

Abstract
Importance of the field
The cytochrome P450 (CYP) isoforms that are selectively induced following exposure to structurally-diverse chemicals often are the ones capable of metabolizing these chemicals. However, the molecular mechanism underlying this apparent functional coupling is not understood at present.
Areas covered in this review
Three hypotheses are developed to explain the complex process of selective chemical induction of CYPs: (i) each inducible CYP may have a corresponding intracellular receptor that interacts with the inducer chemical and mediates the selective induction of this CYP, (ii) each inducible CYP and its corresponding receptor may share a highly similar steric structure for their substrate/inducer binding sites, and (iii) each chemically-inducible CYP gene may have distinct genomic response element(s) that interact selectively with the corresponding receptor.
What the reader will gain
The readers are introduced to a novel theoretical framework that offers a plausible mechanistic explanation at the molecular levels concerning the complex process of how an organism selectively activates the biosynthesis of certain CYP isoform(s) that can effectively metabolize a chemical to which the organism is exposed.
Take home message
The theoretical framework developed herein seeks to ignite additional critical thinking on this important research subject as well as to promote experimental testing of the proposed theories in the future. Undoubtedly, these studies will enhance the understanding of the molecular mechanisms for the selective induction of CYP enzymes by chemicals.
Keywords: Cytochrome P450 isoform, chemical inducers, selective enzyme induction, Ah receptor, nuclear receptors, mechanism of enzyme induction
1. INTRODUCTION
It is estimated that at least several dozens of cytochrome P450 (CYP1) isoforms exist in animals and humans [1, 2]. These structurally-related hemeprotein monooxygenases fulfill many important biological functions, which include, but are not limited to, synthesis of bioactive compounds (such as steroid hormones) and biotransformation of a vast array of endogenous and exogenous chemicals [3-9]. One of the most well-known features of this enzyme system perhaps is the selective induction (i.e., induced de novo biosynthesis) of certain CYP isoforms in animal or human cells following exposure to chemicals such as phenobarbital, tetrachlorodibenzo-p-dioxin (TCDD), or polycyclic aromatic hydrocarbons [8-13]. The purpose of the induction of drug-metabolizing enzymes is primarily for accelerated metabolism of the chemicals being exposed to, which often inactivates and/or detoxifies these chemicals.
Of all the CYPS, the mechanism of CYP1A1 induction by chemicals has been most extensively studied over the past two decades and is perhaps best understood at present [8-15]. Inducer chemicals, such as TCDD, first bind the intracellular aryl hydrocarbon receptor (AhR), and then the liganded AhR is translocated into the nucleus in a heterodimeric complex with the AhR translocator protein. This nuclear AhR complex, acting as a ligand-dependent transcription factor, binds specifically to the aryl hydrocarbon responsive element (AhRE) in the CYP1A1 gene and subsequently activates its expression. In recent years, similar mechanisms were also observed for some of the other inducible CYP isoforms [16-26].
Notably, there are many unique features that are generally associated with the inducer-stimulated biosynthesis of CYP isoforms: (i) the CYP isoform that is induced after exposure to a chemical very often is the one capable of effectively metabolizing this chemical; (ii) usually more than one CYP isoform are induced to varying degrees; (iii) although exposure to an inducer chemical stimulates the biosynthesis of some CYP isoforms, often the biosynthesis of some other isoform(s) is simultaneously inhibited; and (iv) the inducibility and the magnitude of induction of a CYP isoform often differ dramatically in different tissues/cells following exposure to the same inducer. Although great advances have been made in the past decade in the study of nuclear receptors and their roles in mediating CYP isozyme induction, the molecular mechanisms for these as well as other characteristics associated with chemical induction of various CYP isoforms are still not fully understood at present. In this paper, some theoretical aspects concerning the underlying molecular mechanisms are discussed alongside a discussion of some of the available supporting evidence. The theoretical framework developed in this paper seeks to ignite more critical thinking on this research subject as well as to promote experimental testing of the proposed theoretical elements in the future. Undoubtedly, these additional studies will enhance the understanding of the molecular mechanisms for the selective induction of CYP enzymes by chemicals.
2. THEORETICAL CONSIDERATIONS
It is hypothesized that the biological process of selective induction of a CYP isoform by an inducer chemical is primarily determined by the following three core elements (schematically illustrated in Fig. 1):
Each chemically-inducible CYP isoform may have a corresponding intracellular receptor. This receptor interacts with the inducer chemicals and mediates the selective induction of this CYP isoform.
Each chemically-inducible CYP isoform and its corresponding receptor may share a highly similar steric structure for their substrate/inducer binding sites.
Each inducible CYP gene may have a distinct genomic response element that interacts selectively with the corresponding receptor.
Figure 1.
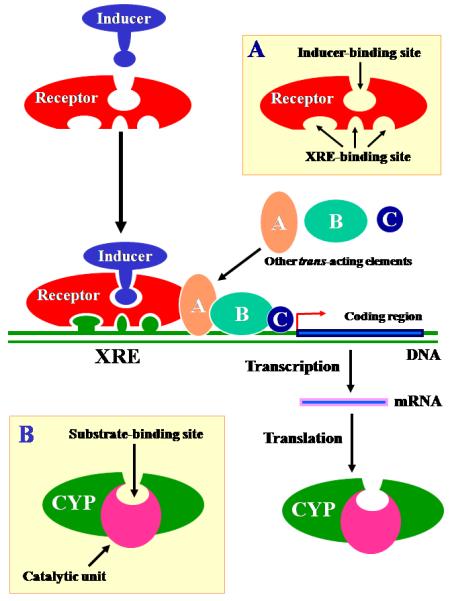
A schematic illustration of the three theoretical elements (see text) that govern the selective chemical induction of CYP isoforms. Inset A depicts a mediating receptor that has an inducer-binding site and a highly selective xenobiotic response element (XRE)-binding site. Inset B depicts a CYP isoform with a substrate-binding site that is identical (or highly similar) in steric structure to the inducer-binding site of its corresponding receptor.
In this paper, the term induction refers only to the commonly-known biological process involving chemical-stimulated transcription of a CYP gene followed by translation of its mRNA, which results in increased de novo biosynthesis and elevated intracellular levels of the CYP protein. Although a chemical may also increase the steady-state concentrations of a CYP isoform in a cell by affecting the degradation of a CYP protein or its mRNA [27-29], the proposed theory is not intended to explain these particular phenomena.
Here it should also be noted that the term “selective induction” was used on the basis of the following two main reasons: (i) Exposure of experimental animals or human subjects to most of the chemical inducers does not result in an equal or similar induction of the various CYP enzymes, rather it almost always predominantly induces one or a few CYP enzymes when most of the other inducible CYP enzymes in the same tissue or cells are only very mildly affected. (ii) The CYP enzyme that is induced after exposure to an inducer chemical very often is the one capable of effectively metabolizing this chemical. While the use of the “selective induction” terminology may help more readily understand some of the biological purposes behind the proposed theories as outlined above, recall that generally the selectivity of the CYP enzyme induction by various inducer chemicals is rather low (when compared to many other biological systems such as hormone and neurotransmitter receptors or antibodies), and thus this term should be applied only in relative terms.
3. LITERATURE REVIEW AND DISCUSSION
As discussed below, the proposed theory is in accordance with many experimental findings. Despite the fact that some of the evidence discussed below appears to be rather unsystematic and scattered in unrelated scientific literature, these fragments of evidence, when viewed collectively, lend considerable support to each of the theoretical elements proposed.
3.1. Hypothesis 1: Each chemically-inducible CYP isoform has a corresponding intracellular receptor
A widely-accepted alternative to this premise is that one receptor system may mediate the induction of multiple CYP isoforms. For instance, it is generally accepted that the AhR system mediates the induction of several CYP1 family isozymes (CYP1A1, 1A2, and 1B1). If this is correct, then we should expect to see a highly consistent induction pattern for each of the co-inducible CYP isoforms in a given tissue or cell following exposure to a series of inducers (i.e., different agonists for the same mediating receptor). However, previous studies with almost any co-inducible CYP isoforms have found little or no support for this prediction.
Let us use the chemical induction of CYP1A1 and CYP1A2 as an example. These two CYP isoforms are highly homologous in their amino acid sequences, similar in their catalytic activities, and often co-induced after exposure to a variety of AhR agonists (e.g., TCDD and 3-methylcholanthrene). For years, most investigators believed that the same AhR system mediated the induction of both CYP isoforms [10, 30]. However, studies have shown that while TCDD induced both CYP1A1 and CYP1A2 in the liver, it only induced CYP1A1 (but not CYP1A2) in the kidney and lung [31]. Moreover, the induction ratios of CYP1A1 to CYP1A2 in the same tissue or cell line often differed markedly with different inducers. For instance, inducers (such as 2-methoxy-4-nitroaniline, piperonyl butoxide, isosafrole, 2,2-dimethyl-5-t-butyl-1,3-benzodioxole, and several methylenedioxyphenyl compounds) could induce CYP1A2 mRNA and/or protein without inducing CYP1A1 [32-35]. Additional studies also showed that piperonyl butoxide, isosafrole, and 2,2-dimethyl-5-t-butyl-1,3-benzodioxole (inducers of CYP1A2) were unable to displace TCDD and 3-methylcholanthrene from the AhR in vitro [36, 37], and the induction of CYP1A2 by methylenedioxyphenyl compounds was not correlated with their activity at the aromatic hydrocarbon-responsive locus [38, 39].
Earlier studies using the AhR knock-out mice further showed that some of the chemical inducers, such as piperonyl butoxide and acenaphthylene, could still induce CYP1A2 expression, independent of the AhR [40]. Similarly, it was shown that while phenobarbital failed to induce CYP1A1 in the AhR knock-out mice, it induced CYP1A2 [41]. These data appear to provide some support for the hypothesis that the AhR does not mediate the induction of CYP1A2.
In various inducible human cell lines in culture, similar observations have also been made. For instance, the extent of CYP1A1 and CYP1A2 induction by polycyclic aromatic hydrocarbons (PAHs) or by various nitro-PAHs was highly CYP isoform-dependent, and the induction of each isoform was not related to the levels of AhR present [42].
In the same context, it is of note that most researchers consider that the AhR also mediates the chemical induction of CYP1B1. However, the induction pattern of CYP1B1 is very different from that of CYP1A1 or 1A2 in different human tissues or cells examined [43-47]. For instance, CYP1A1 was induced by TCDD to a much higher level (45-fold) than CYP1B1 in human HepG2 cells, but CYP1A1 was not induced by TCDD in ACHN cells where only CYP1B1 was selectively induced by TCDD [48]. In JEG-3 cells, while the CYP1A1 mRNA was induced by up to 9000-fold, the expression of CYP1B1 was unaffected [49]. In animal studies, low levels of CYP1B1 expression could still be induced in the liver of AhR-/- mice after treatment with some of the chemical inducers [50]. These observations, along with many others, likely suggest that the inducibility of CYP1B1 is regulated independently of CYP1A1.
The proposed hypothesis speculates that each inducible CYP isoform may have a corresponding intracellular receptor that mediates its induction by chemicals. This premise logically leads to several deducible principles pertaining to the receptor-mediated chemical induction of CYP isoforms: (i) The presence of the mediating receptor in a cell would be a major determinant for the chemical inducibility of the respective CYP isoform. (ii) The density of the mediating receptor in a cell would be another important determinant of the magnitude of the maximum induction of the respective CYP isoform in that cell, although it is not the sole determinant of the absolute magnitude of CYP enzyme induction following exposure to a chemical inducer (discussed in a later section). (iii) The affinity with which a chemical binds to and activates the mediating receptor would be an important determinant of the chemical’s potency for inducing a CYP isoform. Stated differently, the induction potency of a chemical for a given CYP isoform should largely parallel its binding affinity for the corresponding intracellular receptor. (iv) The rank order of potency for the induction of a CYP isoform by a series of inducers (i.e., different agonists for the same mediating receptor) would be highly similar in different responsive cells. (v) Certain chemicals may function as selective inhibitors for the induction of a given CYP isoform if they can function as antagonists for the mediating receptor.
Notably, almost each of the above predictions is strongly supported by experimental evidence in the case of CYP1A1 induction by chemical inducers, a process that is known to be mediated by the AhR. For instance, many previous studies have shown that the binding affinity of AhR agonists correlated closely with their ability to induce CYP1A1. TCDD had a very high binding affinity for the AhR (~ 1 to 10 nM) and correspondingly it had a very high potency for inducing CYP1A1 [51, 52]. In comparison, 3-methylcholanthrene had a much lower affinity than TCDD for the AhR and, correspondingly, it had a weaker inducing potency for CYP1A1 [53]. Similarly, studies with various peroxisome proliferators have shown that their binding affinity for the peroxisome proliferator receptors correlated very closely with their ability to stimulate liver peroxisome proliferation and particularly the induction of a CYP4A isoform [23-25].
Although the ratio of CYP1A2 to CYP1A1 induction or the ratio of CYP1B1 to CYP1A1 induction in different tissues or cells may vary, considerable experimental evidence scattered in the literature consistently shows that the induction of CYP1A2 or CYP1B1 individually by different chemical inducers agrees well with the principles predicted above. This would be in agreement with the suggestion that the induction of CYP1A2 or CYP1B1 is predominantly mediated by their respective specific receptors, although there may be certain degrees of coordination in the induction of functionally-similar metabolizing CYP enzymes (discussed later).
Most of the above-predicted features for the receptor-mediated induction process have also been observed with the chemical induction of other CYP isoforms, in particular the CYP3A and CYP4A family isoforms. For instance, induction of CYP3A isoforms by various chemicals and induction of CYP4A by various peroxisome proliferators each showed highly consistent rank order of induction potency in different responsive tissues or cells. Moreover, their induction by different chemicals showed a high degree of tissue or cell selectivity. Studies in the past decade have shown that the nuclear receptor PXR selectively mediates the chemical induction of a CYP3A family isoform [18-22, 25, 26] and the peroxisome proliferator-activated receptors (PPARs) may selectively mediate the induction of CYP4A family isoforms [54]. There are many recent review articles describing these developments. These new findings with CYP enzymes of the 3A and 4A families provide further support for the general concept that specific receptors may mediate the induction of each chemically-inducible CYP isoform.
Similarly, studies have shown that the nuclear receptor CAR mediates the induction of certain CYP2B family isoform. The discovery of the CAR receptor as a mediator of the induction of a CYP2B family isoform provides further support for the general concept that each inducible CYP isoform may have its own specific receptor. It is of interest to review some of the earlier indirect evidence that suggested a receptor-based mechanism for the induction of CYP2B1 by phenobarbital and related chemicals: (i) The induction of this CYP isoform by inducer chemicals exhibited a high degree of tissue and cell specificity. For instance, CYP2B1 was readily inducible in small intestine and liver by a number of inducers, but little or no induction was seen in some other tissues (such as lung and testis) with the same inducers [55-57]. (ii) The induction of CYP2B1 by phenobarbital showed saturable sigmoidal dose-response curves both in vivo and in cultured hepatocytes [58, 59], which are characteristic of a receptor-mediated process. (iii) Although CYP2B1 inducers include chemicals from many different classes, there is a clear structure-activity relationship for inducers within each individual class (e.g., phenobarbital and phenobarbital-like chemicals). (iv) Several studies showed a highly consistent rank order of potency for CYP2B1 induction in liver and intestine by different inducers of the same class. (v) There is evidence for the selective antagonism of CYP2B1 induction by certain chemicals [59]. Taken together, all of these earlier observations were highly indicative of a receptor-mediated mechanism for the induction of CYP2B1 by the inducer chemicals.
In addition, there is also evidence that supports the notion that the induction of CYP2B1 and 2B2 may be mediated by different receptors. For instance, the magnitude of induction of CYP2B1 and 2B2 isoforms often varies with different chemical inducers and in different tissues, similar to the differential induction of CYP1A family isoforms (CYP1A1, CYP1A2, and CYP1B1) as discussed above. Many chemicals induce CYP2B1 to a much greater extent than they induce CYP2B2 [57, 60, 61], but 4-n-alkyl-methylenedioxybenzene-type inducers induced CYP2B2 more than CYP2B1 [62]. Interestingly, although the responsiveness of CYP2B2 to phenobarbital in the QdJ:SD strain of the Sprague-Dawley rats was selectively impaired, the responsiveness of CYP2B1 to phenobarbital was not impaired [63]. These observations are consistent with the hypothesis that each CYP2B isoform likely has its own mediating receptor.
Lastly, it should be noted that the hypothesis that each inducible CYP isoform may have a mediating receptor also provides an explanation for a commonly observed, yet poorly understood, phenomenon that while many chemicals function as selective inducers of CYP isoform biosynthesis, some other chemicals may function as selective inhibitors of the same process. Typically, an agonist interacts specifically with the receptor resulting in its activation, and an antagonist often also interacts specifically with the same receptor, but its binding does not activate the receptor, instead it blocks the receptor from interacting with the available agonist. If each inducible CYP isoform has a mediating receptor, then it can be understood that, in accordance with the general principals, some chemicals that function as inducers would activate the mediating receptor and stimulate the biosynthesis of a CYP isoform, while some others that function as inhibitors may block the mediating receptor from interacting with the existing inducer and thereby inhibit the biosynthesis of this CYP isoform.
In summary, many of the experimental observations discussed above are in agreement with the premise that selective chemical induction of the CYP1A1, CYP1A2, CYP1B1, CYP2B1, CYP2B2, CYP3A4, CYP3A5, and CYP4A isoforms is mediated by specific receptors, in a mechanism likely analogous to the known AhR-mediated induction of CYP1A1.
3.2. Hypothesis 2: Each inducible CYP isoform and its mediating intracellular receptor may have highly similar steric structures in their substrate/ligand binding sites
This hypothesis predicts that the mediating receptor for a given CYP isoform will recognize the same steric structure of a chemical at which this CYP isoform catalyzes the metabolic reaction. Consequently, the mediating receptor that is effectively activated by the chemical will lead to the induction of a CYP isoform that usually can effectively metabolize it (as depicted in Fig. 1).
There are many examples of CYP enzymes that can be selectively induced by their substrates. Using CYP3A4 as an example, many clinically-used drugs (such as amprenavir, carbamazepine, dexamethasone, lovastatin, mifepristone, nifedipine, rifabutin, ritonavir, and simvastatin) are substrates of CYP3A4-mediated metabolism, and these chemicals are also strong inducers of this CYP enzyme [64-66]. Another classical example is phenobarbitals, which act as both substrates and inducers of CYP2B1 [13].
The above prediction that a substrate of an inducible CYP enzyme should also be an inducer of this enzyme seems to be not supported by the experience we have with the uses of various therapeutic drugs in humans. We know that while the metabolism of most drugs involves CYP enzymes, only a relatively small number of the drugs actually cause significant CYP induction in clinical settings. To better understand this apparent discrepancy, it is important to understand the physiological conditions under which chemical induction of CYP enzymes may take place. Hepatocytes in the body normally express a mixture of various metabolizing enzymes at rather high levels, with a combined high capacity for metabolizing many xenobiotics. Therefore, under normal conditions, most chemicals can be effectively metabolized by the existing enzymes, and these drugs will not accumulate in the liver and other parts of the body for extended length of time. As a result, there is no need to further increase the levels of drug-metabolizing enzymes in the liver and/or other organs to accelerate their metabolic disposition because the corresponding receptors will not be activated at sufficient intensity for a sustained period of time. This is the main reason that most pharmacological agents at the recommended safe doses would not substantially induce CYP enzymes in humans. However, if the doses of these drugs continue to increase, eventually they will exceed the metabolic capacity of the existing CYP enzymes. Subsequently, the concentrations of the chemicals will be persistently elevated, and will result in activation of the mediating receptors and increased expression of the CYP enzymes. In comparison, for those drugs that happen to have a very high binding affinity for the mediating receptors, they may elicit a strong induction of the CYP enzyme expression even when they are used at relatively low doses. In fact, some of the clinically-used drugs (such as rifampin, rifapentine, troglitazone, carbamazepine, and paclitaxel) that are well-known strong inducers of the CYP enzymes all share this property. Given the fact that drugs with strong CYP-inducing activity often are more likely to cause hepatotoxicity, these drugs would have a lower chance of surviving the preclinical and clinical stage testings. This is another underlying reason that, in reality, only a relatively small fraction of the clinically-used drugs are strong inducers of the CYP enzymes in humans at the recommended doses.
The proposed hypothesis also logically leads to another suggestion: a chemical’s binding affinity for an inducible CYP isoform ought to reflect its binding affinity for the mediating receptor. In fact, a wealth of experimental data (mostly qualitative or semi-quantitative) in the literature concurs with this suggestion. For instance, benzo[a]pyrene, 3-methylcholanthrene, and several other polyaromatic hydrocarbons all have high binding affinities (low KD values) for the AhR [51], and they all also have high binding affinities for CYP1A1 (as reflected by the low KM values) [52]. In comparison, some chemicals have relatively low binding affinities for the AhR, and they also have low affinities for CYP1A1-mediated metabolism. It is known that CYP1A1 in rats has a low KM (high affinity) for benzo[a]pyrene hydroxylation, whereas CYP1A2 has a high KM (low affinity) for benzo[a]pyrene hydroxylation [67-69]. As expected, benzo[a]pyrene administration induced CYP1A1 to a much greater extent than CYP1A2 in the same tissues of these animals [70, 71]. This differential induction of CYP1A1 over CYP1A2 in certain tissues by benzo[a]pyrene agrees with the predicted differential binding affinity of this inducer for their corresponding receptors (assuming that each CYP isoform has a corresponding receptor).
Similar characteristics have also been observed with other CYP isoforms such as CYP2B1 and CYP2B2: (i) CYP2B1 has a high KM (low affinity) for the metabolism of phenobarbital, and as expected, the concentrations of phenobarbital required for CYP2B1 induction in cultured rat hepatocytes are also very high (EC50 = 15 μM [60]). (ii) p-Hydroxyphenobarbital, a major metabolite of phenobarbital formed by CYP2B1, understandably has a much lower affinity than phenobarbital (the substrate) for CYP2B1, and has little or no activity for the induction of CYP2B1 [72]. (iii) Purified CYP2B1 has a much higher affinity and a higher catalytic activity than purified CYP2B2 for metabolizing many of the prototypic substrates [73, 74], and as expected, all these substrate chemicals induce CYP2B1 more strongly than they induce CYP2B2 [57, 60-62]. The high degree of correlation between the inducing activity of the different substrates for CYP2B1 and CYP2B2 and their rates of metabolism by these two CYP isoforms is in accord with the proposed receptor-mediated induction mechanism. It is possible that the past failure to identify the phenobarbital receptor(s) by using radiolabeled phenobarbital as a probe might be due to the fact that its binding affinity was too low to allow successful detection of the mediating receptor(s).
It is known that many inducible CYP isoforms (e.g., isoforms of the CYP3A and CYP2B families) have low substrate specificities, i.e., one CYP isoform often metabolizes multiple substrates and the same reaction can often be catalyzed by multiple CYP isoforms. The proposed theory predicts that the mediating receptors would also have low specificity for their inducers, i.e., one inducer chemical may bind multiple types of receptors, or one receptor may interact with various inducers. This suggestion, in general, is consistent with numerous experimental findings showing that one inducer chemical can often induce multiple CYP enzymes and, likewise, a given inducible CYP enzyme can almost always be induced by a variety of chemicals.
Mechanistically, when a chemical binds and activates more than one type of mediating receptors, it is almost certain that the binding affinities of this chemical for the multiple types of receptors will differ vastly. Consequently, the induction profile of each of the multiple CYP isoforms by different chemicals would also be very different. This prediction is in agreement with numerous experimental observations that consistently showed that: (i) when multiple CYP isoforms were co-induced, usually one CYP isoform was predominantly induced while other isoform(s) was only induced to a much smaller extent, and (ii) the induction pattern (ratio) for multiple CYP isoforms was almost always different with different inducers. Based on the mechanistic explanation tendered above, it is readily understood that some CYP1A1 inducers, which are AhR agonists, may also activate the mediating receptor for CYP1A2, thereby resulting in the co-induction of both CYP enzymes. However, because different chemicals would have different binding affinities for these two mediating receptors (as these chemicals would have different Km values for CYP1A1 and 1A2), the ratios of CYP1A1 induction to CYP1A2 induction in the same cell or tissue are expected to vary with different inducers. The same explanation can be used to explain the co-induction of CYP2B1 and 2B2 by various chemical inducers.
It is conceivable that, in some cases, an inducer chemical may interact with multiple receptors in very different ways: activating one or several type(s) of the mediating receptors but inhibiting other type(s) of the mediating receptors. Many previous studies with various chemical inducers have shown that while the chemicals would stimulate the biosynthesis of one or several CYP isoform(s), they often also simultaneously inhibit the biosynthesis of certain other CYP isoform(s). For the ease of understanding of some of the complex interactions discussed above, a schematic depiction is presented in Fig. 2.
Figure 2.
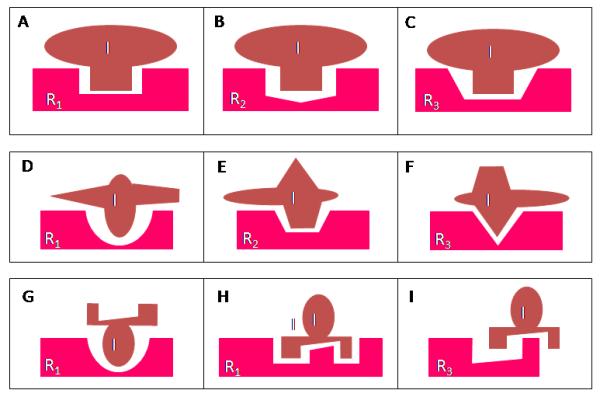
Some deducible pharmacological characteristics that are associated with the hypothesis that each chemically-inducible CYP isoform is mediated by a corresponding receptor. The upper panels (A, B, C) depict a possible situation where an inducer chemical (I) can bind and then activate a number of structurally-similar receptors (R1, R2, R3) with different binding affinities. The induction potency of a chemical for a given CYP isoform will largely depend on its binding affinity for its mediating receptor. As depicted, the binding affinity of inducer I for R1, R2, and R3 is assumed to follow the rank order of R1 > R2 > R3. It is expected that inducer I is a more potent inducer for the induction of R1-mediated CYP isoform than R2- or R3-mediated CYP isoforms. In comparison, the maximal induction of a given isoform (achieved at high inducer concentrations) may be, in a significant part, determined by the levels of the inducer receptor present. Note that in this hypothetical case since the receptors R1, R2, and R3 bind to the same chemical moiety of the inducer, it is expected that the induced CYP isoforms will also have the ability to catalyze the same metabolic reaction (albeit they may have rather different KM values).
The middle panels (D, E, F) depict a situation where multiple chemical moieties of the same inducer chemical may interact with different inducer receptors. The binding affinity of each moiety for the corresponding receptor will basically determine the relative potency of induction of a corresponding CYP isoform, and the total receptor levels may, in part, determine the magnitude of induction of a given CYP isoform. There is considerable experimental evidence showing that a chemical inducer (such as phenobarbital) can induce multiple CYP isoforms with widely different potencies and efficacies. Notably, since in this case different moieties of the inducer chemical interact with different receptors to induce different CYP isoforms, it is expected that each of the induced CYP isoforms will also have a preferential ability to metabolize the corresponding moiety of the inducer chemical.
The bottom panels (G, H, I) depict a situation where an inducer chemical may activate multiple inducer receptors (such as R1 and R2), resulting in the induction of corresponding CYP isoforms, but it may also competitively block some other inducer receptors (such as R3), resulting in a reduced expression of the corresponding CYP isoform. This phenomenon has been observed with some inducer chemicals, such as phenobarbital.
Lastly, it is worth mentioning that the current CYP isoform classification is based on the degree of homology of the amino acid sequences. Different CYP isoforms of the same subfamily share a greater degree of homology in their amino acid sequences than those in different subfamilies, and often isoforms within a subfamily also can catalyze the same reactions. However, the degree of similarity of their amino acid sequences may not always reflect the degree of their functional similarity, which is largely determined by their steric structures. Therefore, the steric structures of the inducer-binding sites of the receptors for the CYP isoforms of the same subfamily may not always share a higher degree of similarity compared to those receptors for the CYP isoforms of different subfamilies.
3.3. Hypothesis 3: Each inducible CYP gene may have a different genomic response element that interacts selectively with the corresponding receptor
An alternative premise to this hypothesis is that the same genomic response element for a mediating receptor is present in multiple inducible CYP genes. If this is the case, then the transcription of multiple CYP genes would always be simultaneously activated by a series of inducers (i.e., agonists for the same mediating receptor), and the rank order of potency for the induction of these CYP isoforms with various inducers should be identical or very similar. As discussed earlier, most experimental observations in the literature strongly disagreed with this alternative premise. In addition, earlier studies have shown that the same aryl hydrocarbon response element (AhRE) identified for the CYP1A1 gene did not appear to be present in the CYP1A2 and CYP1B1 gene promoters. This finding disagrees with this alternative premise.
The proposed hypothesis, however, does not rule out the possibility that an activated mediating receptor may interact with a homologous regulatory region in other functionally-similar CYP genes. Such cross interactions may provide certain degrees of coordination for the induction of other functionally-similar CYP isoforms.
In addition, it is known that the AhRE for CYP1A1 gene is also present in genes encoding several Phase II enzymes, such as glucuronosyltransferase and glutathione S-transferase [14, 15]. Similarly, a recent study [75] showed that the nuclear PXR not only mediated the induction of certain CYP3A isoform, but it also mediated the induction of certain Phase II conjugating enzymes, such as dehydroepiandrosterone sulfotransferase [75]. These interesting findings demonstrated a general mechanism for the coordinated induction of certain Phase I oxidative enzymes, Phase II conjugative enzymes, and possibly Phase III drug transporters that are functionally coupled. Understandably, if elevated oxidative metabolism of a drug (primarily catalyzed by cellular CYP isoforms) is uncoupled to a selective increase of its conjugative metabolism, it may be potentially hazardous to cells, whereas coordinated upregulation of CYP isoforms (such as CYP1A1), certain conjugation enzymes that can effectively metabolize the oxidation products, and transporters would favorably detoxify lipophilic chemicals such as polycyclic aromatic hydrocarbons and their oxidative metabolites.
In brief, the proposed hypothesis suggests that a mediating receptor protein would have both ligand specificity and certain degrees of target gene selectivity, and these two features would enable the receptor to mediate the selective induction of a CYP isoform by the inducer chemicals. The chemically-activated receptors will act as transcription factors leading to the initiation of selective transcriptional activation of target CYP genes. The relative selectivity at the target gene is enabled by the specific response element usually present in the promoter of an inducible CYP gene. Although this mechanism has been demonstrated with the AhR-mediated chemical induction of CYP1A1 as well as the receptor-mediated induction of CYP3A and CYP4A family members, it is believed that the same mechanism is shared in the chemical induction of other CYP isoforms.
4. CONCLUDING REMARKS AND EXPERT OPINIONS
Selective induction of CYP isoforms by inducer chemicals has attracted intense research interest throughout the past half century. Such inductive responses of various organs or cells in the body can have a major impact on drug metabolism, on pharmacokinetics and drug-drug interactions, on the toxicity and carcinogenicity of foreign chemicals, and on the potency and disposition of circulating hormones [3-9]. In this paper, a general theoretical framework is proposed concerning the mechanisms of selective induction of CYP isoforms by chemicals. Specifically, it is suggested that: (i) each chemically-inducible CYP isoform may have a receptor that interacts with the inducer chemical and mediates the selective induction of this CYP isoform; (ii) each chemically-inducible CYP isoform and its corresponding receptor may share a highly similar steric structure in their substrate/inducer binding sites; and (iii) each inducible CYP gene may have a distinct genomic response element that interacts selectively with the activated receptor.
While the proposed theories are strongly supported by the existing experimental data on CYP1A1 induction (known to be mediated by the AhR), they are also largely in agreement with a considerable body of experimental observations scattered in scientific literature concerning the chemical induction of several other CYP enzymes. For some of the CYP enzymes, their mediating receptors are already known, but for many others their mediating receptors are presently unknown. Ultimately, for the verification of each of the proposed theoretical elements, one needs to identify the inducer receptor for each of the inducible CYP isoforms, and one also needs to directly compare the steric structures of the inducer-binding site of the mediating receptor with the substrate-binding site of the corresponding CYP enzyme. These key pieces of experimental evidence are needed to verify the proposed mechanistic explanation for the complex process of selective CYP enzyme induction by many structurally-diverse chemicals.
For the fairness of the discussion of the proposed theories, it should be noted that there are also experimental and clinical observations concerning the chemical induction of CYP enzymes that may not be readily explained by the proposed theories. In some of the cases, it appears that other non-receptor mediated processes may be involved. For instance, omeprazole is a strong inducer of the CYP1A1 and 1A2, but this chemical does not appear to have a binding affinity for AhR [76]. The exact mechanism for the induction of the CYP1A family enzymes by omeprazole is still poorly understood at present, although it has been suggested that it may depend on its effect on tyrosine kinase activity [77]. In many other cases, however, the apparent inconsistencies may not contradict the theories proposed; rather they are largely due to the involvement of many other regulatory pathways and factors that could affect the expression of the CYP genes. The presence of these confounding factors sometimes obscures the proposed mechanistic core elements. A brief discussion of some of the confounding factors using concrete examples would be helpful and is provided below.
Theoretically, a high-affinity substrate for an inducible CYP enzyme (usually determined in vitro with microsomes or recombinant CYP enzymes) would also be a high-affinity activator of its mediating receptor. However, there are also other underlying factors that, at times, can cause discrepancies when these inducers are tested in vivo. The poor absorption of an inducer chemical, its slow transport across the plasma membrane into the cells, or its high-affinity binding to plasma proteins, for example, are among many common causes of the low intracellular concentrations of the inducer chemicals, and consequently, for their low in vivo enzyme-inducing activity.
Based on the proposed theory, the density of the mediating receptor in a cell would be an important determinant of the magnitude of the maximal induction of the respective CYP isoform in that cell. This would be true if we compare two cells that have different levels of the mediating receptor but everything else remains exactly the same. However, in real biological situations, often other factors that are involved in transcriptional regulation of a CYP gene may not be exactly the same. Besides the ligand-activated receptor (which functions as a gene-specific transcriptional factor), the transcriptional activation of an inducible CYP gene also involves other trans-acting factors (such as co-activators and co-repressors). These accessory factors are usually less cell-specific and less gene-specific but their levels could be altered under different conditions. Tissue- or cell-specific differences in these regulators are expected to affect the rate of constitutive as well as inducer-stimulated transcription of a given CYP gene [31]. This notion is exemplified in many recent studies of the selective estrogen receptor modulators (SERMs), which highlights the complexity of the transcriptional regulation of intracellular receptor-controlled genes [78, 79]. As a result of these complex regulations, the magnitude of the maximal induction of a given CYP isoform by a chemical in two different types of cells or tissues may not be exactly proportional to the density of the mediating receptor present. However, it is expected that the rank order of potency for a given series of chemical inducers will still be highly similar in different responsive cells, which would reflect the critical role of a specific receptor in determining the inducibility of a given CYP gene following exposure to chemical inducers.
Chemical induction of CYP isoforms is also subject to modulation by a variety of factors that alter the overall growth status of a cell, which include, but are not limited to, growth hormones, thyroid hormone, certain interleukins, intraceullular levels of cAMP, and many others. It is believed, however, that modulation of chemical induction of CYP enzymes by these growth-related factors is generally nonspecific (or far less specific) with respect to the inducer chemicals used or the CYP isoforms involved. For instance, studies have shown that rapidly proliferating hepatocytes would have much lower ability to express CYP enzymes compared to well-differentiated, slow-proliferating hepatocytes. Therefore, in the presence of these growth-regulating factors, the magnitude of induction of various CYP enzymes following exposure to various chemical inducers would be greatly reduced, which has been repeatedly observed in many studies. However, in these cells, the rank order of potency for the induction of a given CYP enzyme almost always remain nearly the same, which agrees with the speculated role of the corresponding receptor in selectively mediating the induction of a target CYP enzyme.
There were experimental observations showing that the binding affinity of a substrate to the catalytic site of a CYP enzyme may not always parallel its rate of metabolism. TCDD is a typical example in this regard. TCDD is a very potent inducer of CYP1A1 (because it is a very potent, high-affinity agonist for the AhR), and it also has a high affinity for binding to CYP1A1 [51, 53]. However, TCDD is essentially not metabolized by CYP1A1. This unusual discrepancy between its high binding affinity for CYP1A1 and its low rate of metabolism by this isoform may lie in the possibility that the unusually strong electron-withdrawing effect of the multiple halogen atoms in this rare, man-made molecule makes its oxidative reaction via the electrophilic attack of the substrate molecule catalyzed by the CYP1A1 enzyme exceptionally difficult (nearly impossible).
Many other vitally-important biological systems usually have a built-in redundant mechanism. The proposed theory shows that there is also a built-in redundancy in the chemical induction of CYP enzymes. Here let us use benzo[a]pyrene as an example, which is a well-known chemical carcinogen that can be metabolized by several CYP1A family enzymes, including 1A1, 1A2, and 1B1. When benzo[a]pyrene is given to an animal (such as i.p. injection into rats), it will induce the expression of all three CYP1A isoforms (1A1, 1A2, and 1B1) to varying degrees. In fact, there are considerable data in the literature showing that the induction ratio of these three CYP1A isoforms matches very closely their preference for metabolizing benzo[a]pyrene as a substrate. The possible explanation (based on the proposed theory) is that the receptors for CYP1A1, 1A2, and 1B1 also have similar binding sites as the corresponding enzymes such that these receptors can all bind the inducer chemicals (albeit with varying binding affinities). It is apparent that there is redundancy here in the induction of the metabolizing enzymes, i.e., three CYP enzymes that possess similar catalytic functions are all induced to metabolize the same inducer chemical. Such redundancy is crucial when one CYP enzyme system cannot be induced due to genetic defects, other functionally-similar enzyme systems can be induced to compensate. Understandably, when three functionally similar enzymes (such as CYP1A1, 1A2, and 1B1) are all induced, each one may be induced at a relatively low level in order to achieve the intended biological purpose, but when one of the enzyme systems cannot be induced due to genetic defects, then the other two systems may need to be induced to a higher level to achieve the same goal.
Lastly, as a unique case that may stimulate further thinking, it is worth noting that there is a phenomenon in antibody immunology that bears some resemblance to the selective induction of CYP isoforms by chemicals. It is known that all antibody molecules produced by a B cell (or a clone of B cells) have the same antigen-binding site. In fact, the first set of antibodies produced by a newly-formed B cell are not secreted, instead they are modified and inserted into the plasma membrane where they serve as the receptor for recognizing antigens. In this way, each clone of the antibodies has a corresponding clone of membrane receptors with exactly the same antigen-binding site. When the antigen binds specifically to the receptor antibodies present in the surface of a virgin B cell or a memory B cell, it initiates a series of intracellular events culminating in cell proliferation to produce antibody-secreting B cells. In this case, because the receptor antibodies present in the surface of the antibody-secreting B cells have exactly the same antigen-binding site as the antibodies to be secreted, this assures that the secreted antibodies can selectively recognize the antigen to which the organism is exposed. The overall resemblance between the antigen induction of specific antibodies and the chemical induction of CYP isoforms is intriguing, and it adds biological credence to the hypothesis that a CYP isoform and its corresponding receptor may share a highly similar steric structure in their substrate/inducer binding sites. However, the specificity of a CYP enzyme for various substrates generally is far lower than an antibody’s specificity for the antigens. The high degrees of specificity of antibodies are primarily due to the existence of very large numbers of different clones of B cells that can be selectively stimulated for antibody production. In comparison, the numbers of CYP isoforms are far less. In fact, the ability of such small numbers of CYP isoforms to metabolize almost unlimited numbers of endogenous or exogenous chemicals is fulfilled by the relatively low substrate specificity of many of the inducible CYP isoforms together with their suitable biochemical properties as having high metabolic capacity (i.e., high Vmax) but generally low binding affinity (i.e., high Km).
Article highlights box.
A general theoretical framework regarding the mechanisms of selective induction of CYP enzymes by chemical inducers is presented. Briefly, this theory suggests that each chemically-inducible CYP isoform may have a specific receptor that can interact with the inducer chemical to mediate the selective induction of this CYP isoform. Moreover, each chemically-inducible CYP isoform and its corresponding receptor may share a highly similar steric structure in their substrate/inducer binding sites.
Each of the proposed theoretical elements is formulated on the basis of strong scientific rationale and is also supported by a large body of direct or indirect experimental observations.
There are also experimental and clinical observations of the chemical induction of CYP enzymes that may not be readily explained by the proposed theories. While in some of the cases non-receptor mediated processes may be involved, most of the other inconsistencies actually do not contradict the theories proposed; rather they are largely due to the involvement of many other regulatory pathways and factors that could affect the expression of the CYP genes. These confounding factors sometimes may obscure the core elements.
ACKNOWLEDGEMENTS
When an earlier version of this manuscript (containing the same three theoretical elements) was competed in the Fall of 1996, Dr. Allan H. Conney at Rutgers University (Piscataway, NJ) gave the manuscript a careful reading and offered encouragements as well as some additional literature examples for consideration. The author wishes to thank Dr. Conney for his encouragement and generous help. The author also wishes to express his indebtedness to many researchers in this research field because their equally important research work has been left out of the reference list solely because of space constraint. Lastly, the author would like to thank Stephanie C. Bishop for proof-reading of the manuscript, and the Expert Opinions on Drug Metabolism & Toxicology for providing a rare forum for this theoretical/opinion piece, which otherwise may never see the light of the day.
DECLARATION OF INTEREST The author was supported, in part, by grants (ES015242, CA092391 and CA097109) from the National Institutes of Health.
Abbreviations used
- CYP
cytochrome P450
- AhR
aryl hydrocarbon receptor
- AhRE
AhR response element
- PXR
Pregnane X receptor
- CAR
Constitutive androstane receptor.
Footnotes
The author does not have any competing financial interests to declare.
BIBLIOGRAPHY
Papers of special note have been highlighted as either of interest (•) to readers.
- 1.Gonzalez FJ. Molecular genetics of the P-450 superfamily. Pharmacol Ther. 1990;45:1–38. doi: 10.1016/0163-7258(90)90006-n. [DOI] [PubMed] [Google Scholar]
- 2.Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW. P450 superfamily: update on new sequences, gene mapping, accession numbers, and nomenclature. Pharmacogenetics. 1996;6:1–43. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Esterbrook R. An introduction to the cytochrome P450s. Mol Aspects Med. 1999;20:5–137. [PubMed] [Google Scholar]
- 4.Coon MJ. Cytochrome P450: Nature’s most versatile biological catalyst. Ann Rev Pharmacol Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- 5.de Montellano PR Ortiz., editor. Cytochrome P-450: Structure, metabolism and biochemistry. Plenum Press; NY: 1986. pp. 1–556. [Google Scholar]
- 6.Schuster I, editor. Cytochrome P-450: Biochemistry and Biophysics. Taylor & Francis; London: 1989. pp. 1–902. [Google Scholar]
- 7.Guengerich FP. Enzymatic oxidation of xenobiotic chemicals. Crit Rev Biochem Mol Biol. 1990;25:97–153. doi: 10.3109/10409239009090607. [DOI] [PubMed] [Google Scholar]
- 8.Conney AH. Pharmacological implications of microsomal enzyme induction. Pharmacol Rev. 1967;19:317–366. [PubMed] [Google Scholar]
- 9.Conney AH. Induction of drug-metabolizing enzymes: a path to the discovery of multiple cytochromes P450. Ann Rev Pharmacol Toxicol. 2003;43:1–30. doi: 10.1146/annurev.pharmtox.43.100901.135754. • Review of the research on the chemical induction of cytochrome P450 as well as other drug-metabolizing enzymes.
- 10.Okey AB. Enzyme induction in the cytochrome P-450 system. Pharmacol Ther. 1990;45:241–298. doi: 10.1016/0163-7258(90)90030-6. [DOI] [PubMed] [Google Scholar]
- 11.Safe SH. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther. 1995;67:247–281. doi: 10.1016/0163-7258(95)00017-b. [DOI] [PubMed] [Google Scholar]
- 12.Denison MS, Whitlock JP. Xenobiotic-inducible P450 genes. J Biol Chem. 1995;270:18175–18178. doi: 10.1074/jbc.270.31.18175. [DOI] [PubMed] [Google Scholar]
- 13.Waxman DJ, Azaroff L. Phenobarbital induction of cytochrome P-450 gene expression. Biochem J. 1992;281:577–592. doi: 10.1042/bj2810577. • An interesting review paper that discussed the hypothesis that the induction of cytochrome P450 2B family enzymes by pehnobarbital likely was receptor-mediated.
- 14.Whitlock J., Jr Induction of cytochrome P4501A1. Ann Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. • A review paper that summarized the general molecular mechanism underlying the induction of cytochrome P450 1A family enzymes.
- 15.Whitlock J, Jr, Okino ST, Dong L, Ko HP, Clarke-Katzenberg R, Ma Q, Li H. Cytochromes P450. Induction of cytochrome P4501A1: A model for analyzing mammalian gene transcription. FASEB J. 1996;10:809–818. doi: 10.1096/fasebj.10.8.8666157. [DOI] [PubMed] [Google Scholar]
- 16.Yamada H, Ishii Y, Yamamoto M, Oguri K. Induction of the hepatic cytochrome P450 2B subfamily by xenobiotics: research history, evolutionary aspect, relation to tumorigenesis, and mechanism. Curr Drug Metab. 2006;7:397–409. doi: 10.2174/138920006776873508. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Negishi M. Transcriptional regulation of cytochrome p450 2B genes by nuclear receptors. Curr Drug Metab. 2003;4:515–525. doi: 10.2174/1389200033489262. • A review paper that summarized the role of nuclear receptor CAR in the induction of cytochrome P450 2B family enzymes.
- 18.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci USA. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol. 1999;56:1329–1339. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 20.Hamaoka N, Oda Y, Hase I, Asada A. Cytochrome P4502B6 and 2C9 do not metabolize midazolam: kinetic analysis and inhibition study with monoclonal antibodies. Br J Anaesth. 2001;86:540–544. doi: 10.1093/bja/86.4.540. [DOI] [PubMed] [Google Scholar]
- 21.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 23.Johnson EF, Hsu MH, Savas U, Griffin KJ. Regulation of P450 4A expression by peroxisome proliferator activated receptors. Toxicology. 2002;181-182:203–6. doi: 10.1016/s0300-483x(02)00282-2. [DOI] [PubMed] [Google Scholar]
- 24.Johnson EF, Palmer CN, Griffin KJ, Hsu MH. Role of the peroxisome proliferator-activated receptor in cytochrome P450 4A gene regulation. FASEB J. 1996;10:1241–1248. doi: 10.1096/fasebj.10.11.8836037. [DOI] [PubMed] [Google Scholar]
- 25.Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab. 2002;3:481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- 26.Waxman DJ. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- 27.Shirak H, Guengerich FP. Turnover of membrane protein: kinetics of induction and degradation of seven forms of rat liver microsomal cytochrome P-450, NADPH-dependent P-450 reductase, and epoxide hydrolase. Arch Biochem Biophys. 1984;235:86–96. doi: 10.1016/0003-9861(84)90257-1. [DOI] [PubMed] [Google Scholar]
- 28.Watkins PB, Wrighton SA, Schuetz EG, et al. Macrolide antibiotics inhibit the degradation of the glucocorticoid-responsive cytochrome P-450p in rat hepatocytes in vivo and in primary monolayer culture. J Biol Chem. 1986;261:6264–6271. [PubMed] [Google Scholar]
- 29.Watkins PB, Bond JS, Guzelian PS. Degradation of the hepatic cytochrome P-450. In: Guengerich FP, editor. Mammalian Cytochrome P-450. vol. 2. CRC Press; Boca Raton, FL: 1987. p. 173. [Google Scholar]
- 30.Swanson HI, Bradfield CA. The Ah receptor: genetics, structure and function. Pharmacogenetics. 1993;3:213–230. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Dey A, Jones JE, Nebert DW. Tissue- and cell type-specific expression of cytochrome P450 1A1 and cytochrome P450 1A2 mRNA in the mouse localized in situ hybridization. Biochem Pharmacol. 1999;58:525–537. doi: 10.1016/s0006-2952(99)00110-0. [DOI] [PubMed] [Google Scholar]
- 32.Degawa M, Nakayama M, Yoshinari K, Hashimoto Y. 2-Methoxy-4-nitroaniline is a selective inducer of cytochrome P450IA2 (CYP1A2) in rat liver. Cancer Lett. 1995;96:95–98. doi: 10.1016/0304-3835(95)03910-o. [DOI] [PubMed] [Google Scholar]
- 33.Adams NH, Levi PE, Hodgson E. Regulation of cytochrome P-450 isozymes by methylenedioxyphenyl compounds. Chem Biol Interact. 1993;86:255–274. doi: 10.1016/0009-2797(93)90101-4. [DOI] [PubMed] [Google Scholar]
- 34.Ryu DY, Levi PE, Hodgson E. Regulation of cytochrome P-450 isozymes CYP1A1, CYP1A2 and CYP2B10 by three benzodioxole compounds. Chem Biol Interact. 1995;96:235–247. doi: 10.1016/0009-2797(94)03594-x. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Bondoc FY, Lee MJ, Hussin AH, Thomas PE, Yang CS. Caffeine induces cytochrome P4501A2: induction of CYP1A2 by tea in rats. Drug Metab Dispos. 1996;24:529–533. [PubMed] [Google Scholar]
- 36.Cook JC, Hodgson E. The induction of cytochrome P-450 by isosafrole and related methylenedioxyphenyl compounds. Chem Biol Interact. 1985;54:299–305. doi: 10.1016/s0009-2797(85)80171-x. [DOI] [PubMed] [Google Scholar]
- 37.Cook JC, Hodgson Cytochrome P-450 induction by 3-methylcholanthrene and its antagonism by 2,2-dimethyl-5-t-butyl-1,3-benzodioxole. Biochem Pharmacol. 1986;35:167–176. doi: 10.1016/0006-2952(86)90510-1. [DOI] [PubMed] [Google Scholar]
- 38.Cook JC, Hodgson Induction of cytochrome P-450 in congenic C57BL/6J mice by isosafrole: lack of correlation with the Ah locus. Chem Biol Interact. 1986;58:233–240. doi: 10.1016/s0009-2797(86)80100-4. [DOI] [PubMed] [Google Scholar]
- 39.Adams NH, Levi PE, Hodgson E. Regulation of cytochrome P-450 isozymes by methylenedioxyphenyl compounds. Chem Biol Interact. 1993;86:255–274. doi: 10.1016/0009-2797(93)90101-4. [DOI] [PubMed] [Google Scholar]
- 40.Ryu DY, Levi PE, Fernandez-Salguero P, Gonzalez FJ, Hodgson E. Piperonyl butoxide and acenaphthylene induce cytochrome P450 1A2 and 1B1 mRNA in aromatic hydrocarbon-responsive receptor knock-out mouse liver. Mol Pharmacol. 1996;50:443–446. [PubMed] [Google Scholar]
- 41.Zaher H, Yang TJ, Gelboin HV, Fernandez-Salguero P, Gonzalez FJ. Effect of phenobarbital on hepatic CYP1A1 and CYP1A2 in the Ahr-null mouse. Biochem Pharmacol. 1998;55:235–238. doi: 10.1016/s0006-2952(97)00476-0. [DOI] [PubMed] [Google Scholar]
- 42.Iwanari M, Nakajima M, Kizu R, Hayakawa K, Yokoi T. Induction of CYP1A1, CYP1A2, and CYP1B1 mRNAs by nitropolycyclic aromatic hydrocarbons in various human tissue-derived cells: chemical-, cytochrome P450 isoform-, and cell-specific differences. Arch Toxicol. 2002;76:287–298. doi: 10.1007/s00204-002-0340-z. [DOI] [PubMed] [Google Scholar]
- 43.Hakkola J, Pasanen M, Pelkonen O, Hukkanen J, Evisalmi S, Anttila S, Rane A, Mantyla M, Purkunen R, Saarikoski S, Tooming M, Raunio H. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis. 1997;18:391–397. doi: 10.1093/carcin/18.2.391. [DOI] [PubMed] [Google Scholar]
- 44.Pottenger LH, Jefcoate CR. Characterization of a novel cytochrome P450 from the transformable cell line, C3H/10T1/2. Carcinogenesis. 1990;11:321–327. doi: 10.1093/carcin/11.2.321. [DOI] [PubMed] [Google Scholar]
- 45.Spink DC, Spink BC, Cao JQ, DePasquale JA, Pentecost BT, Fasco MJ, Li Y, Sutter TR. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19:291–298. doi: 10.1093/carcin/19.2.291. [DOI] [PubMed] [Google Scholar]
- 46.Kress S, Greenlee WF. Cell-specific regulation of human CYP1A1 and CYP1B1 genes. Cancer Res. 1997;57:1264–1269. [PubMed] [Google Scholar]
- 47.Spink DC, Spink BC, Cao JQ, Gierthy JF, Hayes CL, Li Y, Sutter TR. Expression of aryl hydrocarbon receptor repressor in normal human tissues and inducibility by polycyclic aromatic hydrocarbons in human tumor-derived cell lines. J Steroid Biochem Mol Biol. 1997;62:223–232. [Google Scholar]
- 48.Hakkola J, Pasanen M, Pelkonen O, Hukkanen J, Evisalmi S, Anttila S, Rane A, Mäntylä M, Purkunen R, Saarikoski S, Tooming M, Raunio H. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis. 1997;18:391–397. doi: 10.1093/carcin/18.2.391. [DOI] [PubMed] [Google Scholar]
- 49.Hakkola J, Pasanen M, Pelkonen O, Hukkanen J, Evisalmi S, Anttila S, Rane A, Mäntylä M, Purkunen R, Saarikoski S, Tooming M, Raunio H. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis. 1997;18:391–397. doi: 10.1093/carcin/18.2.391. [DOI] [PubMed] [Google Scholar]
- 50.Shimada T, Inoue K, Suzuki Y, Kawai T, Azuma E, Nakajima T, Shindo M, Kurose K, Sugie A, Yamagishi Y, Fujii-Kuriyama Y, Hashimoto M. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis. 2002;23:1199–1207. doi: 10.1093/carcin/23.7.1199. [DOI] [PubMed] [Google Scholar]
- 51.Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem. 1976;251:4936–4946. [PubMed] [Google Scholar]
- 52.Riddick DS, Huang Y, Harper PA, Okey AB. 2,3,7,8-Tetrachlorodibenzo-p-dioxin versus 3-methylcholanthrene: comparative studies of Ah receptor binding, transformation, and induction of CYP1A1. J Biol Chem. 1994;269:12118–12128. [PubMed] [Google Scholar]
- 53.Poland A, Glover E. Comparison of 2,3,7,8-tetrachlorodibenzo-p-dioxin, a potent inducer of aryl hydrocarbon hydroxylase, with 3-methylcholanthrene. Mol Pharmacol. 1974;10:349–359. [PubMed] [Google Scholar]
- 54.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 55.Traber PG, Wang W, McDonnell M, Gumucio JJ. P450IIB gene expression in rat small intestine: cloning of intestinal P450IIB1 mRNA using the polymerase chain reaction and transcriptional regulation of induction. Mol Pharmacol. 1990;37:810–819. [PubMed] [Google Scholar]
- 56.Omiecinski CJ. Tissue-specific expression of rat mRNAs homologous to cytochromes P-450b and P-450e. Nucleic Acids Res. 1986;14:1525–1539. doi: 10.1093/nar/14.3.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christou M, Wilson NM, Jefcoate CR. Expression and function of three cytochrome P-450 isozymes in rat extrahepatic tissues. Arch Biochem Biophys. 1987;258:519–534. doi: 10.1016/0003-9861(87)90374-2. [DOI] [PubMed] [Google Scholar]
- 58.Kocarek TA, Schuetz EG, Guzelian PS. Differentiated induction of cytochrome P450b/e and P450p mRNAs by dose of phenobarbital in primary cultures of adult rat hepatocytes. Mol Pharmacol. 1990;38:440–444. [PubMed] [Google Scholar]
- 59.Shaw PM, Adesnik M, Weiss MC, Corcos L. The phenobarbital-induced transcriptional activation of cytochrome P-450 genes is blocked by the glucocorticoid-progestoerone antagonist RU486. Mol Pharmacol. 1993;44:775–783. [PubMed] [Google Scholar]
- 60.Dannan GA, Guengerich FP, Kaminsky LS, Aust SD. Regulation of cytochrome P-450. Immunochemical quantitation of eight isozymes in liver microsomes of rats treated with polybrominated biphenyl congeners. J Biol Chem. 1983;258:1282–1288. [PubMed] [Google Scholar]
- 61.Yamazoe Y, Shimada M, Murayama N, Kato R. Suppression of levels of phenobarbital-inducible rat liver cytochrome P-450 by pituitary hormone. J Biol Chem. 1987;262:7423–7428. [PubMed] [Google Scholar]
- 62.Marcus CB, Wilson NM, Jefcoate CR, Wilkinson CF, Omiecinski CJ. Selective induction of cytochrome P450 isozymes in rat liver by 4-n-alkyl-methylenedioxybenzenes. Arch Biochem Biophys. 1990;277:8–16. doi: 10.1016/0003-9861(90)90543-8. [DOI] [PubMed] [Google Scholar]
- 63.Hashimoto T, Matsumoto T, Nishizawa M, Kawabata S, Morohashi K, Handa S, Omura T. A mutant rat strain deficient in induction of a phenobarbital-inducible form of cytochrome P-450 in liver microsomes. J Biochem (Tokyo) 1988;10:487–492. doi: 10.1093/oxfordjournals.jbchem.a122297. [DOI] [PubMed] [Google Scholar]
- 64.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9:310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 65.Liu YT, Hao HP, Liu CX, Wang GJ, Xie HG. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab Rev. 2007;39:699–721. doi: 10.1080/03602530701690374. [DOI] [PubMed] [Google Scholar]
- 66.Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, Hamilton G, Rizzo C, Jolley S, Gilbert D, Downey A, Mudra D, Graham R, Carroll K, Xie J, Madan A, Parkinson A, Christ D, Selling B, LeCluyse E, Gan LS. CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Dispos. 2002;30:795–804. doi: 10.1124/dmd.30.7.795. [DOI] [PubMed] [Google Scholar]
- 67.Wood AW, Levin W, Lu AY, Yagi H, Hernandez O, Jerina DM, Conney AH. Metabolism of benzo(a)pyrene and benzo (a)pyrene derivatives to mutagenic products by highly purified hepatic microsomal enzymes. J Biol Chem. 1976;251:4882–4890. [PubMed] [Google Scholar]
- 68.Shou M, Korzekwa KR, Crespi CL, Gonzalez FJ, Gelboin HV. The role of 12 cDNA-expressed human, rodent, and rabbit cytochromes P450 in the metabolism of benzo[a]pyrene and benzo[a]pyrene-trans-7,8-dihydrodiol. Mol Carcinogen. 1994;10:159–168. doi: 10.1002/mc.2940100307. [DOI] [PubMed] [Google Scholar]
- 69.Guengerich FP, Shimada T. Activation of procarcinogens by human cytochrome P450 enzymes. Mutat Res. 1998;400:201–213. doi: 10.1016/s0027-5107(98)00037-2. [DOI] [PubMed] [Google Scholar]
- 70.Shimada T, Inoue K, Suzuki Y, Kawai T, Azuma E, Nakajima T, Shindo M, Kurose K, Sugie A, Yamagishi Y, Fujii-Kuriyama T, Hashimoto M. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis. 2002;23:1199–1207. doi: 10.1093/carcin/23.7.1199. [DOI] [PubMed] [Google Scholar]
- 71.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 72.Cresteil T, Mahu JL, Dansette PM, Leroux JP. In vivo administration of hydroxylated phenobarbital metabolites: effect on rat hepatic cytochromes P-450, epoxide hydrolase and UDP-glucuronosyltransferase. Biochem Pharmacol. 1980;29:1127–1133. doi: 10.1016/0006-2952(80)90407-4. [DOI] [PubMed] [Google Scholar]
- 73.Waxman DJ, Walsh C. Cytochrome P-450 isozyme 1 from phenobarbital-induced rat liver: purification, characterization, and interactions with metyrapone and cytochrome b5. Biochemistry. 1982;22:4846–4855. doi: 10.1021/bi00289a035. [DOI] [PubMed] [Google Scholar]
- 74.Ryan DE, Thomas PE, Levin W. Purification of characterization of a minor form of hepatic microsomal cytochrome P-450 from rats treated with polychlorinated biphenyls. Arch Biochem Biophys. 1982;216:272–288. doi: 10.1016/0003-9861(82)90212-0. [DOI] [PubMed] [Google Scholar]
- 75.Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR) Proc Natl Acad Sci USA. 2002;99:13801–13806. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Daujat M, Peryt B, Lesca P, Fourtanier G, Domergue J, Maurel P. Omeprazole, an inducer of human CYP1A1 and 1A2, is not a ligand for the Ah receptor. Biochem Biophys Res Commun. 1992;188:820–825. doi: 10.1016/0006-291x(92)91130-i. [DOI] [PubMed] [Google Scholar]
- 77.Kikuchi H, Hossain A, Yoshida H, Kobayashi S. Induction of cytochrome P-450 1A1 by omeprazole in human HepG2 cells is protein tyrosine kinase-dependent and is not inhibited by alpha-naphthoflavone. Arch Biochem Biophys. 1998;358:351–358. doi: 10.1006/abbi.1998.0869. [DOI] [PubMed] [Google Scholar]
- 78.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal. 2008;6:e003. doi: 10.1621/nrs.06003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]