide up and down the microvillar membrane now appears evident, although it hasn't been visualized directly in vivo. The identity and nature of the motor(s) driving this response are not entirely clear. It is possible that the opposite polarity motors myosin VI and myosin II-A work in tandem to effect translocation of the transporter complex both up and down the microvilli, analogous to how kinesin and dynein motors translocate organelles (2).
Perspectives and Significance
In summary, ECFV and BP homeostasis are, at least in part, maintained by continuous and acute redistribution of a transporter complex up and down the PT microvilli, which regulates PT sodium reabsorption in response to fluctuations in ECFV, BP, SNS, and RAS. These stimuli and signals appear to converge on a very similar response. As a result, it is not surprising that responses to natriuretic hormones are blunted during chronic hypertension since the hypertension itself effects a redistribution of the transporters out of the microvilli. The coordinate complex of transporters, regulators, motors, and cytoskeleton suggests that a mutation in any one component could influence the pressure natriuresis response and, thus, the baseline blood pressure. The precedent of the effects of mutations in the cytoskeleton-associated protein adducin illustrate this point (4). The next era of investigation will, ideally, integrate the systems physiology approaches of Starling with the systems biology approaches that are now possible to understand the real-time integration of the effects of BP, ECFV, SNS, and RAS on renal sodium transport along the nephron and use this information to solve the riddles so elegantly articulated by Starling (38) and to understand how the overall pattern is disrupted in disease.
GRANTS
This work was supported by National Institutes of Health Grants DK-34316 and HL-085388.
ACKNOWLEDGMENTS
This work could not have been accomplished without the substantive creative contributions of Li E. Yang, Patrick K. Leong, Anne Riquier-Brison, Monica B. Sandberg, Yibin Zhang, Clara E. Magyar, Arvid B. Maunsbach, and Daniel K. P. Yip. Harvey Kaslow provided a critical reading of the manuscript.
REFERENCES
- 1. Ahmed F, Kemp BA, Howell NL, Siragy HM, Carey RM. Extracellular renal guanosine cyclic 3'5'-monophosphate modulates nitric oxide and pressure-induced natriuresis. Hypertension 50: 958–963, 2007 [DOI] [PubMed] [Google Scholar]
- 2. Ally S, Larson AG, Barlan K, Rice SE, Gelfand VI. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol 187: 1071–1082, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Azuma KK, Balkovetz DF, Magyar CE, Lescale-Matys L, Zhang Y, Chambrey R, Warnock DG, McDonough AA. Renal Na+/H+ exchanger isoforms and their regulation by thyroid hormone. Am J Physiol Cell Physiol 270: C585–C592, 1996 [DOI] [PubMed] [Google Scholar]
- 4. Bianchi G. Genetic variations of tubular sodium reabsorption leading to “primary” hypertension: from gene polymorphism to clinical symptoms. Am J Physiol Regul Integr Comp Physiol 289: R1536–R1549, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Biemesderfer D, Mentone SA, Mooseker M, Hasson T. Expression of myosin VI within the early endocytic pathway in adult and developing proximal tubules. Am J Physiol Renal Physiol 282: F785–F794, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Birkenfeld LW, Leibman J, O'Meara MP, Edelman IS. Total exchangeable sodium, total exchangeable potassium, and total body water in edematous patients with cirrhosis of the liver and congestive heart failure. J Clin Invest 37: 687–698, 1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blaine J, Okamura K, Giral H, Breusegem S, Caldas Y, Millard A, Barry N, Levi M. PTH-induced internalization of apical membrane NaPi2a: role of actin and myosin VI. Am J Physiol Cell Physiol 297: C1339–C1346, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campese VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl 75: S2–S6, 2000 [PubMed] [Google Scholar]
- 9. Chou CL, Marsh DJ. Role of proximal convoluted tubule in pressure diuresis in the rat. Am J Physiol Renal Fluid Electrolyte Physiol 251: F283–F289, 1986 [DOI] [PubMed] [Google Scholar]
- 10. Chou CL, Marsh DJ. Time course of proximal tubule response to acute arterial hypertension in the rat. Am J Physiol Renal Fluid Electrolyte Physiol 254: F601–F607, 1988 [DOI] [PubMed] [Google Scholar]
- 11. Dos Santos EA, Dahly-Vernon AJ, Hoagland KM, Roman RJ. Inhibition of the formation of EETs and 20-HETE with 1-aminobenzotriazole attenuates pressure natriuresis. Am J Physiol Regul Integr Comp Physiol 287: R58–R68, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Evans RG, Majid DS, Eppel GA. Mechanisms mediating pressure natriuresis: what we know and what we need to find out. Clin Exp Pharmacol Physiol 32: 400–409, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Hensley CB, Bradley ME, Mircheff AK. Parathyroid hormone-induced translocation of Na-H antiporters in rat proximal tubules. Am J Physiol Cell Physiol 257: C637–C645, 1989 [DOI] [PubMed] [Google Scholar]
- 14. Hensley CB, Bradley ME, Mircheff AK. Subcellular distribution of Na+/H+ antiport activity in rat renal cortex. Kidney Int 37: 707–716, 1990 [DOI] [PubMed] [Google Scholar]
- 15. Hiatt A, McDonough AA, Edelman IS. Assembly of the (Na+ + K+)-adenosine triphosphatase. Post-translational membrane integration of the alpha subunit. J Biol Chem 259: 2629–2635, 1984 [PubMed] [Google Scholar]
- 16. Hirano K, Nagasawa M, Saito K, Tomino Y, Koide H. Adaptation of low-phosphate diet in renal brush borders of spontaneously hypertensive rats. Contrib Nephrol 90: 59–64, 1991 [DOI] [PubMed] [Google Scholar]
- 17. Hong SK, Claybaugh JR, Frattali V, Johnson R, Kurata F, Matsuda M, McDonough AA, Paganelli CV, Smith RM, Webb P. Hana Kai II: a 17-day dry saturation dive at 18.6 ATA. III. Body fluid balance. Undersea Biomed Res 4: 247–265, 1977 [PubMed] [Google Scholar]
- 18. Kocinsky HS, Dynia DW, Wang T, Aronson PS. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am J Physiol Renal Physiol 293: F212–F218, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Kocinsky HS, Girardi AC, Biemesderfer D, Nguyen T, Mentone S, Orlowski J, Aronson PS. Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol 289: F249–F258, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Lee DH, Riquier AD, Yang LE, Leong PK, Maunsbach AB, McDonough AA. Acute hypertension provokes acute trafficking of distal tubule Na-Cl cotransporter (NCC) to subapical cytoplasmic vesicles. Am J Physiol Renal Physiol 296: F810–F818, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leong PK, Devillez A, Sandberg MB, Yang LE, Yip DK, Klein JB, McDonough AA. Effects of ACE inhibition on proximal tubule sodium transport. Am J Physiol Renal Physiol 290: F854–F863, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Leong PK, Yang LE, Holstein-Rathlou NH, McDonough AA. Angiotensin II clamp prevents the second step in renal apical NHE3 internalization during acute hypertension. Am J Physiol Renal Physiol 283: F1142–F1150, 2002 [DOI] [PubMed] [Google Scholar]
- 23. Leong PK, Yang LE, Landon CS, McDonough AA, Yip KP. Phenol injury-induced hypertension stimulates proximal tubule Na+/H+ exchanger activity. Am J Physiol Renal Physiol 290: F1543–F1550, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Leong PK, Zhang Y, Yang LE, Holstein-Rathlou NH, McDonough AA. Diuretic response to acute hypertension is blunted during angiotensin II clamp. Am J Physiol Regul Integr Comp Physiol 283: R837–R842, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Leyssac PP, Christensen P. A comparison between endogenous and exogenous lithium clearance in the anaesthetized rat. Acta Physiol Scand 151: 173–179, 1994 [DOI] [PubMed] [Google Scholar]
- 26. Magyar CE, Zhang Y, Holstein-Rathlou NH, McDonough AA. Proximal tubule Na transporter responses are the same during acute and chronic hypertension. Am J Physiol Renal Physiol 279: F358–F369, 2000 [DOI] [PubMed] [Google Scholar]
- 27. Maunsbach AB. Absorption of I-125-labeled homologous albumin by rat kidney proximal tubule cells. A study of microperfused single proximal tubules by electron microscopic autoradiography and histochemistry. J Ultrastruct Res 15: 197–241, 1966 [DOI] [PubMed] [Google Scholar]
- 28. Maunsbach AB. The influence of different fixatives and fixation methods on the ultrastructure of rat kidney proximal tubule cells. I. Comparison of different perfusion fixation methods and of glutaraldehyde, formaldehyde and osmium tetroxide fixatives. J Ultrastruct Res 15: 242–282, 1966 [DOI] [PubMed] [Google Scholar]
- 29. McDonough AA. Motoring down the microvilli. Focus on “PTH-induced internalization of apical membrane NaPi2a: role of actin and myosin VI”. Am J Physiol Cell Physiol 297: C1331–C1332, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDonough AA, Hiatt A, Edelman IS. Characteristics of antibodies to guinea pig (Na+ + K+)-adenosine triphosphatase and their use in cell-free synthesis studies. J Membr Biol 69: 13–22, 1982 [DOI] [PubMed] [Google Scholar]
- 31. McDonough AA, Magyar CE, Komatsu Y. Expression of Na+-K+-ATPase alpha- and beta-subunits along rat nephron: isoform specificity and response to hypokalemia. Am J Physiol Cell Physiol 267: C901–C908, 1994 [DOI] [PubMed] [Google Scholar]
- 32. Panico C, Luo Z, Damiano S, Artigiano F, Gill P, Welch WJ. Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension 54: 1291–1297, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peti-Peterdi J, Morishima S, Bell PD, Okada Y. Two-photon excitation fluorescence imaging of the living juxtaglomerular apparatus. Am J Physiol Renal Physiol 283: F197–F201, 2002 [DOI] [PubMed] [Google Scholar]
- 34. Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2 and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol 298: F177–F186, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riquier AD, Lee DH, McDonough AA. Renal NHE3 and NaPi2 partition into distinct membrane domains. Am J Physiol Cell Physiol 296: C900–C910, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandberg MB, Maunsbach AB, McDonough AA. Redistribution of distal tubule Na+-Cl− cotransporter (NCC) in response to a high-salt diet. Am J Physiol Renal Physiol 291: F503–F508, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB. ANG II provokes acute trafficking of distal tubule Na+-Cl− cotransporter to apical membrane. Am J Physiol Renal Physiol 293: F662–F669, 2007. [DOI] [PubMed] [Google Scholar]
- 38. Starling EH. The Fluids of the Body. London: Archibald Constable, 1909 [Google Scholar]
- 39. Takahashi K, Capdevila J, Karara A, Falck JR, Jacobson HR, Badr KF. Cytochrome P-450 arachidonate metabolites in rat kidney: characterization and hemodynamic responses. Am J Physiol Renal Fluid Electrolyte Physiol 258: F781–F789, 1990 [DOI] [PubMed] [Google Scholar]
- 40. Wang X, Armando I, Upadhyay K, Pascua A, Jose PA. The regulation of proximal tubular salt transport in hypertension: an update. Curr Opin Nephrol Hypertens 18: 412–420, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weinman EJ, Biswas RS, Peng G, Shen L, Turner CL, EX , Steplock D, Shenolikar S, Cunningham R. Parathyroid hormone inhibits renal phosphate transport by phosphorylation of serine 77 of sodium-hydrogen exchanger regulatory factor-1. J Clin Invest 117: 3412–3420, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Weinman EJ, Cunningham R, Wade JB, Shenolikar S. The role of NHERF-1 in the regulation of renal proximal tubule sodium-hydrogen exchanger 3 and sodium-dependent phosphate cotransporter 2a. J Physiol 567: 27–32, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Welling LW, Welling DJ. Surface areas of brush border and lateral cell walls in the rabbit proximal nephron. Kidney Int 8: 343–348, 1975 [DOI] [PubMed] [Google Scholar]
- 44. Williams JM, Sarkis A, Lopez B, Ryan RP, Flasch AK, Roman RJ. Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 49: 687–694, 2007 [DOI] [PubMed] [Google Scholar]
- 45. Yang L, Leong PK, Chen JO, Patel N, Hamm-Alvarez SF, McDonough AA. Acute hypertension provokes internalization of proximal tubule NHE3 without inhibition of transport activity. Am J Physiol Renal Physiol 282: F730–F740, 2002 [DOI] [PubMed] [Google Scholar]
- 46. Yang LE, Leong PK, McDonough AA. Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. Am J Physiol Renal Physiol 293: F1197–F1208, 2007 [DOI] [PubMed] [Google Scholar]
- 47. Yang LE, Leong PK, Ye S, Campese VM, McDonough AA. Responses of proximal tubule sodium transporters to acute injury-induced hypertension. Am J Physiol Renal Physiol 284: F313–F322, 2003 [DOI] [PubMed] [Google Scholar]
- 48. Yang LE, Maunsbach AB, Leong PK, McDonough AA. Differential traffic of proximal tubule Na+ transporters during hypertension or PTH: NHE3 to base of microvilli vs. NaPi2 to endosomes. Am J Physiol Renal Physiol 287: F896–F906, 2004 [DOI] [PubMed] [Google Scholar]
- 49. Yang LE, Maunsbach AB, Leong PK, McDonough AA. Redistribution of myosin VI from top to base of proximal tubule microvilli during acute hypertension. J Am Soc Nephrol 16: 2890–2896, 2005 [DOI] [PubMed] [Google Scholar]
- 50. Yang LE, Sandberg MB, Can AD, Pihakaski-Maunsbach K, McDonough AA. Effects of dietary salt on renal Na+ transporters' subcellular distribution, abundance, and phosphorylation status. Am J Physiol Renal Physiol 295: F1003–F1016, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang LE, Zhong H, Leong PK, Perianayagam A, Campese VM, McDonough AA. Chronic renal injury-induced hypertension alters renal NHE3 distribution and abundance. Am J Physiol Renal Physiol 284: F1056–F1065, 2003 [DOI] [PubMed] [Google Scholar]
- 52. Ye S, Gamburd M, Mozayeni P, Koss M, Campese VM. A limited renal injury may cause a permanent form of neurogenic hypertension. Am J Hypertens 11: 723–728, 1998 [DOI] [PubMed] [Google Scholar]
- 53. Ye S, Ozgur B, Campese VM. Renal afferent impulses, the posterior hypothalamus, and hypertension in rats with chronic renal failure. Kidney Int 51: 722–727, 1997 [DOI] [PubMed] [Google Scholar]
- 54. Ye S, Zhong H, Yanamadala S, Campese VM. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension 48: 309–315, 2006 [DOI] [PubMed] [Google Scholar]
- 55. Ye S, Zhong H, Yanamadala V, Campese VM. Renal injury caused by intrarenal injection of phenol increases afferent and efferent renal sympathetic nerve activity. Am J Hypertens 15: 717–724, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Yip KP, Tse CM, McDonough AA, Marsh DJ. Redistribution of Na+/H+ exchanger isoform NHE3 in proximal tubules induced by acute and chronic hypertension. Am J Physiol Renal Physiol 275: F565–F575, 1998 [DOI] [PubMed] [Google Scholar]
- 57. Yip KP, Wagner AJ, Marsh DJ. Detection of apical Na+/H+ exchanger activity inhibition in proximal tubules induced by acute hypertension. Am J Physiol Regul Integr Comp Physiol 279: R1412–R1418, 2000 [DOI] [PubMed] [Google Scholar]
- 58. Zhang Y, Magyar CE, Norian JM, Holstein-Rathlou NH, Mircheff AK, McDonough AA. Reversible effects of acute hypertension on proximal tubule sodium transporters. Am J Physiol Cell Physiol 274: C1090–C1100, 1998 [DOI] [PubMed] [Google Scholar]
- 59. Zhang Y, Mircheff AK, Hensley CB, Magyar CE, Warnock DG, Chambrey R, Yip KP, Marsh DJ, Holstein-Rathlou NH, McDonough AA. Rapid redistribution and inhibition of renal sodium transporters during acute pressure natriuresis. Am J Physiol Renal Fluid Electrolyte Physiol 270: F1004–F1014, 1996 [DOI] [PubMed] [Google Scholar]
- 60. Zhang Y, Norian JM, Magyar CE, Holstein-Rathlou NH, Mircheff AK, McDonough AA. In vivo PTH provokes apical NHE3 and NaPi2 redistribution and Na-K-ATPase inhibition. Am J Physiol Renal Physiol 276: F711–F719, 1999 [DOI] [PubMed] [Google Scholar]
- 61. Zhang YB, Magyar CE, Holstein-Rathlou NH, McDonough AA. The cytochrome P-450 inhibitor cobalt chloride prevents inhibition of renal Na+,K+-ATPase and redistribution of apical NHE-3 during acute hypertension. J Am Soc Nephrol 9: 531–537, 1998 [DOI] [PubMed] [Google Scholar]
- 62. Zhao D, Navar LG. Acute angiotensin II infusions elicit pressure natriuresis in mice and reduce distal fractional sodium reabsorption. Hypertension 52: 137–142, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhou Y, Boron WF. Role of endogenously secreted angiotensin II in the CO2-induced stimulation of HCO3 reabsorption by renal proximal tubules. Am J Physiol Renal Physiol 294: F245–F252, 2008 [DOI] [PubMed] [Google Scholar]
- 64. Zou AP, Imig JD, Kaldunski M, Ortiz de Montellano PR, Sui Z, Roman RJ. Inhibition of renal vascular 20-HETE production impairs autoregulation of renal blood flow. Am J Physiol Renal Fluid Electrolyte Physiol 266: F275–F282, 1994. [DOI] [PubMed] [Google Scholar]