


Abstract
Gram-positive bacteria utilize a tRNA-responsive transcription antitermination mechanism, designated the T box system, to regulate expression of many amino acid biosynthetic and aminoacyl-tRNA synthetase genes. The RNA transcripts of genes controlled by this mechanism contain 5′ untranslated regions, or leader RNAs, that specifically bind cognate tRNA molecules through pairing of nucleotides in the tRNA anticodon loop with nucleotides in the Specifier Loop domain of the leader RNA. We have determined the solution structure of the Specifier Loop domain of the tyrS leader RNA from Bacillus subtilis. Fifty percent of the nucleotides in the Specifier Loop domain adopt a loop E motif. The Specifier Sequence nucleotides, which pair with the tRNA anticodon, stack with their Watson–Crick edges rotated toward the minor groove and exhibit only modest flexibility. We also show that a Specifier Loop domain mutation that impairs the function of the B. subtilis glyQS T box RNA disrupts the tyrS loop E motif. Our results suggest a mechanism for tRNA–Specifier Loop binding in which the phosphate backbone kink created by the loop E motif causes the Specifier Sequence bases to rotate toward the minor groove, which increases accessibility for pairing with bases in the anticodon loop of tRNA.
INTRODUCTION
The expression of many aminoacyl-tRNA synthetase genes and genes involved in amino acid metabolism and uptake in Gram-positive bacteria is regulated at the level of transcription attenuation by the T box riboswitch, an RNA element in the 5′ untranslated (or leader) region of the gene (1,2). The expression of genes in this family is determined by the relative levels of charged and uncharged tRNAs in the cell, with each gene in the family responding individually to a specific tRNA species. If the corresponding tRNA is highly aminoacylated, a transcriptional attenuator in the leader region is active, and the gene is not expressed. Readthrough of the termination site is dependent on binding of uncharged tRNA to the T box element, which is 200–300 nt in length and contains several conserved primary and secondary structure elements, including three helices (Stems I, II and III), the Stem IIA/B pseudoknot and the terminator and antiterminator structures (Figure 1A). The secondary structures of the terminator and antiterminator helices are mutually exclusive and the terminator helix is thermodynamically more favorable (3). Stabilization of the antiterminator helix, and subsequent transcriptional readthrough, is accomplished by pairing of the four 3′ terminal nucleotides of the tRNA molecule with residues in a 7-nt bulge of the antiterminator helix (4).
Figure 1.

Sequence and proposed secondary structures of (A) the leader RNA of the B. subtilis tyrS gene and (B) the 38-nt RNA molecule, tyrSSD, corresponding to the Specifier Loop domain of the tyrS leader RNA.
Specific tRNA recognition is dependent primarily on the identity of three nucleotides, the Specifier Sequence, within the Specifier Loop domain, a conserved internal loop of variable size located in Stem I (3,5). The Specifier Sequence nucleotides are complementary to the anticodon nucleotides of the cognate tRNA. Changes in the Specifier Sequence can in some cases result in a switch in the specificity of the T box riboswitch to allow recognition of a new tRNA (3,5,6). The residue 3′ to the Specifier Sequence is conserved as a purine, and pairs with the conserved U at position 33 in tRNA, 5′ to the anticodon (7). The functional importance of these tRNA–leader RNA interactions have been demonstrated for the Bacillus subtilis tyrS and glyQS leader RNAs (3,8) and reproduced in vitro using a purified transcription assay and the glyQS leader RNA (9,7). In addition, interaction between glyQS Specifier Loop domain and tRNAGly anticodon stem–loop was demonstrated with fluorescence assays (10).
In addition to the overall secondary structure and nucleotide conservation of T box leader RNAs (3), a loop E (or S-turn) motif is predicted to form in the Specifier Loop (11). The loop E motif is a common RNA structural element found in many RNAs including ribosomal RNAs (12–14), the hairpin ribozyme (15), and the nucleolin-recognition element in eukaryotes (16,17). The sequences and structures of these motifs are very similar among different RNAs and feature a group of three non-canonical base pairs (18). This motif frequently is located proximal to multi-helix junctions and has crucial roles in protein-binding sites and in mediating RNA–RNA interactions (14). The high conservation of this motif within the Specifier Loop suggests an important role in leader RNA function, including tRNA binding.
The Specifier Loop domain of the B. subtilis tyrS leader RNA is an internal loop of 14 nt, including the 4 nt that pair with the tRNATyr anticodon loop, flanked by two short helices. We have used heteronuclear nuclear magnetic resonance (NMR) spectroscopy to determine the solution structure and dynamics of this domain. Our results confirm the presence of the predicted loop E structural element within the internal loop, adjacent to the Specifier Sequence. We also show that a mutation within the loop E nucleotides that leads to loss of function in the context of the B. subtilis glyQS T box leader (Green,N.J., Grundy,F.J. and Henkin,T.M., unpublished data), destabilizes the motif, but does not significantly alter the conformation of the internal loop, confirming the importance of this motif for proper function.
MATERIALS AND METHODS
Materials
All enzymes were purchased from Sigma Chemical (St. Louis, MO, USA) except for T7 RNA polymerase, which was prepared as described (19). Deoxyribonuclease I type II, pyruvate kinase, adenylate kinase and nucleotide monophosphate kinase were obtained as powders, dissolved in 15% glycerol, 1 mM dithiothreitol and 10 mM Tris–HCl, pH 7.4, and stored at –20°C. Guanylate kinase and nuclease P1 were obtained as solutions and stored at –20°C. Unlabeled 5′ nucleoside triphosphates (5′-NTPs) were purchased from Sigma, phosphoenolpyruvate (potassium salt) was purchased from Bachem, and 99% [15N]-ammonium sulfate and 99% [13C]-glucose were purchased from Spectra Stable Isotopes (Branchburg, NJ, USA).
Preparation of RNA samples
The RNA sequence depicted in Figure 1B was prepared by in vitro transcription with T7 RNA polymerase using a synthetic DNA template (20) and either unlabeled 5′-NTPs or 13C/15N -labeled 5′-NTPs as previously described (21). The RNA molecules were purified using 20% (w/v) preparative polyacrylamide gels, electroeluted (Schleicher & Schuell), and precipitated with ethanol. The purified RNA molecules were resuspended in 1.0 M KCl, 20 mM KPi, pH 6.8 and 2.0 mM EDTA and extensively dialyzed against 10 mM KCl, 5 mM KPi, pH 6.7 and 0.02 mM EDTA using a Centricon-3 concentrator (Millipore, Bedford, MA, USA). All RNA samples were concentrated to a volume of 320 µl, lyophilized to a powder, and either resuspended in 90% H2O/10% D2O or 99.96% D2O. The samples were then heated to 80°C for 60 s and snap-cooled on ice. The sample concentrations varied between 120 and 180 A260 O.D. in 320 µl (∼1.5–2.5 mM). Partial alignment of 13C/15N-labeled RNA for residual dipolar coupling measurements was achieved by adding RNA to concentrated Pf1 filamentous phage in 99.96% D2O NMR buffer, yielding final concentrations of 19 mg/ml Pf1 and 0.35 mM RNA. The degree of alignment was quantified using the quadrupole splitting of the 2H2O resonance (22).
NMR spectroscopy
All spectra were acquired on a Varian Inova 500-MHz spectrometer equipped with a 1H–(13C, 15N, 31P) probe and Inova 600 and 800-MHz spectrometers equipped with cryogenically cooled 1H–(13C, 15N) probes. Solvent suppression for 1H homonuclear spectra collected in 90% H2O was achieved using the WATERGATE scheme. Typically, the data points were extended by 25% using linear prediction for the indirectly detected dimensions. NMR spectra were processed and analyzed using Felix 2007 (Felix NMR Inc., San Diego, CA, USA).
Two-dimensional (2D) 13C-1H HSQC spectra were collected to identify 13C-1H chemical shift correlations. Sugar spin systems were assigned using 3D HCCH-TOCSY (16 ms and 24 ms DIPSI-3 spin lock) experiments collected in D2O. A 3D HCCH-TOCSY (56 ms DIPSI-3 spin lock) was collected to establish the intra-base H2-C2-C8-H8 correlations in adenine residues. A 3D HCN experiment (23) was used to identify intra-residue base–ribose correlations.
Sequential assignments and distance constraints for the non-exchangeable resonances were derived at 28°C from 2D 1H -1H nuclear Overhauser effect spectroscopy (NOESY) spectra (τm = 120, 160 and 360 ms) and 3D 13C-edited NOESY spectra (τm = 120 and 360 ms). Pyrimidine C2 and C4 resonances were assigned from H6-C2 and H5-C4 correlations using 2D H(CN)C and 2D CCH-COSY experiments. 2D 15N-1H HSQC spectra optimized for 2-bond HN couplings were collected to identify purine N7 and adenine N1 and N3 resonances. For the exchangeable resonances, 2D 15N-1H HSQC spectra were collected to identify 15N-1H chemical shift correlations. 2D NOESY spectra (τm = 180 and 400 ms) were acquired in H2O and at 12°C to obtain distance restraints involving exchangeable protons.
1H-13C residual dipolar coupling constants (RDCs) were determined from the measured frequency difference between corresponding proton doublets in HSQC spectra acquired for isotropic and partially aligned samples. Thirty RDC values from base CH bond vectors and 16 from ribose 1′ CH vectors were obtained in this manner. The axial and rhombic terms were determined within Xplor-NIH using an extensive grid search (24), and yielded values of DaH = 31.67 and RhH = 0.19.
3JHH coupling constants were estimated from DQF-COSY experiments. 3JC-P coupling constants were determined using the 13C-1H ct-HSQC spin-echo difference method. 3JP-H couplings were estimated using 31P-1H HetCor experiments.
13C T1ρ relaxation times were measured using 2D 13C-1H ct-HSQC-based experiments optimized for C2, for C1′ and for C6 and C8 resonances. A 2.1-kHz 13C spin-lock field was used with delays of 5, 10, 15, 20, 30, 40, 50, 60, 70, 90 and 120 ms. The 5-ms experiment was collected twice to provide an estimate of the error of the measured intensities. The 13C-1H cross-peak volumes were fit to a single exponential decay.
Distance and torsion angle constraints
Interproton distance estimates were obtained from cross-peak intensities in 2D NOESY and 3D 13C-edited NOESY spectra. Cross-peak intensities were calibrated using the pyrimidine H5–H6 fixed distance of 2.54 Å. NOE cross-peak intensities were classified into five categories assigned upper distance bounds of 3.0, 4.0, 5.0, 6.0 or 7.0 Å and a common lower bound of 1.8 Å. Base pairs were identified by direct detection of hydrogen bonds (25) or by observation of strong G•C NH–NH2 or A•U H2–NH NOEs. Hydrogen bond distances restraints and planarity constraints were introduced for residues that form base pairs.
Ribose ring pucker and backbone dihedral constraints were derived from 3JHH, 3JHP and 3JCP couplings (26). Ribose rings with 3JH1′-H2′>7 Hz a′nd with C3′ and C4′ resonances between 76–80 and 85–86 p.p.m., respectively, were constrained to C2′-endo. Residues with 3JH1′-H2′ < 5 Hz and couplings were constrained to C3′-endo. Residues with intermediate 3JH1′-H2′ couplings were left unconstrained. For stem residues 1–6 and 33–38, γ was constrained to the gauche+ conformation (60 ± 30°) (26); γ was left unconstrained for all other residues. For stem residues, β was constrained to the trans conformation (170 ± 40°). β was loosely constrained to the trans conformation (160 ± 50°) for internal loop residues except G11 and A27 that were constrained loosely to gauche+ (80 ± 50°) as determined from examination of loop E containing crystal structures (27,28). ε was constrained to exclude the gauche+ conformation (–150 ± 50°) for residues with 3JP-H3′>5 Hz or 3JP-C2′>5 Hz. α and ζ were constrained to –70 ± 30° for the stem residues and were constrained to exclude the trans conformation (0 ± 120°) for residues 7–14 and 25–32 based on the absence of down-field shifted 31P resonances (29).
Structure refinement
Structure refinement was carried out with simulated annealing and restrained molecular dynamics (rMD) calculations were performed using Xplor-NIH v2.19 (24). Starting coordinates for the tyrSSD model were generated using Insight II (Accelrys, San Diego, CA, USA) and were based on standard A-form helical geometry. The structure calculations were performed in two stages. Beginning with the energy-minimized starting coordinates, 100 structures were generated during the first round of structure calculation by 80 ps of rMD at 1200 K with hydrogen bond, NOE-derived distance and base-pairing restraints. The system then was cooled to 25 K in 47 cycles of rMD corresponding to a total of 12 ps. During this stage, RDC constraints and repulsive van der Waals forces were introduced into the system and the SANI force const0raint used for RDCs was gradually increased from 0.010 kcal mol–1 Hz–2 to 1.000 kcal mol–1 Hz–2. Other force constants used for the calculations were increased—from 2 kcal mol–1 Å–2 to 30 kcal mol–1 Å–2 for the NOE and from 2 kcal mol–1 rad–2 to 30 kcal mol–1 rad–2 for the dihedral angle constraints. Once the temperature reached the target, each structure was then subjected to another round of constrained minimization (30). Ten structures were selected for the final refinement. The criteria for final structure selection included lowest energies, fewest constraint violations and fewest predicted unobserved NOEs (1H pairs <3.5 Å apart, but no corresponding cross-peak in the NOE spectra). A second round of rMD was performed on these structures using protocols similar to those used in the first round of structure calculation. The major difference was the starting temperature of 300 K followed by cooling to 25 K over 28 ps of rMD. The 10 refined structures were analyzed using Xplor-NIH and Insight II.
RESULTS
The RNA molecule used in this study, tyrSSD (Figure 1B), corresponds to the Specifier Loop from Stem I of B. subtilis tyrS leader RNA. Cross-peaks in the NH 15N-1H HSQC spectrum and the resonance linewidths are consistent with the predicted secondary structure, and the NH spectra at low concentration (5 µM) and high concentration (1.8 mM) are nearly identical, supporting the prediction that the RNA is monomeric. The addition of Mg2+ or the reduction of pH from 6.8 to 5.5 caused slight broadening of a few NH resonances, but did not lead to the appearance of new peaks.
Chemical-shift assignments
The sequence-specific resonance assignment of tyrSSD was accomplished using 1H-1H NOESY and 2D and 3D heteronuclear experiments. The NH resonances were assigned using the NOE connectivities between NH proton resonances of neighboring base pairs. These connectivities are continuous in the lower helix from G2 to G33 and the upper helix from G14 to G21. The cytidine and adenine NH2 resonances were assigned using NOESY and HNCCH experiments (31). Additional weak NH resonances are present in the 1D 1H spectrum between 10.5 and 11.8 p.p.m., but cannot be uniquely assigned to U12, U19, U29, G1, G11 or G26.
The non-exchangeable 1H and 13C resonances of tyrSSD were assigned using standard heteronuclear techniques (32,33). Most of the base and ribose 1′ 1H-13C correlations are resolved, with four of the base resonances (A13, G14 and G26 H8-C8 and A13 H2-C2) having spectral characteristics indicative of intermediate exchange (Figure 2A). All residues except U12, A13 and G14 yielded base–ribose correlations in 3D HCN spectra, and 37 of the ribose spin systems were identified using 3D HCCH-TOCSY experiments (G1 was only partially labeled).
Figure 2.

(A) Two-dimensional 13C-1H HSQC spectrum of the base C6/8 and C2 regions of tyrSSD RNA molecule. The sequence-specific resonance assignments are shown. The A13 C2H2 cross-peak is significantly weakened by chemical exchange. The in vitro transcription reaction was primed with unlabeled 5′-GMP, therefore the G1 C8H8 does not appear in this spectrum. (B) Sequential connectivities through the base-1′ region of the 220-ms mixing time two-dimensional NOE spectrum. The dotted lines trace the connectivities among the specifier codon nucleotides U29–A31. The sequential connectivity is disrupted between steps A10–G11 (gray box). The H1′ resonances of G14 and G22 are shifted upfield to 4.25 and 4.42 p.p.m., respectively. These chemical shifts are characteristic of the nucleotides flanking the 3′ side of the adenine of a sheared G–A base pair and the guanine of a UNCG tetraloop (59). See also Supplementary Table S1.
Assignments for the non-exchangeable resonances were made using 2D NOESY (Figure 2B) and 3D 13C-edited NOESY experiments to identify sequential H6/8-H1′ NOE connectivities (32). The sequential H6/8-H1′ NOE connectivities are continuous in the 180-ms NOESY spectrum except at steps A10–G11 and G11–U12. However, G11H8–U12H6 and G11H4′–U12H6 cross-peaks are present in the spectrum. Interestingly, i to i+2 NOE cross-peaks between A10H1′/H2′ and U12H6 are observed (Figure 2B) and suggest that the G11 base bulges from the strand. The chemical shifts of G14H1′ (4.25 p.p.m.) and G11H4′ (5.85 p.p.m.) are unusual and are discussed below.
Most inter-nucleotide 31P resonances are dispersed between –3.4 and –5.1 p.p.m., but the G21pG22 resonance has a chemical shift of –2.19 p.p.m. as previously noted for the UNCG tetraloop motif (34). Several 31P resonances could be assigned using the HCP experiment (35) or the H8/6-P and H1′-P correlations from 2D 31P-1H hetero-TOCSY-NOESY spectra (36). The chemical shifts for tyrSSD are listed in Supplementary Table S1.
Structure of the tyrSSD molecule
The structure of tyrSSD was calculated using a restrained molecular dynamics routine. The calculations used a total of 296 conformationally restrictive distance constraints, 189 dihedral angle constraints and 46 RDC constraints (Table 1) to produce 10 converged structures (Figure 3). The converged structures had an average of 5.8 distance constraint violations between 0.3 and 0.6 Å that were randomly distributed throughout the hairpins. None of the converged structures had NOE constraint violations >0.6 Å. The heavy atoms of the converged structures superimpose on the average structure with an average root mean square deviation (RMSD) of 1.25 Å. The local RMSDs for the loop (A7–A13 and G26–A32) and stems are 0.54 Å and 1.22 Å, respectively.
Table 1.
Summary of experimental distance and dihedral angle constraints and refinement statistics for tyrSSD
| Constraint | tyrSSD |
|---|---|
| NOE distance constraints | |
| Intra-residuea | 87 |
| Inter-residue | 135 |
| Mean number per residue | 16 |
| NOE constraints by category | |
| Very strong (1.8–3.0 Å) | 34 |
| Strong (1.8–4.0 Å) | 180 |
| Medium (1.8–5.0 Å) | 215 |
| Weak (1.8–6.0 Å) | 117 |
| Very weak (1.8–7.0 Å) | 24 |
| Base pair constraints | |
| Total | 32 |
| Dihedral angle constraints | |
| Ribose ringb | 104 |
| Backbone | 189 |
| Mean number per residue | 7.7 |
| Residual dipolar coupling constraints | |
| Base CH | 30 |
| Ribose 1′ CH | 16 |
| Violations | |
| Average distance constraints > 0.5 Åc | 5.8 |
| Average dihedral constraints > 0.5°d | 1.6 |
| RMSD from ideal geometrye | |
| Heavy atoms (Å) | 1.24 |
| Backbone atoms (Å) | 1.27 |
aOnly conformationally restrictive constraints are included.
bThree torsion angles within each ribose ring were used to constrain the ring to either the C2′-endo or C3′-endo conformation; the ring pucker of residues G11, A13, G26 and A28 were not constrained.
cA distance violation of 0.5 Å corresponds to 5.0 kcal energy penalty.
dA dihedral angle violation of 0.5° corresponds to 0.05 kcal energy penalty.
eCalculated against the minimized average structure.
Figure 3.

(A) Structure of the tyrSSD RNA with view into the minor groove at the center of the helix. (B) Superposition of 11 lowest energy structures for the full tyrSSD, and the Specifier Loop domain only. Views are into the minor groove at the center of the Specifier Loop domain. The RMSDs between the individual structures and the average structure are listed in Table 1. The internal loop and stem regions are generally well defined. The disorder of the bulged G11 base reflects the paucity of constraints for this residue.
The abundance of constraints for the internal loop and flanking stem nucleotides (residues C6–G14 and C25–G33) defines the conformation of these nucleotides with good precision (0.60 Å rmsd) (Figure 3B and Table 1). The ribose ring puckers of several residues in the internal loop adopt non-A-form, C3′-endo, conformations. Residues A9, A10 and A32 adopt C2′-endo ring puckers and residues G11, A13, G26 and A28 exhibit conformations intermediate between C2′-endo and C3′-endo.
The tyrSSD Specifier Loop domain contains a loop E motif
The nucleotide sequence within the tyrSSD RNA corresponding to the loop E motif is similar to that found in several eukaryotic 5S rRNAs (37) and the hairpin ribozyme (15,38). The core of this element typically contains three nonstandard base–base interactions, a sheared A–G pair, a U–A trans-Hoogsteen interaction and a parallel A–A base pair, that stack on one another (13). In some cases, a bulged G or C nucleotide is present between the non-canonical U–A and A–A interactions. The spectral data indicate that the loop E motif structure is present but adopts a more open conformation than is found in other contexts. A characteristic spectral signature of the sheared A–G pair is an upfield shift of the H1′ resonance of the residue 3′ to the A (13,17,39). This H1′ resonance shift is observed for G14 and is consistent with a sheared A13–G26 orientation. The sheared A–G base pair forms two hydrogen bonds, AN6H2–GN3 and AN7–GN2H2 (Figure 4), and the A13 N7 and N6 15N chemical shifts provide additional support for the A13–G26 interaction. The trans-Hoogsteen U–A interaction adjacent to the sheared A–G pair also produces a very unusual spectral feature, an A10H8–U12H1′ NOE cross-peak. The trans-Hoogsteen U–A arrangement rotates the uridine ribose ring so that the H1′ is repositioned proximal to the 5′ (i-2) residue rather than the 3′ (i + 1) residue. The U–A interaction involves hydrogen bonds between UO2 and AN6H2 and UN3H and AN7 (14). The U12N3H resonance is exchange-broadened and could not be observed, indicative of a weak hydrogen bond or solvent-accessible U12N3H. Adjacent to the trans-Hoogstein base pair is a parallel A–A interaction (Figure 4). The parallel A–A base pair is facilitated by an S-turn in the phosphate backbone and stabilized by two symmetric N6H2–N7 inter-base hydrogen bonds (14,28). The 15N chemical shifts are consistent with the A28N6H2–A10N7 hydrogen bond, but the chemical shifts do not support the second (symmetric) interaction (Supplementary Table S1; Figure 4). The G11 base is bulged and its conformation is supported by NOE data. Several i, i + 2 NOE cross-peaks connect A10 with U12 and no inter-residue NOE cross-peaks are observed between G11 and the A10 ribose. These NOE cross-peaks indicate stacking of the A10 and U12 bases. Efforts to confirm the predicted hydrogen bonds via through-bond correlation experiments (25) were unsuccessful. This result is consistent with the 15N chemical-shift data that suggest weak hydrogen bonds.
Figure 4.

Stereoview of the loop E motif bases in the tyrSSD RNA molecule showing the arrangement of base–base interactions. Hydrogen bonds, supported by 15N chemical shifts, are indicated by dashed black lines. Hydrogen bonds present in other loop E motifs but for which there is no evidence in the tyrSSD RNA are shown as dashed red lines. The sheared G–A base pair (pink), reverse Hoogstein A–U base pair (blue), parallel A–A base pair (red) stack on each other. The bulged G11 base (orange) makes no hydrogen bonds with either the phosphate backbone or the flanking bases.
A hallmark of the loop E motif is the turn in the phosphate backbone 5′ to the bulged nucleotide (between A10 and G11) (28). This turn permits the parallel A–A interaction without the adenine residues adopting the syn orientation about the glycosidic bond. The turn itself is accommodated by the flipping of the ribose moiety of one of the adenine residues relative to other ribose groups along the phosphate backbone. This ribose orientation is supported by a characteristic NOE pattern (13,17) that includes weak intensity A10 H4′–U12 H6 and A10 H4′–U12 H5 cross-peaks and moderate intensity A10 H4′–U12 H1′ and A10 H8–U12 H1′ cross-peaks.
The Specifier Sequence bases are stacked
The remaining nucleotides in the Specifier Loop domain are asymmetrically distributed, four on the Specifier Sequence strand and three on the opposite strand (Figures 3 and 5). The calculated structures show intra-strand stacking and slight rotation of the Specifier Sequence (U29, A30 and C31) bases toward the minor groove. A32 also is rotated toward the minor groove, but stacks with G33 of the lower helix. These structures are consistent with the experimental data, i.e. base–base and ‘standard’ base–ribose NOEs and 15N/13C chemical shifts characteristic of unpaired bases. This conformation may facilitate pairing of the Specifier Sequence bases with the tRNA anticodon loop through the ordered presentation of the Watson–Crick edges of the bases.
Figure 5.

Stereoview of the minimized average structure of the Specifier Loop domain. The specifier nucleotide bases are colored green and the loop E motif nucleotide bases are colored red. The functional groups on the Watson–Crick edges of the specifier nucleotides are colored pink. The S-turn of the sugar-phosphate backbone can be seen between residues A8 and G11.
Localization of metal ion binding sites in the tyrSSD RNA
Metal ions play important roles in RNA structure and function by facilitating RNA folding, stabilizing tertiary structure and participating directly in catalysis. Mg2+ is crucial for the correct folding of glyQS leader RNA (7), but the specific structural changes and exact sites of coordination associated with Mg2+ binding are not known.
The effects of Mg2+ on different RNAs that contain loop E motifs are variable. NMR spectra show that the loop E motif in eukaryotic 5S rRNA is stabilized by Mg2+ (40), but spectra of the same motif sequence located in the sNRE and the hairpin ribozyme are unaffected by Mg2+ (41–43). In addition, no metal ions are associated with the loop E motif in the crystal structure of the sarcin/ricin loop of 5S rRNA (44).
The NH and NH2 chemical-shift perturbations by Mg2+ are limited to the G2–U37 NH resonances in tyrSSD. However, the chemical shifts of several CH base resonances are altered by up to 0.15 p.p.m. (Supplementary Figure S1). These include the base C8–H8 and C6–H6 resonances of loop residues G33, C8, A27, A9 and C31 and adenine C2–H2 resonances A13, A7, A9 and A32. The intensity of the A13 H8–C8 resonance increases, but the G11 H8–C8 resonance broadens. Significantly, the pattern and intensities of NOE cross-peaks of the Mg2+-free sample are largely preserved upon addition of Mg2+, indicating little or no structural impact.
13C Relaxation measurements
The reorientation of a 13C-1H bond vector on the picosecond timescale can be assessed through its carbon T1ρ relaxation: the longer the relaxation time, the more mobile the 13C-1H pair. The T1ρ relaxation times for the base C6 and C8 and ribose C1′ positions of tyrSSD were measured. Cross-peak overlap and chemical exchange prevented accurate measurement of a few adenine C2 nuclei, pyrimidine C6 and several C1′ nuclei. The majority of base resonances from internal loop residues exhibited relaxation times between 40 and 50 ms, which are comparable to stem region nucleotides. However, the T1ρ values for nucleotides A10, G26, A27 and A28 within the loop E motif and the proximal stem nucleotides G14, and C25 were increased by 20%, indicative of increased mobility. Chemical exchange is dominant among the base resonances of G11 U12 and A13 and prevents accurate T1ρ measurements for these residues. A similar pattern is not exhibited by the C1′ nuclei. Only the A9, A10, A28 and A30 residues from the internal loop have relaxation times 30–50% longer than other residues in the internal loop and the stems. The increased relaxation times of nucleotide base resonances and exchange broadening for residues in the region of the loop E motif are consistent with a structural element that is moderately rigid. The addition of Mg2+ did not significantly alter the relaxation profile of the tyrSSD RNA.
DISCUSSION
The T-box mechanism for regulation of amino acid-related genes is widely utilized in Gram-positive bacteria. Extensive mutagenesis studies of the leader RNA sequence have defined sequence requirements for tRNA binding in the 5′ and 3′ regions of the leader RNA (4–6,11,45). Residues within the Specifier Loop domain of the leader RNA bind directly to the anticodon nucleotides of cognate tRNA molecules and confer specificity to the leader RNA–tRNA interaction. The Specifier Loop domain also contains conserved nucleotides that correspond to the loop E RNA secondary structure motif.
Structure of conserved residues in the tyrS leader RNA Specifier Loop domain
The 14 nt corresponding to the Specifier Loop domain of the B. subtilis tyrS leader RNA are well ordered and only moderately dynamic. The results show that the nucleotides in the upper half of the domain adopt a loop E motif (Figure 3). This structural element includes an S-turn of the phosphate backbone at residue A10 resulting in an inverted orientation of the A10 ribose. Although well ordered, the loop E element in the Specifier Loop domain has a more open and possibly more flexible fold than loop E motifs in other contexts (13,17,44). The hydrogen-bonding pattern among the loop E motif bases could be inferred only from 15N chemical shifts and could not be directly confirmed through hydrogen bond-mediated inter-base correlations. These results point to a fold that is open and may be partially accessible to solvent (Figure 5). Flexibility on the intermediate timescale in this region is reflected in the exchange-broadened base resonances of residues G11–A13 and the relaxation data support the modest dynamic nature. The limited mobility of these residues may allow for an organized target to be presented to the incoming tRNA anticodon but still allow sufficient flexibility to optimize Specifier Sequence nucleotide–anticodon pairing.
To determine if the more open structure results from an insufficient number of experimental constraints, a set of calculations was performed that explicitly imposed base–base interactions present in canonical loop E motifs identified in other studies (13,17,44). The structures calculated using these constraints have approximately twice as many NOE violations (36 per structure) and greater RDC errors (16.6 Hz on average) than the more open structures. Most of the new violations are in the internal loop region and include restraints involving the loop E motif and Specifier Sequence nucleotides. These results support the experimental constraints as the driving force for the more open conformation.
While the upper half of the Specifier Loop domain exhibits extensive cross-strand interactions, nucleotides in the lower half present no evidence for cross-strand pairing. The Specifier Sequence nucleotides maintain regular base stacking with their Watson–Crick edges rotated toward the minor groove (Figure 5). The residue 3′ to the Specifier Sequence, A32, also is single stranded, but does not form a continuous stack with the Specifier Sequence bases. Instead, A32, which pairs with U33 of tRNA, stacks on G33 of the lower helix. The 5′ strand residues are less well stacked and are slightly rotated toward the major groove. The unpaired nature of this region is consistent with biochemical data that indicate the susceptibility of these nucleotides to Mg2+ cleavage (7).
The structure and dynamics of the Specifier Loop domain contrast with those of the 7-nt bulge within the antiterminator hairpin (46). The antiterminator hairpin is one of two mutually exclusive stem–loop structures that can form near the 3′-end of the leader RNA (Figure 1). The antiterminator hairpin contains a bulge of 7 nt, four of which pair with the 3′ terminal nucleotides of uncharged tRNA (4). The three invariant nucleotides (UGG) that pair with tRNA are at the 5′-end of the bulge and display considerable flexibility. These residues appear to sample multiple conformations and do not have a unique structure in the absence of tRNA (46). Nucleotides at the 3′-end of the bulge are highly conserved and well ordered, and are stabilized primarily by base–base stacking. While the flexibility associated with the invariant residues is an important component of their tRNA binding function, the more rigid 3′-end of the bulge may limit the conformations sampled by the loop nucleotides and facilitate tRNA binding (46). The significant dynamics among the tRNA-pairing nucleotides in the antiterminator hairpin is not shared by the Specifier Sequence nucleotides. Although Specifier Sequence–tRNA pairing requires additional rotation of the Specifier Sequence bases away from the helix axis, these residues are well ordered and appear to be stabilized by stacking. The ribose groups of these residues also exhibit limited dynamics and adopt the regular A-form 3′-endo conformation. The three residues of the Specifier Loop domain that are dynamic (G11–A13) may provide flexibility to optimize tRNA binding.
Role of the loop E motif in tRNA binding
The loop E motif is a common RNA structural element that is well represented in ribosomal RNAs (13,37,47–50), loop B of the hairpin ribozyme (15,51), the specificity domains of some type B RNase P molecules (52,53), the internal ribosome entry site (IRES) element found in the 5′ untranslated region of hepatitis C virus (HCV) RNA (54,55), the central domain of potato spindle tuber viroid (PSTV) (37), and in in vitro selected nucleolin recognition element (sNRE) mutants (17). Loop E nucleotides participate directly in protein–RNA interactions in most of the rRNA molecules and in the sNRE RNA (13,16,17,47,49,56) and, in the hairpin ribozyme, the motif forms part of a corridor for active site water migration (57). In the case of the loop E structures found in domains IIb and IIId of the IRES element of HCV RNA, it is still not clear whether the loop E structure provides a platform for protein or RNA binding (54,55).
The functional importance of the loop E motif in the Specifier Loop domain is suggested by nucleotide conservation and mutagenesis (11, Green,N.J., Grundy,F.J. and Henkin,T.M., unpublished data). Comparison of the Specifier Loop domains of several T-box leader RNAs across multiple bacterial species shows the conservation of two elements, the Specifier Sequence nucleotides at the 3′ end of the 3′ strand followed by an unpaired adenine nucleotide and a group of 7 nt in the upper part of the internal loop that correspond to the loop E motif (11,58). The number and identities of the remaining nucleotides in the Specifier Loop domain are variable, but nucleotide changes within the loop E motif can be deleterious. For example, an uridine-to-cytidine mutation in the Specifier Loop domain of the glyQS leader RNA at a position corresponding to residue 12 in the tyrSSD RNA leads to loss of function in in vivo and in vitro transcriptional attenuation assays (Green,N.J., Grundy,F.J. and Henkin,T.M., unpublished data). Figure 6 shows that resonances primarily affected by the U-to-C mutation correspond to nucleotides that comprise the loop E motif and indicate that the structure of the motif has been disrupted. Interestingly, the structural perturbations appear limited to the loop E motif. The chemical shifts corresponding to the residues of the Specifier Loop domain outside of the loop E motif are not significantly affected by the mutation, suggesting minimal perturbation of their local structure.
Figure 6.

Overlay of the base C6/8 and C2 regions of 13C-1H HSQC spectra of tyrSSD RNA molecule (gray) and the tyrSSD RNA molecule with the substitution U12 to C12 (black). Peaks from the native sequence shifted by the mutation are labeled. Peaks from the sequence containing the U12C mutation that do not overlap peaks from the native sequence are indicated by an arrow. The resonances most affected by the single base substitution correspond to the residues of the loop E motif. Nucleotides in the opposite end of the internal loop, including specifier nucleotides A30 and C31 show much smaller affects.
The structure of tyrSSD and the effects caused by the U-to-C mutation suggest a model for tRNA binding to the Specifier Sequence on the minor groove side of the Specifier Loop domain (Figure 7). The Specifier Sequence bases are well ordered in the solution structure and are only moderately more dynamic than the helix residues, suggesting that they are primed for pairing with the tRNA anticodon loop. In order for the Specifier Sequence nucleotides to pair with tRNA, the bases must rotate out toward the major or minor groove to present their Watson–Crick edges. Rotation toward the major groove would result in under-winding of the sugar-phosphate backbone, whereas rotation toward the minor groove would entail over-winding of the backbone. In the structure of tyrSSD, the 4 Specifier Sequence bases turn toward the minor groove, whereas the 3 bases of the partner strand turn out toward the major groove.
Figure 7.
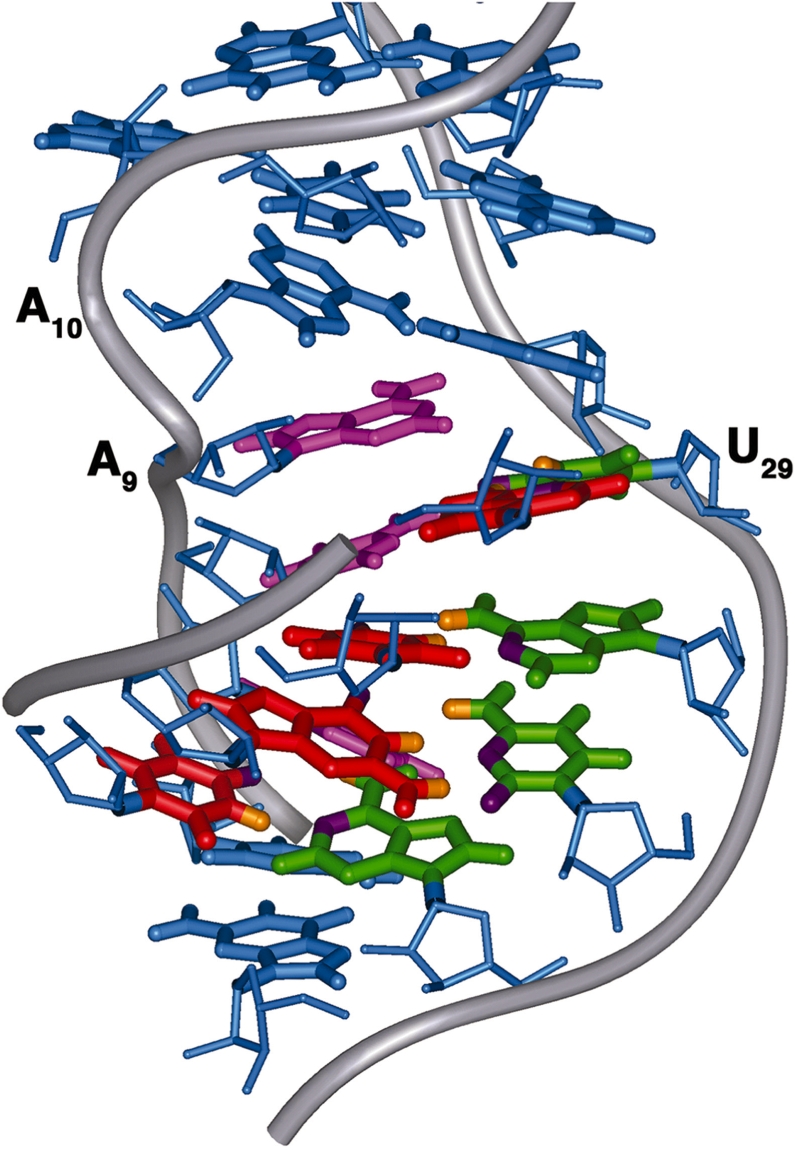
Proposed model of the interaction between the Specifier Loop domain and the tRNA anticodon loop. Nucleotide bases in the anticodon loop (red) approach from the minor groove side of the Specifier Domain and form Watson–Crick base pairs with the Specifier Sequence bases (green). Nucleotides of the partner strand (pink) rotate toward the major groove. The S-turn of the phosphate backbone on the partner strand is introduced by the loop E motif (blue) and facilitates the minor groove displacement of the Specifier Sequence bases.
We propose that the S-turn of the loop E motif is responsible for this base positioning and that it acts through the phosphate backbone. The S-turn, centered on A10, causes a differential shortening of the distance between the lower helix and the A10–A28 base pair of the loop. To accommodate the constraints on the phosphate backbone imposed by the S-turn and the asymmetric number of residues in this region, bases on the partner strand rotate toward the major groove and the Specifier Sequence bases rotate toward the minor groove. Although the U-to-C mutation does not appear to affect the local structure around the Specifier Sequence nucleotides, their propensity to rotate toward the minor groove would be impaired by loss of the S-turn at A10. Thus, in this model for tRNA binding, the loop E motif positions a specific structural component, the S-turn, that kinks the sugar-phosphate backbone of the partner strand and promotes rearrangement of the Specifier Sequence bases toward the minor groove for pairing with the tRNA (Figure 7). The loop E motif may additionally provide a platform to help stabilize the duplex formed by the Specifier Sequence and anticodon base pairs through stacking interactions.
ACCESSION NUMBER
Atomic coordinates for the refined structures have been deposited with the Protein Data Bank under accession code 2KHY.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Robert A. Welch Foundation (C-1277 to E.P.N.) and by the National Institutes of Health (GM73969 to E.P.N.; GM47823 to T.M.H.). Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Malgorzata Michnicka for preparation of the T7 RNA polymerase and synthesis of the labeled 5′-nucleotide triphosphates. The 800 MHz NMR spectrometer was purchased with funds from the W. M. Keck Foundation and the John S. Dunn Foundation.
REFERENCES
- 1.Henkin TM. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008;22:3383–3390. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henkin TM, Grundy FJ. Sensing metabolic signals with nascent RNA transcripts: the T box and S box riboswitches as paradigms. Cold Spring Harbor Symp. Quant. Biol. 2006;71:231–237. doi: 10.1101/sqb.2006.71.020. [DOI] [PubMed] [Google Scholar]
- 3.Grundy FJ, Henkin TM. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell. 1993;74:475–482. doi: 10.1016/0092-8674(93)80049-k. [DOI] [PubMed] [Google Scholar]
- 4.Grundy FJ, Rollins SM, Henkin TM. Interaction between the acceptor end of tRNA and the T box stimulates antitermination in the Bacillus subtilis tyrS gene: a new role for the discriminator base. J. Bacteriol. 1994;176:4518–4526. doi: 10.1128/jb.176.15.4518-4526.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grundy FJ, Hodil SE, Rollins SM, Henkin TM. Specificity of tRNA-mRNA interactions in Bacillus subtilis tyrS antitermination. J. Bacteriol. 1997;179:2587–2594. doi: 10.1128/jb.179.8.2587-2594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grundy FJ, Moir TR, Haldeman MT, Henkin TM. Sequence requirements for terminators and antiterminators in the T box transcription antitermination system: disparity between conservation and functional requirements. Nucleic Acids Res. 2002a;30:1646–1655. doi: 10.1093/nar/30.7.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yousef MR, Grundy FJ, Henkin TM. Structural transitions induced by the interaction between tRNAGly and the Bacillus subtilis glyQS T box leader RNA. J. Mol. Biol. 2005;349:273–287. doi: 10.1016/j.jmb.2005.03.061. [DOI] [PubMed] [Google Scholar]
- 8.Grundy FJ, Henkin TM. Conservation of a transcription antitermination mechanism in aminoacyl-tRNA synthetase and amino acid biosynthesis genes in Gram-positive bacteria. J. Mol. Biol. 1994;235:798–804. doi: 10.1006/jmbi.1994.1038. [DOI] [PubMed] [Google Scholar]
- 9.Grundy FJ, Winkler WC, Henkin TM. tRNA-mediated transcription antitermination in vitro: codon-anticodon pairing independent of the ribosome. Proc. Natl Acad. Sci. USA. 2002b;99:11121–11126. doi: 10.1073/pnas.162366799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson AR, Henkin TM, Agris PF. tRNA regulation of gene expression: interactions of an mRNA 5′-UTR with a regulatory tRNA. RNA. 2006;12:1254–1261. doi: 10.1261/rna.29906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rollins SM, Grundy FJ, Henkin TM. Analysis of cis-acting sequence and structural elements required for antitermination of the Bacillus subtilis tyrS gene. Mol. Microbiol. 1997;25:411–421. doi: 10.1046/j.1365-2958.1997.4851839.x. [DOI] [PubMed] [Google Scholar]
- 12.Szewczak AA, Moore PB. The sarcin/ricin loop, a modular RNA. J. Mol. Biol. 1995;247:81–98. doi: 10.1006/jmbi.1994.0124. [DOI] [PubMed] [Google Scholar]
- 13.Dallas A, Moore PB. The loop E-loop D region of Escherichia coli 5S rRNA: the solution structure reveals an unusual loop that may be important for binding ribosomal proteins. Structure. 1997;5:1639–1653. doi: 10.1016/s0969-2126(97)00311-0. [DOI] [PubMed] [Google Scholar]
- 14.Leontis NB, Westhof E. A common motif organizes the structure of multi-helix loops in 16 S and 23 S ribosomal RNAs. J. Mol. Biol. 1998;283:571–583. doi: 10.1006/jmbi.1998.2106. [DOI] [PubMed] [Google Scholar]
- 15.Butcher SE, Burke JM. A photo-cross-linkable tertiary structure motif found in functionally distinct RNA molecules is essential for catalytic function of the hairpin ribozyme. Biochemistry. 1994a;33:992–999. doi: 10.1021/bi00170a018. [DOI] [PubMed] [Google Scholar]
- 16.Bouvet P, Allain FH, Finger LD, Dieckmann T, Feigon J. Recognition of pre-formed and flexible elements of an RNA stem-loop by nucleolin. J. Mol. Biol. 2001;309:763–775. doi: 10.1006/jmbi.2001.4691. [DOI] [PubMed] [Google Scholar]
- 17.Johansson C, Finger LD, Trantirek L, Mueller TD, Kim S, Laird-Offringa IA, Feigon J. Solution structure of the complex formed by the two N-terminal RNA-binding domains of nucleolin and a pre-rRNA target. J. Mol. Biol. 2004;337:799–816. doi: 10.1016/j.jmb.2004.01.056. [DOI] [PubMed] [Google Scholar]
- 18.Brunel C, Romby P, Westhof E, Ehresmann C, Ehresmann B. Three-dimensional model of Escherichia coli ribosomal 5 S RNA as deduced from structure probing in solution and computer modeling. J. Mol. Biol. 1991;221:293–308. doi: 10.1016/0022-2836(91)80220-o. [DOI] [PubMed] [Google Scholar]
- 19.Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl Acad. Sci. USA. 1984;81:2035–2039. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikonowicz EP, Sirr A, Legault P, Jucker FM, Baer LM, Pardi A. Preparation of 13C and 15N labelled RNAs for heteronuclear multi-dimensional NMR studies. Nucleic Acids Res. 1992;20:4507–4513. doi: 10.1093/nar/20.17.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- 23.Marino JP, Diener JL, Moore PB, Griesinger C. Multiple-quantum Coherence dramatically enhances the sensitivity of CH and CH2 correlations in uniformly 13C-labeled RNA. J. Am. Chem. Soc. 1997;119:7361–7366. [Google Scholar]
- 24.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 25.Dingley AJ, Grzesiek S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 1998;120:8293–8297. [Google Scholar]
- 26.Varani G, Aboul-ela F, Allain FHT. NMR investigation of RNA structure. Prog. Nucl. Magn. Reson. Spec. 1996;29:51–127. [Google Scholar]
- 27.Szep S, Wang J, Moore PB. The crystal structure of a 26-nucleotide RNA containing a hook-turn. RNA. 2003;9:44–51. doi: 10.1261/rna.2107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Correll CC, Freeborn B, Moore PB, Steitz TA. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell. 1997;91:705–712. doi: 10.1016/s0092-8674(00)80457-2. [DOI] [PubMed] [Google Scholar]
- 29.Gorenstein DG. Phosphorus-31 NMR: Principles and Applications. New York: Academic Press; 1984. [Google Scholar]
- 30.Clore GM, Kuszewski J. Improving the accuracy of NMR structures of RNA by means of conformational database potentials of mean force as assessed by complete dipolar coupling cross-validation. J. Am. Chem. Soc. 2003;125:1518–1525. doi: 10.1021/ja028383j. [DOI] [PubMed] [Google Scholar]
- 31.Simorre JP, Zimmermann GR, Mueller L, Pardi A. Triple-resonance experiments for assignment of adenine base resonances in 13C/15N-labeled RNA. J. Am. Chem. Soc. 1996;118:5316–5317. [Google Scholar]
- 32.Pardi A. Multidimensional heteronuclear NMR experiments for structure determination of isotopically labeled RNA. Methods Enzymol. 1995;261:350–380. doi: 10.1016/s0076-6879(95)61017-0. [DOI] [PubMed] [Google Scholar]
- 33.Dieckmann T, Feigon J. Assignment methodology for larger RNA oligonucleotides: application to an ATP-binding RNA aptamer. J. Biomol. NMR. 1997;9:259–272. doi: 10.1023/a:1018622708674. [DOI] [PubMed] [Google Scholar]
- 34.Allain FHT, Varani G. Divalent metal ion binding to a conserved wobble pair defining the upstream site of cleavage of group I self-splicing introns. Nucleic Acids Res. 1995;23:341–350. doi: 10.1093/nar/23.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heus HA, Wijmenga SS, van de Ven FJM, Hilbers CW. Sequential backbone assignment in 13C-labeled RNA via through-bond coherence transfer using three-dimensional triple resonance spectroscopy (1H,13C,31P) and two-dimensional hetero TOCSY. J. Am. Chem. Soc. 1994;116:4983–4984. [Google Scholar]
- 36.Kellogg GW, Schweitzer BI. Two- and three-dimensional 31P-driven NMR procedures for complete assignment of backbone resonances in oligodeoxyribonucleotides. J. Biomol. NMR. 1993;3:577–595. doi: 10.1007/BF00174611. [DOI] [PubMed] [Google Scholar]
- 37.Branch AD, Benenfeld BJ, Robertson HD. Ultraviolet light-induced crosslinking reveals a unique region of local tertiary structure in potato spindle tuber viroid and HeLa 5S RNA. Proc. Natl Acad. Sci. USA. 1985;82:6590–6594. doi: 10.1073/pnas.82.19.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butcher SE, Burke JM. Structure-mapping of the hairpin ribozyme. Magnesium-dependent folding and evidence for tertiary interactions within the ribozyme-substrate complex. J. Mol. Biol. 1994b;244:52–63. doi: 10.1006/jmbi.1994.1703. [DOI] [PubMed] [Google Scholar]
- 39.Tolbert BS, Kennedy SD, Schroeder SJ, Krugh TR, Turner DH. NMR structures of (rGCUGAGGCU)2 and (rGCGGAUGCU)2: probing the structural features that shape the thermodynamic stability of GA pairs. Biochemistry. 2007;46:1511–1522. doi: 10.1021/bi061350m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serra MJ, Baird JD, Dale T, Fey BL, Retatagos K, Westhof E. Effects of magnesium ions on the stabilization of RNA oligomers of defined structures. RNA. 2002;8:307–323. doi: 10.1017/s1355838202024226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butcher SE, Allain FH, Feigon J. Determination of metal ion binding sites within the hairpin ribozyme domains by NMR. Biochemistry. 2000;39:2174–2182. doi: 10.1021/bi9923454. [DOI] [PubMed] [Google Scholar]
- 42.Rupert PB, Ferre-D'A;mare AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 43.Finger LD, Trantirek L, Johansson C, Feigon J. Solution structures of stem-loop RNAs that bind to the two N-terminal RNA-binding domains of nucleolin. Nucleic Acids Res. 2003;31:6461–6472. doi: 10.1093/nar/gkg866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Correll CC, Wool IG, Munishkin A. The two faces of the Escherichia coli 23 S rRNA sarcin/ricin domain: the structure at 1.11 Å resolution. J. Mol. Biol. 1999;292:275–287. doi: 10.1006/jmbi.1999.3072. [DOI] [PubMed] [Google Scholar]
- 45.Grundy FJ, Collins JA, Rollins SM, Henkin TM. tRNA determinants for transcription antitermination of the Bacillus subtilis tyrS gene. RNA. 2000;6:1131–1141. doi: 10.1017/s1355838200992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerdeman MS, Henkin TM, Hines JV. Solution structure of the Bacillus subtilis T-box antiterminator RNA: seven nucleotide bulge characterized by stacking and flexibility. J. Mol. Biol. 2003;326:189–201. doi: 10.1016/s0022-2836(02)01339-6. [DOI] [PubMed] [Google Scholar]
- 47.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 48.Endo Y, Gluck A, Wool IG. Ribosomal RNA identity elements for recognition by ricin and by alpha-sarcin: mutation in the putative CG pair that closes a GAGA tetraloop. Nucleic Acids Symp Ser. 1993;29:165–166. [PubMed] [Google Scholar]
- 49.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 50.Wimberly B, Varani G, Tinoco I., Jr The conformation of loop E of eukaryotic 5S ribosomal RNA. Biochemistry. 1993;32:1078–1087. doi: 10.1021/bi00055a013. [DOI] [PubMed] [Google Scholar]
- 51.Butcher SE, Allain FH, Feigon J. Solution structure of the loop B domain from the hairpin ribozyme. Nat. Struct. Biol. 1999;6:212–216. doi: 10.1038/6651. [DOI] [PubMed] [Google Scholar]
- 52.Krasilnikov AS, Yang X, Pan T, Mondragon A. Crystal structure of the specificity domain of ribonuclease P. Nature. 2003;421:760–764. doi: 10.1038/nature01386. [DOI] [PubMed] [Google Scholar]
- 53.Massire C, Jaeger L, Westhof E. Derivation of the three-dimensional architecture of bacterial ribonuclease P RNAs from comparative sequence analysis. J. Mol. Biol. 1998;279:773–793. doi: 10.1006/jmbi.1998.1797. [DOI] [PubMed] [Google Scholar]
- 54.Klinck R, Westhof E, Walker S, Afshar M, Collier A, Aboul-Ela F. A potential RNA drug target in the hepatitis C virus internal ribosomal entry site. RNA. 2000;6:1423–1431. doi: 10.1017/s1355838200000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukavsky PJ, Kim I, Otto GA, Puglisi JD. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Biol. 2003;10:1033–1038. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- 56.Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 57.Salter J, Krucinska J, Alam S, Grum-Tokars V, Wedekind JE. Water in the active site of an all-RNA hairpin ribozyme and effects of Gua8 base variants on the geometry of phosphoryl transfer. Biochemistry. 2006;45:686–700. doi: 10.1021/bi051887k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutierrez-Preciado A, Henkin TM, Grundy FJ, Yanofsky C, Merino E. Biochemical features and functional implications of the RNA-based T box regulatory mechanism. Microbiol. Mol. Biol. Rev. 2009;73:36–61. doi: 10.1128/MMBR.00026-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varani G, Cheong C, Tinoco I., Jr Structure of an unusually stable RNA hairpin. Biochemistry. 1991;30:3280–3289. doi: 10.1021/bi00227a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.