

Abstract
Innate immune responses are critical for the immediate protection against microbial infection. In Drosophila, infection leads to the rapid and robust production of antimicrobial peptides, through two NF-κB signaling pathways - IMD and Toll. The IMD pathway is triggered by DAP-type peptidoglycan, common to most Gram-negative bacteria. Signaling downstream from the peptidoglycan receptors is thought to involve K63-ubiquitination and caspase-mediated cleavage, but the molecular mechanisms remain obscure. We now show that PGN-stimulation causes caspase-mediated cleavage of the imd protein, exposing a highly conserved IAP-binding motif (IBM) at its neo-N-terminus. A functional IBM is required for the association of cleaved-IMD with the ubiquitin E3 ligase DIAP2. Through its association with DIAP2, IMD is rapidly conjugated with K63-linked polyubiquitin chains. These results mechanistically connect caspase-mediated cleavage and K63-ubiquitination in immune-induced NF-κB signaling.
Keywords: IMD, DIAP2, DREDD, Ubiquitin
Introduction
Activation of the Drosophila IMD pathway by DAP-type peptidoglycan (PGN) leads to the robust and rapid production of a battery of antimicrobial peptides (AMPs) and other immune responsive genes (Ferrandon et al., 2007; Lemaitre and Hoffmann, 2007). Two peptidoglycan recognition protein (PGRP) receptors are responsible for the recognition of DAP-type PGN, the cell surface receptor PGRP-LC and the cytosolic receptor PGRP-LE (Kaneko et al., 2006). DAP-type PGN binding causes these receptors to multimerize or cluster (Chang et al., 2006; Lim et al., 2006) triggering signal transduction. IMD signaling culminates in activation of the NF-κB precursor Relish and transcriptional induction of AMP genes.
Currently, the molecular mechanisms linking these PGN-binding receptors and activation of Relish remain unclear. Genetic experiments suggest that the most receptor proximal component of the pathway is the imd protein (Georgel et al., 2001), while the MAP3 kinase TAK1 appears to function downstream (Silverman et al., 2003; Vidal et al., 2001). In turn, TAK1 is required for activation of the Drosophila IKK complex, which is essential for the immune-induced cleavage and activation of the NF-κB precursor Relish, the key transcription factor required for immune-responsive AMP gene expression (Silverman et al., 2000). In addition to NF-κB signaling, TAK1 also mediates immune-induced JNK signaling (Silverman et al., 2003).
Other major components in the IMD pathway include the caspase-8 like DREDD and its adapter FADD. RNAi-based studies suggest that these proteins have two distinct roles in IMD pathway signaling, one relatively early in the cascade and the second further downstream. Using RNAi, DREDD and FADD were shown to be required for immune-induced activation of the IKK complex (Zhou et al., 2005). These data suggested that DREDD and FADD function downstream of IMD but upstream of TAK1, however, it was not established if this upstream role for DREDD involves its protease activity. In its second role, DREDD is thought to proteolytically cleave Relish, (Stöven et al., 2003; Erturk-Hasdemir et al., 2009).
In addition to the components outlined above, several studies have suggested that ubiquitination plays a critical role in the IMD signaling cascade. Recently, Drosophila Inhibitor of Apoptosis 2 (DIAP2) was shown to be a crucial component of the IMD pathway (Gesellchen et al., 2005; Huh et al., 2007; Kleino et al., 2005; Leulier et al., 2006). Typical of IAP proteins, DIAP2 has three N-terminal BIR domains, which are involved in interactions with proteins carrying conserved IAP binding motifs (IBMs) (Wu et al., 2000). In addition, some IAPs, including DIAP2, carry a C-terminal RING finger domain that provides these proteins with ubiquitin E3 ligase activity (Vaux and Silke, 2005). Although it is unclear where in the pathway DIAP2 functions, one study showed that the RING finger is indispensable for its role in the immune response, suggesting it operates as an E3 ubiquitin ligase (Huh et al., 2007). Also, Zhou et al. (2005) showed, using RNAi-based approaches, that the E2 ubiquitin conjugating enzymes Uev1a and Ubc13 (bendless) are critical components of the IMD pathway. Notably, Ubc13 and Uev1a function together in a complex to generate K63-linked polyubiquitin chains. K63-polyubiquitin chains are not linked to proteasomal degradation but, instead, are thought to play regulatory roles (Chiu et al., 2009; Mukhopadhyay and Riezman, 2007; Xia et al., 2009). However, no K63-ubiquitinated target protein(s) have been identified in the IMD pathway. Although no connection between DIAP2 and the Ubc13/Uev1a E2 complex has been established, one attractive scenario is that DIAP2 functions as an E3 together with the Ubc13-Uev1a E2 complex.
The imd1 allele is a strong hypomorphic mutation that impairs innate immune responses. Surprisingly, this allele encodes a conservative amino acid substitution, alanine (A) to valine (V) at position 31 and is positioned in a region with no obvious structural motifs (Georgel et al., 2001). The reason for the strong hypomorphic phenotype associated with the A31V substitution remains unclear. In this work, we demonstrate that imd protein is rapidly cleaved following PGN-stimulation. Cleavage requires the caspase DREDD and occurs at caspase recognition motif, 27LEKD/A31, creating a neo-N-terminus at A31 that is critical for the immune-induced association of IMD with DIAP2. Substitution of the neo-N-terminus with valine, as in imd1, disrupts the IMD-DIAP2 interaction. Moreover, once associated with DIAP2, cleaved-IMD is rapidly K63-polyubiquitinated. Together, these data resolve a number of outstanding questions in IMD signal transduction and present a clear molecular mechanism linking caspase-mediated-cleavage to NF-κB activation.
Results
Signal-induced modification of IMD
To determine if the proteolytic activity of DREDD is required upstream in the IMD pathway, the caspase inhibitor zVAD-fmk was utilized. Immune responsive Drosophila S2* cells were treated with zVAD-fmk (or vehicle control) prior to stimulation with E. coli peptidoglycan (PGN). Treatment with caspase inhibitor suppressed the activation of TAK1 kinase, as monitored by in-vitro IP-kinase assay. zVAD-fmk also prevented accumulation of phospho-JNK (Figure 1A).
Figure 1. Dredd-dependent Cleavage of IMD.

(A) S2* cells were treated with zVAD-fmk or vehicle control (DMSO) prior to stimulation with peptidoglycan for 30 minutes. TAK1 kinase activity was monitored by TAK1 IP-kinase assay and JNK phosphorylation was monitored by immunoblotting.
(B) S2* cells were stimulated with peptidoglycan from 0 to 120 minutes, as indicated, and endogenous-IMD was monitored in whole cell lysates by immunoblotting.
(C) Endogenous IMD was monitored in S2* cell lysates after pretreatment with zVAD-fmk followed by PGN stimulation.
(D) Endogenous IMD was monitored by IP-immunoblotting from S2* lysates after treatment with RNAi targetting various IMD pathway components, as indicated.
(E) Endogenous IMD was monitored in S2* cell lysates after expression of DREDD-WT, DREDD-CA or DRONC. Caspases were expressed from the metallothionein promoter by addition of copper sulfate for 6 hours. DREDD-CA is a catalytically inactive mutant. All 3 caspases were robustly expressed (data not shown).
In all cases,  marks unmodified full length IMD, ◁ highlights phosphorylated IMD, and
marks unmodified full length IMD, ◁ highlights phosphorylated IMD, and 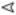 marks the cleaved-IMD products.
marks the cleaved-IMD products.
See also Supplemental Figure 1
Hypothesizing that IMD signaling results in the DREDD-mediated cleavage of an upstream signaling component, we next examined the fate of endogenous imd protein. Peptidoglycan stimulation of S2* cells resulted in the rapid, signal dependent modification of IMD (Figure 1B). The faster migrating IMD product did not accumulate in the presence of caspase inhibitor (Figure 1C), suggesting that it represents an IMD cleavage product. Cleavage of IMD appeared very rapidly, within one minute, and peaked between 10 and 30 minutes. Treatment of S2* cells with RNAi targeting either DREDD and FADD, prior to stimulation with peptidoglycan, resulted in a decrease in IMD cleavage, and accumulation of full length IMD (Figure 1D). Furthermore, over expression of wild type DREDD (WT), but not a non-catalytic mutant (CA) or the caspase DRONC, from the copper inducible metallothionein promoter resulted in the cleavage of IMD, independent of immune stimulation (Figure 1E). Together these results suggest that the proteolytic activity of DREDD is required for the cleavage of IMD. Phosphorylation of IMD was also detected after immune stimulation, with the appearance of a slower migrating form, see Supplemental Figure 1.
DIAP2 functions between IMD and TAK1
To examine how IMD cleavage might be linked to downstream signaling events such as K63-ubiquitination, we sought to more carefully characterize the role of DIAP2, a putative ubiquitin E3-ligase, in this pathway. As previously shown, DIAP2 RNAi markedly inhibited the induction of the antimicrobial peptide gene Diptericin following immune stimulation with DAP-type peptidoglycan, similar to RNAi targeting PGRP-LC or IKKγ, as analyzed by Northern blotting (Figure 2A) (Gesellchen et al., 2005; Huh et al., 2007; Kleino et al., 2005; Leulier et al., 2006). Using a stable S2* cell line that expresses imd from the metallothionein promoter, AMP gene expression can be induced by the addition of copper (Zhou et al., 2005). DIAP2 RNAi also inhibited Diptericin induction in this assay (Figure 2B), similar to RNAi targeting Uev1a and Ubc13 or TAK1, as shown previously (Zhou et al., 2005). In contrast, RNAi mediated knockdown of PGRP-LC did not inhibit IMD-induced signaling. These results suggest that DIAP2, TAK1, Ubc13 and Uev1a, but not PGRP-LC, function downstream of IMD.
Figure 2. DIAP2 functions between IMD and TAK1.

(A) S2* cells were treated with RNAi against various IMD pathway components, as indicated, prior to stimulation with E. coli peptidoglycan (PGN) for 6 hours. Induction of the AMP gene Diptericin was monitored by Northern blotting. rp49 levels were monitored as a loading control.
(B) S2* cells stably expressing metallothionein IMD were treated with RNAi against various IMD pathway components, as indicated. Then cells were stimulated with copper sulfate for 6 hours to induce IMD expression. Expression of the AMP gene Diptericin and rp49 were monitored by Northern blotting.
(C) The activation of TAK1 or IKK kinases was monitored by immunoprecipitation-kinase assays, after stimulation with E. coli PGN for 10 minutes. Recombinant MKK6K28A and recombinant Relish were used as substrates for TAK1 and IKK, respectively. RNAi was used to target various IMD pathway components, as indicated.
In addition, the activation of the two downstream kinases, TAK1 and IKK, was directly assayed by immunoprecipitation-kinase assays. PGN-induced activation of both kinases required DIAP2 (Figure 2C). Together these data argue that DIAP2 functions downstream of IMD but upstream of TAK1, similar to Ubc13 and Uev1a (Zhou et al., 2005).
Cleaved IMD associates with DIAP2 and is ubiquitinated
As IMD is rapidly cleaved following immune stimulation and DIAP2 appears to function immediately downstream, we hypothesized that these two proteins may associate. To that end, endogenous DIAP2 was immunoprecipitated from PGN-stimulated and unstimulated S2* cells. Endogenous IMD co-precipitated with DIAP2 in a signal dependent fashion (Figure 3A). Strikingly, DIAP2 preferentially associated with cleaved-IMD. Overall, the amount of IMD cleavage observed in whole cell lysates varied, ranging from 5% to 15% of the total, while the cleaved IMD was preferentially (50–70%) associated with immunoprecipitated DIAP2.
Figure 3. Association of DIAP2 and cleaved-IMD leads to IMD ubiquitination.

(A) Endogenous DIAP2 was immunoprecipitated from whole cell lysates prepared from S2* cells before or after a 10 minute stimulation with E. coli PGN, and associated IMD was monitored by immunoblotting (left lanes). For comparison, levels of full length and cleaved IMD in whole cell lysates are shown on the right. marks unmodified full length IMD, ◁ highlights phosphorylated IMD, and marks the cleaved-IMD products.
(B) Ubiquitination of endogenous IMD was monitored in lysates from S2* cells after stimulation with E. coli PGN for various times (as indicated) by anti-IMD immunoprecipitation followed by immunoblotting with anti-UbPan (top). An anti-IMD immunoblot serves as a loading control (bottom).
(C) Ubiquitination of endogenous DIAP2 was monitored in lysates from S2* cells after stimulation with E. coli PGN for various times (as indicated) by anti-DIAP2 immunorecipitation followed by immunoblotting with anti-UbPan. An anti-DIAP2 immunoblot serves as a loading control (bottom).
(D) Ubiquitination of endogenous IMD in PGN-stimulated S2* cells was monitored after RNAi targeting various pathway members (top). An anti-IMD immunoblot serves as a loading control (bottom).
(E) Ubiquitination of endogenous IMD after PGN-stimulation was monitored after treatment with RNAi targeting various E2-ubiquitin conjugating enzymes (top). An anti-IMD immunoblot serves as a loading control (middle). In parallel, IMD pathway activation was also monitored by Northern blotting for the AMP gene Diptericin. The ratio of Diptericin to Rp49 message was quantified by phosphoimager and plotted (bottom). Y-axis is arbitrary normalized phosphoimager units.
(F) Ubiquitination of endogenous IMD in S2* cells was monitored after treatment with zVAD-fmk, or vehicle control (DMSO) (top). An anti-IMD immunoblot serves as a loading control (bottom).
See also Supplemental Figures 2 and 3
These results, linking cleaved IMD and the E3-ligase DIAP2, suggest that one or both of these proteins may be conjugated with ubiquitin. Using immunoprecipitation followed by immunoblotting for total ubiquitin, IMD was found to be ubiquitinated rapidly and robustly after PGN stimulation of S2* cells (Figure 3B). DIAP2 was also ubiquitinated, but to a lesser extent (Figure 3C, Supplemental Figure 2A). This modification occurs within one minute of stimulation, reaching a maximum at 5–10 minutes, and is lost in approximately 30 minutes. IMD ubiquitination is stable after boiling in 1% SDS, further arguing that it is directly conjugated (Supplemental Figure 2B). Analysis of IMD cleavage, DIAP2 association, and ubiquitination from the same samples demonstrates that ubiquitination peaks shortly after cleavage and maximal DIAP2 association (Supplemental Figure 3).
In order to determine which members of the IMD pathway are required for PGN-induced IMD ubiquitination, S2* cells were treated with RNAi targeting various pathway components, and analyzed by IMD immunoprecipitation and ubiquitin immunoblotting. PGRP-LC, IMD, DREDD, FADD, and DIAP2 RNAi reduced IMD ubiquitination. Conversely, targeting downstream components, such as TAK1 or IKKγ, did not robustly affect IMD ubiquitination (Figure 3D). Previously, the E2 complex of Ubc13, and Uev1a was implicated in IMD pathway ubiquitination. RNAi treatment targeting both of these proteins (separately and together), reduced but did not eliminate the ubiquitination of IMD after stimulation (Figure 3E, lanes 6–8) similar to their partial effect on immune-induced Diptericin expression (Zhou et al., 2005 and Figure 3E). Recently, it has been suggested that another E2, Ubc5, may also form K63-ubiquitin chains during mammalian NF-κB signaling (Xia et al., 2009, Xu et al., 2009). Therefore, we determined if the Drosophila Ubc5 homologue, Effete, was also required for IMD ubiquitination. RNAi targeting Effete, in concert with Ubc13, or Uev1a (together or separately) were able to completely inhibit ubiquitination of IMD (Figure 3E, lanes 3–5). Similarly, when Diptericin induction was analyzed, RNAi targeting Uev1a, Bend or Effete alone or in pairs only partially inhibited IMD signaling, while treatment with all three RNAis nearly abolished immune-induced Diptericin expresion. (Figure 3E, bottom)
Consistent with above data, pretreatment with the caspase inhibitor zVAD-fmk reduced the amount of ubiquitinated IMD observed (Figure 3F). DIAP2 ubiquitination similarly required these same factors (Supplemental Figure 2C and 2D). Together, these results indicate that cleaved IMD and DIAP2 associate, triggering their Uev1a/Bend/Ubc5 dependent ubiquitination.
DIAP2 is the E3 for IMD K63-Ubiquitination in vivo
Strikingly, infection-induced IMD ubiquitination was readily detectable when IMD was immunoprecipitated from lysates extracted from adult flies, 30 minutes after septic infection with E. coli. As expected, diap2 null flies, diap27c, show no IMD-ubiquitination following infection with E. coli (Figure 4A). Previously, the RING finger of DIAP2 was shown to play an important role in the IMD pathway (Huh et al., 2007). Therefore, we next sought to determine if this domain was required for IMD-ubiquitination. Transgenic rescue flies, expressing wild type DIAP2 from the daughterless-gal4/UAS system were able to rescue the diap27c IMD ubiquitination phenotype. However, expression of a RING finger mutant of DIAP2 (DIAP2C466Y) was unable to rescue this phenotype (Figure 4A, right panels). These data demonstrate that IMD is ubiquitinated in a signal-dependent manner in flies, and that the DIAP2 RING finger is critical for this ubiquitination. These results strongly suggest that DIAP2 is the E3-ligase involved in IMD modification.
Figure 4. IMD is K63-polyubiquitinated.

(A) Ubiquitination of endogenous IMD from adult flies was monitored after infection with live E. coli (top). An anti-IMD immunoblot serves as a control (middle, marks unmodified full length IMD, ◁ highlights phosphorylated IMD, and marks the cleaved-IMD product). IMD ubiquitination was monitored in wildtype (DD1), diap27c (null), and diap27c expressing transgenic wildtype (WT) or RING-finger mutated (RF) DIAP2. DIAP2 immunoprecipitation/immunoblot verifies the presence/absence of DIAP2 protein in the various mutant Drosophila lines, as indicated.
(B) Total K63-linked (left) or K48-linked (right) polyubiquitin chains were immunoprecipiated from denatured cell lysates prepared from S2* cells stimulated with PGN for various times, as indicated. The presence of IMD was then monitored by IMD immunoblotting (top). Subsequently the same membranes were probed for total ubiquitin as a control (bottom).
The data implicating Ubc13 (bendless), Uev1a and Effete in this pathway (Zhou et al., 2005, work herein), suggest that IMD may be modified with K63-linked polyubiquitin chains. In order to clarify if IMD is conjugated with K48- or K63-polyubiquitin chains, we took advantage of two monoclonal antibodies that are specific to either K48- or K63-polyubiquitin chains (Newton et al., 2008). After denaturing lysis, these antibodies were used to immunoprecipitate ubiquitin conjugated proteins from PGN-stimulated S2 cells. Immunoprecipitated samples were then analyzed by immunoblotting for IMD. These experiments demonstrate that IMD is strongly K63-ubiquitinated in a PGN-inducible manner, peaking at approximately 10 minutes after stimulation (Figure 4B), while negligible IMD was detected in the K48-immunoprecipitated samples. Control probing of these blots, with an antibody that recognizes all ubiquitin forms, showed that the K63- and K48-specific antibodies immunopurified similar amounts of ubiquitin-conjugated material. These data demonstrate that IMD is conjugated with K63-linked polyubiquitin chains.
Non-cleavable IMD prevents signaling
We next sought to identify the IMD cleavage site and determine how IMD cleavage might be linked to association with DIAP2 and K63-ubiquitination. Based on experiments with either N- or C-terminally epitope-tagged IMD (data not shown) and the size of the cleaved product (~30 kDa), the site of cleavage was tentatively mapped to the N-terminal region of IMD. The involvement of the caspase DREDD further suggested a candidate cleavage site after residue 30, in the motif 27LEKD/A31. Wild type or D30A mutant versions of IMD, with epitope tags at both the C- and N-termini, were expressed in S2* cells by stable transfection, using the constitutive actin promoter. Double-tagged wild type IMD showed the expected cleavage and ubiquitination kinetics after stimulation with PGN, as detected with the C-terminal FLAG-tag (Figure 5A). The N-terminal T7-tag did not detect cleaved-IMD (data not shown). With the aspartate at the putative cleavage site substituted with alanine (D30A), no PGN-induced cleavage was observed (Figure 5A, lower panel, right lanes), suggesting that caspase-mediated cleavage of IMD occurs at this position. Furthermore, IMDD30A acted as a dominant negative, blocking the PGN-induced ubiquitination of endogenous IMD (Figure 5A, upper panel, right lanes), and blocking downstream IMD signaling as monitored by analysis of Diptericin expression (Figure 5B). Similarly, transgenic flies, over expressing IMDD30A show a marked inhibition of Diptericin induction following E. coli challenge (Figure 5C). These data strongly argue that cleavage of IMD occurs between residues 30 and 31, and that this cleavage is required for ubiquitination and activation of downstream target genes.
Figure 5. Uncleavable IMD is a dominant negative.

(A) Total ubiquitinated IMD was monitored by immunoprecipitation/Ubpan immublotting, in cells stably expressing either WT or mutant (D30A) IMD from the actin promoter (top). The lower anti-FLAG blot monitors cleavage of the WT or D30A IMD.
(B) Northern blot analysis of Diptericin expression was used to monitor IMD pathway signaling in cells over expressing WT or D30A IMD, before and after stimulation with E. coli PGN.
(C) Northern blot analysis of Diptericin expression was used to monitor IMD pathway signaling in flies expressing a transgenic copy of UAS-IMDD30A under the control of ubiquitous daughterless-Gal4 driver (top). Quantitation of these data is presented below.
(D) A map of IMD deletions used to analyze signaling activity is shown. The caspase cleavage (LEKD) and IAP binding motif (AAPV) domains are indicated. The C-terminal death domain is indicated in grey.
(E) Wile type IMD or IMDΔ5 was stably expressed in S2* cells from the copper inducible metallothionein promoter. Activation of immune signaling was monitored by Northern blotting for Diptericin. Rp49 blot serves as a loading control.
(F) Ubiquitination of wild type IMD or IMDΔ5 was monitored in two conditions. (Left) IMD protein was expressed at high level by addition of 500μM copper for 5 hours, identical to that seen in panel E. (Right) IMD protein was moderately expressed, with lower copper (250μM) for only 1 hour, and then cells were stimulated with PGN. In both cases, IMD was then immunoprecipitated with FLAG antibody, and ubiquitination levels were assayed by immunoblotting for ubiquitin. FLAG blot (bottom) serves as a control for the levels of IMD immunoprecipitated.
Using a series of IMD deletion mutants we also analyzed which regions of IMD where required for signal induced ubiquitination (Figure 5D). As shown previously, over expression of wild type IMD drives pathway signaling (Figure 2B). Interestingly only IMDΔ1 and IMDΔ5 were unable to drive signaling after over expression (Figure 5E and data not shown). The region removed in the IMDΔ1 construct contains the IMD cleavage site, which is critical for signaling, as shown above, via its involvement in DIAP2 interaction (see below for more details). In order to determine if the region removed in IMDΔ5 functions in ubiquitination, imd protein was analyzed in two ways. First, IMD was strongly over expressed from the metallothionein promoter, at a level strong enough to induce AMP expression (Figure 5F, left). Second, IMD was expressed at lower levels, before cells were stimulated with PGN (Figure 5F, right). In both cases wild type IMD shows robust ubiquitination after activation, while IMDΔ5 shows no ubiquitination over background. These data indicated that this region of IMD is crucial for signal induced ubiquitination and the induction of downstream target genes.
IMD Cleavage Exposes an IAP-Binding Motif
BIR domains, such as those found in DIAP2, preferentially bind to unblocked N-terminal alanines, generated either by removal of the initiating methionine or endoproteolytic cleavage. These exposed neo-N-terminal alanines are invariant components of IAP-binding motifs (IBMs), which also include a strong preference for proline at position 3 (Shi, 2002, 2004). In particular, the DIAP1 BIR1 domain associates with the neo-N-termini IBM of the cleaved caspases drICE and DCP-1 (Tenev et al., 2005). Likewise, cleavage of IMD at D30 exposes a putative IBM sequence with an initial alanine and a proline at position three (AAPV). Moreover, both the proposed caspase cleavage site and this IBM are highly conserved in the imd protein from 12 Drosophila species and the Anopheles mosquito (Figure 6A, highlighted in blue and red respectively).
Figure 6. IMD A31 is required for DIAP2 association.

(A) An alignment of IMD from 12 Drosophila species and the Anopheles mosquito show conservation in the caspase cleavage site (blue), and IAP-binding motif (red).
(B) The binding of WT or mutant cleaved-IMD proteins to DIAP2 was monitored using the ubiquitin-fusion technique. HA tagged full length (FL), A31-IMD31-273 (AAPV) or V31-IMD31-273 (VAPV) were co-expressed in cells along with GST-DIAP2. GST purification and immunoblotting for anti-HA were used to monitor IMD/DIAP2 association.
(C) Northern blot analysis of Diptericin expression was used to monitor IMD signaling in cells expressing WT, D30A or A31V IMD after treatment with RNAi to LacZ or imd 3′ UTR.
(D) Ubiquitination of endogenous IMD from WT (DD1) and various mutant adult flies was motioned after infection with live E. coli via IMD immunoprecipitation followed by UbPan immunoblotting (top). IMD immunoblot is shown as a loading control (bottom). marks unmodified full length IMD, ◁ highlights phosphorylated IMD, and marks the cleaved-IMD product.
(E) Association of cleaved IMD and DIAP2 mutants was monitored via GST-coprecpitation, as in panel B.
See also Supplemental Figure 4
Interestingly, the imd1 allele substitutes alanine 31 with valine, altering the key residue of the IBM. Although a fairly conservative change, this substitution generates a strong hypomorphic phenotype through unknown mechanisms (Georgel et al., 2001). To test whether cleaved IMD exposes a bona fide IBM we assessed whether DIAP2 interacted with cleaved-wild type IMD (A31) or cleaved-mutant IMD (V31). To this end, we used the ubiquitin-fusion technique to express IMD31-273 (Varshavsky, 2000), which generates IMD with an unblocked N-terminal IBM (See Supplemental Figure 4). Wild type A31-IMD31-273 robustly interacted with DIAP2 (Figure 6B, lane marked AAPV). However, V31-IMD31-273, equivalent to the cleavage product expected from the imd1 allele, showed markedly reduced binding to DIAP2 (Figure 6B, lane marked VAPV). Also, in transient transfections, full length IMD was spontaneously cleaved at a low level (Figure 6B, lower left panels), and only the cleaved product associated with cotransfected DIAP2, consistent with the data examining the endogenous proteins.
In order to verify the importance of the neo-N-terminal alanine on IMD pathway signaling, we over expressed IMD-A31V in S2* cells from the actin promoter. Cells were treated with RNAi against the 3′ UTR region of IMD, to remove the endogenous wild type protein, or LacZ (as a control) prior to stimulation with PGN. As expected, expression of IMD-D30A shows a strong dominant negative phenotype, consistent with previous data. On the other hand, cells expressing IMD-A31V and treated with the control LacZ RNAi showed a strong Diptericin induction. Treatment with IMD 3′UTR RNAi, which selectively degrades endogenous, but not ectopically expressed IMD, resulted in a near complete inhibition of Diptericin induction, showing that IMD-A31V is not sufficient for signaling in cells, as observed in flies (Figure 6C).
In order to probe whether or not these same mechanisms are involved in immune signaling in the whole animal, IMD cleavage and ubiquitination was also probed in the imd1 (A31V) strain. In lysates prepared from these flies, cleaved IMD was readily detected following E. coli infection but ubiquitinated IMD was completely absent (Figure 6D). In fact, the cleaved IMD product accumulates in imd1 animals, relative to the wild type control, presumably because cleaved IMD is rapidly and efficiently ubiquitinated in wild type but not imd1 animals. Likewise, cleaved IMD is more easily detected in the diap2 mutant strain (Figure 4A). On the other hand, neither cleaved nor ubiquitinated IMD was detectable in either dredd, or PGRP-LE;PGRP-LC mutant animals, consistent with the results from cell culture.
In order to determine which domains of DIAP2 are required for the binding of cleaved IMD, a series of DIAP2 mutations were generated. A C149G mutation of BIR2, predicted to abrogate its IBM binding (Ribeiro et al., 2007), reduced IMD31-273 association. Under the same conditions, mutation of the BIR3 domain (C249G) did not affect the interaction with IMD31-273. When both BIR2 and BIR3 domains were altered (C149/249G), IMD31-273 completely failed to interact (Figure 6E). As expected, the RING finger domain did not contribute to IMD binding, since its deletion had no noticeable effect on IMD31-273 binding. Together these results indicate that cleaved IMD carries a bona fide IBM at its neo-N-terminus, which preferentially binds to the BIR2 of DIAP2, and to some extent also binds the BIR3 domain. The alanine at the N-terminus of the IMD cleavage product is critical for this binding, while valine at this position, as in imd1, weakens the interaction.
Discussion
In previous work, we demonstrated that the caspase-8 like protease DREDD and its binding partner FADD are required upstream in the IMD pathway, at a position similar to Ubc13 and Uev1a (Zhou et al., 2005). However it was not clear from these studies if the protease activity of DREDD is also required in this role upstream in the IMD pathway. Here, we show that upon immune stimulation the imd protein is rapidly cleaved in a DREDD and FADD dependent manner. In fact, expression of DREDD, without immune stimulation, was sufficient to cause IMD cleavage. A caspase recognition site was identified in IMD, with cleavage predicted to occur after aspartate 30. Substitution of this residue with alanine prevents signal-induced cleavage and creates a dominant-negative allele of imd. This putative cleavage site in IMD (27LEKD/A31) is similar to the Relish cleavage site (542LQHD/G546), consistent with the notion that both proteins are cleaved by the same protease. Likewise, when IMD cleavage was blocked by caspase inhibitors, IMD was no longer ubiquitinated. Alignment of imd protein sequences from 12 Drosophila species and the Anopheles mosquito showed that the cleavage site is highly conserved (LEKD or LETD in all cases). These findings strongly argue that IMD cleavage after position 30 is mediated by DREDD, and that this cleavage is critical for further downstream signaling events.
Cleavage of IMD exposes a highly conserved IAP-binding motif (IBM) (Figure 6A), which then binds the BIR 2/3 domains of DIAP2. In the context of programmed cell death regulation, these IBM motifs are best defined by their neo-N-terminal alanine as well as the proline at position 3, both of which are also present in cleaved-IMD, supporting the notion that IMD includes an IBM starting at position 31. The notion that IMD carries an IBM also provides a molecular explanation for the hypomorphic phenotype observed in the imd1 mutant, which carries a valine substitution for this alanine at position 1 of cleaved IMD. Although several IAP proteins have been implicated in mammalian innate immune/NF-κB signaling (Deveraux and Reed, 1999; Verhagen et al., 2001; Bertrand et al., 2008, Bertrand et al., 2009), the significance of their associated BIR domains, as well as their possible binding to proteins with exposed IBMs, has remained largely unexplored. We show here, for the first time, that the BIR/IBM association plays a crucial role in innate immune NF-κB signaling in Drosophila. These findings present a unique role for the BIR-IBM interaction module outside of the cell death arena.
Furthermore, characterization of signaling in the imd1, diap2, dredd and PGRP-LC/LE mutant flies provides critical in vivo verification of the cell culture data and leads to the model presented in Figure 7. In particular, the molecular mechanism we propose suggests that immune stimulation leads to the DREDD-dependent cleavage of IMD, perhaps by recruiting IMD, FADD and DREDD to a receptor complex. Consistent with this aspect of the model, dredd mutants and receptor mutants failed to cleave (or ubiquitinate IMD) following infection. Once cleaved, the exposed IBM of IMD interacts with BIRs 2 & 3 of DIAP2. Currently, we do not know precisely where in the cell the IMD/DIAP2 association occurs. In figure 7, this interaction is diagramed as occurring away from the receptor proximal complex, but this is only for illustrative purposes. Once associated with DIAP2, cleaved IMD is rapidly K63-ubiquitinated. As the RING mutated version of diap2 failed to support IMD ubiquitination in flies, DIAP2 likely functions as the E3 for this reaction. Furthermore, the imd1 allele, which fails to interact with DIAP2 because of a mutation in the IBM, demonstrates the critical nature of the IMD-DIAP2 interaction for innate immune signaling. Consistent with the notion that cleavage precedes ubiquitination, mutants that fail to generate ubiquitinated IMD (i.e. diap2 and imd1) actually accumulate more cleaved IMD than is observed in wild type flies. Presumably, in wild type animals, cleaved IMD is efficiently ubiquitinated, and thus is difficult to detect in our assays. On the other hand, dredd mutants or mutants lacking the key immunoreceptors (PGRP-LC/LE) failed to cleave and ubiquitinate IMD, consistent with our cell culture data.
Figure 7. IMD pathway model.

DAP-type PGN binding causes multimerization or clustering of PGRP receptors. This likely recruits the adapter proteins IMD, FADD and the caspase DREDD. Once in proximity, DREDD cleaves IMD, generating an exposed neo-N-terminal A31 residue. This neo-N-terminus then binds the E3 ligase DIAP2 via its BIR2/3 domains. In conjuction with the E2-ubiquitin conjugating enzymes UEV1a, Bendless (Ubc13) and Effete (Ubc5), IMD (and to a lesser degree DIAP2) are then K63-polyubiquitinated. These polyubiquitin chains then induce the activation of downstream kinases ultimately leading to the phosphorylation and activation of Relish and induction of downstream targets, like the AMP genes.
Previous work has suggested that ubiquitination plays a critical role in IMD signaling in the Drosophila immune response (Gesellchen et al., 2005; Huh et al., 2007; Kleino et al., 2005; Leulier et al., 2006; Zhou et al., 2005). However, the molecular target(s) of ubiquitination and the mechanisms of its activation have remained elusive. As discussed above, the data presented here indicate that DIAP2 functions as the E3-ligase in the IMD pathway, a function usually attributed to the TRAF or, more recently, cIAP proteins in mammalian NF-κB signaling pathways (Bertrand et al., 2009). The E2 complex of Ubc13 and Uev1a also appears to be involved in IMD ubiquitination. RNAi targeting of these K63-ubiquitinating enzymes reproducibly decreases IMD ubiquitination and the induction of target genes, however the degree of inhibition is variable and never complete (data herein and Zhou et al., 2005). We now show that a third E2 enzyme, Effete, the Drosophila Ubc5 homologue, also plays a vital role in ubiquitination of IMD. RNAi treatment targeting Effete, in concert with Uev1a and/or Bendless reproducibly eliminated IMD ubiquitination and the induction of Diptericin.
Several lines of evidence argue that IMD is the critical target for K63-ubiqutination in this pathway. First, IMD is by far, the most robustly modified component that we have identified, and the only one in which modifications can be detected in whole animals. Second, the protein produced as a result of the imd1 mutation, which does not signal, is also not ubiquitinated. Third, we present a deletion mutant, IMDΔ5, which is not ubiquitinated and fails to signal. Finally, Thevenon et al. (2009) recently identified the Drosophila ubiquitin specific protease, USP36, as a negative regulator of IMD ubiquitination. Functionally, USP36 is able to remove K63-polyubiquitin chains from IMD, promoting K48 mediated polyubiquitination and degradation of IMD. Consistent with our model, animals which over express USP36 show decreased levels of IMD ubiquitination, reduced IMD pathway activation as monitored by Diptericin RNA expression, and are susceptible to bacterial infection. Together, these data strongly argue that IMD is the critical substrate for K63-polyubiqutination in IMD pathway signaling, although other proteins may also be conjugated to lesser degree (as shown here for DIAP2) and could potentially substitute for IMD as the platform for ubiquitin conjugation. Interestingly, Chen and colleagues recently showed that unanchored K63-polyubiquitin chains (i.e. ubiquitin chains which are not conjugated to a target substrate) are sufficient to activate the mammalian TAK1 and IKK kinase complexes. Furthermore they show that unanchored polyubiquitin chains are produced after stimulation of HEK cells with IL-1β (Xia et al., 2009). Thus, the presence (or absence) of K63-polyubiquitin chains may be more important than their conjugation substrate.
K63-polyubiquitin chains are likely to serve as scaffolds to recruit the key kinases TAK1 and IKK, in the IMD pathway. Both of these kinases include regulatory subunits with highly conserved K63-polyubiquitin binding domains. Drosophila TAB2, which complexes with TAK1, and the IKKγ subunit are predicted to contain conserved K63-polyubiquitin binding domains (Ea et al., 2006; Kleino et al., 2005; Zhou et al., 2005; Zhuang et al., 2006). Thus, we hypothesize that K63-polyubiquitin chains will recruit both the TAB2/TAK1 complex and the IKK complex, creating a local environment for optimal kinase activation and signal transduction, however this aspect of our model is still speculative.
Although mammalian caspase-8 and FADD are best known for their role in apoptosis, a growing body of literature indicates that these factors, along with RIP1 (which has some homology to IMD), also function in RIG-I signaling to NF-κB (Balachandran et al., 2004; Takahashi et al., 2006). In addition, caspase-8 has been implicated in NF-κB signaling in B-cell, T-cell and LPS signaling (Bidere et al., 2006; Chun et al., 2002; Lemmers et al., 2007; Salmena et al., 2003; Su et al., 2005; Sun et al., 2008). Cells, from mice or humans, lacking caspase-8 have defects in immune activation, cytokine production and nuclear translocation of NF-κB p50/p65 (Chun et al., 2002; Lemmers et al., 2007). Furthermore, recent evidence also shows that during mammalian NOD signaling the RIP2 protein is ubiquitinated in a cIAP1/2 dependent manner (Bertrand et al., 2009). Given that Drosophila homologs of RIP1, FADD and capase-8 also function in the IMD pathway, the results presented here may help elucidate the mechanism by which these factors function in these mammalian immune signaling pathways.
Experimental Procedures
Fly Stocks
All fly strains, except the IMDD30A line, were previously published (Supplemental Experimental Procedures). The UAS-IMDD30A line was generated by standard P-element mediated transformation.
RNAi
dsRNA used in this work was produced using T7 RiboMAX Express Large Scale RNA Production System (Promega). S2* cells were treated with RNAi delivered by calcium phosphate transfection, see supplemental Experimental Procedures for details.
RNA Analysis
Total RNA was isolated with the TRIzol reagent (Invitrogen) and gene expression was analyzed by Northern blotting, as previously described (Silverman et al., 2000).
IMD Antibody
For the production of anti-IMD polyclonal antibodies, the full-length IMD coding sequence was cloned in the E. coli expression vector pDS56/RBII, 6xhis. His-tagged-IMD was expressed in M15 E. coli (strain M15), purified by Ni-chromatography and used to immunize rabbits.
TAK Antibody
For the production of the anti-TAK polyclonal antibody, the peptide KSDGRERLTVTDTKP was generated and used to immunize rabbits (Open Biosystems, Inc.). Serum was then pooled and affinity purified.
Protein and Immunoprecipitation Assays
Following stimulation S2* cells were lysed in standard lysis buffer (see Supplemental Experimental Procedures). For total protein analysis 50–100μg of total lysate were immunoblotted. For co-immunoprecipitation analysis, 200–600μg of total cell lysate, or 600–900μg of fly lysate, was subjected to precipitation with appropriate agarose cross-linked antibody or with antibody and protein A agarose (Sigma).
K48- and K63-polyubiquitin Immunoprecipitation Assays
Were performed as described previously (Newton et al. 2008) with minor modification for Drosophila cells (Supplemental Experimental Procedures).
Kinase Assays
The kinase activity of IKK and TAK1 was assayed by immunoprecipitating kinases using antisera to TAK1, IKKγ or anti-FLAG (Sigma), as indicated. Standard kinase assay conditions were then used to monitor activity, see Supplemental Experimental Procedures for details
Caspase Inhibitor Treatment
S2* cells were split 0.5×106 cells per ml and 20 hours later 1μM 20-hydroxyecdysone was added. 24 hours later, 100μM caspase inhibitor zVAD-fmk, dissolved in DMSO, was then added to cells for 30 minutes or 4 hours prior to stimulation with E. coli peptidoglycan.
IMD/DIAP2 Pull Down
S2 cells were transfected with pAc myc-Imd-HA or the pMT DHFR-HA-Ub-Imd (31-273) constructs with or without pMT-DIAP2-GTC. IMD/DIAP2 expression was induced by the addition of CuSO4 and cells were lysed (see Supplemental Experimental Procedures). DIAP2 was then purified using GSH-Sepharose (GE Healthcare), eluted and immunoblotted as indicated.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balachandran S, Thomas E, Barber GN. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432:401–405. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Bidere N, Snow AL, Sakai K, Zheng L, Lenardo MJ. Caspase-8 regulation by direct interaction with TRAF6 in T cell receptor-induced NF-kappaB activation. Curr Biol. 2006;16:1666–1671. doi: 10.1016/j.cub.2006.06.062. [DOI] [PubMed] [Google Scholar]
- Chang CI, Chelliah Y, Borek D, Mengin-Lecreulx D, Deisenhofer J. Structure of Trachael Cytotoxin in Complex with a Heterodimeric Pattern-Recognition Receptor. Science. 2006;311:1761–1764. doi: 10.1126/science.1123056. [DOI] [PubMed] [Google Scholar]
- Chiu YH, Zhao M, Chen ZJ. Ubiquitin in NF-kappaB Signaling. Chemical reviews. 2009 doi: 10.1021/cr800554j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Erturk-Hasdemir D, Broemer M, Leulier F, Lane WS, Paquette N, Hwang D, Kim CH, Stoven S, Meier P, Silverman N. Two roles for the Drosophila IKK complex in the activation of Relish and the induction of antimicrobial peptide genes. Proc Natl Acad Sci U S A. 2009;106:9779–9784. doi: 10.1073/pnas.0812022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nature reviews. 2007;7:862–874. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- Georgel P, Naitza S, Kappler C, Ferrandon D, Zachary D, Swimmer C, Kopczynski C, Duyk G, Reichhart JM, Hoffmann JA. Drosophila immune deficiency (IMD) is a death domain protein that activates antibacterial defense and can promote apoptosis. Dev Cell. 2001;1:503–514. doi: 10.1016/s1534-5807(01)00059-4. [DOI] [PubMed] [Google Scholar]
- Gesellchen V, Kuttenkeuler D, Steckel M, Pelte N, Boutros M. An RNA interference screen identifies Inhibitor of Apoptosis Protein 2 as a regulator of innate immune signalling in Drosophila. EMBO Rep. 2005;6:979–984. doi: 10.1038/sj.embor.7400530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Foe I, Muro I, Chen CH, Seol JH, Yoo SJ, Guo M, Park JM, Hay BA. The Drosophila inhibitor of apoptosis (IAP) DIAP2 is dispensable for cell survival, required for the innate immune response to gram-negative bacterial infection, and can be negatively regulated by the reaper/hid/grim family of IAP-binding apoptosis inducers. J Biol Chem. 2007;282:2056–2068. doi: 10.1074/jbc.M608051200. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Yano T, Aggarwal K, Lim JH, Ueda K, Oshima Y, Peach C, Erturk-Hasdemir D, Goldman WE, Oh BH, et al. PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat Immunol. 2006;7:715–723. doi: 10.1038/ni1356. [DOI] [PubMed] [Google Scholar]
- Kleino A, Valanne S, Ulvila J, Kallio J, Myllymaki H, Enwald H, Stoven S, Poidevin M, Ueda R, Hultmark D, et al. Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. Embo J. 2005;24:3423–3434. doi: 10.1038/sj.emboj.7600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B, Hoffmann J. The Host Defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, et al. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J Biol Chem. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- Leulier F, Lhocine N, Lemaitre B, Meier P. The Drosophila inhibitor of apoptosis protein DIAP2 functions in innate immunity and is essential to resist gram-negative bacterial infection. Mol Cell Biol. 2006;26:7821–7831. doi: 10.1128/MCB.00548-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Kim MS, Kim HE, Yano T, Oshima Y, Aggarwal K, Goldman WE, Silverman N, Kurata S, Oh BH. Structural Basis for Preferential Recognition of Diaminopimelic Acid-type Peptidoglycan by a Subset of Peptidoglycan Recognition Proteins. J Biol Chem. 2006;281:8286–8295. doi: 10.1074/jbc.M513030200. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Ribeiro PS, Kuranaga E, Tenev T, Leulier F, Miura M, Meier P. DIAP2 functions as a mechanism-based regulator of drICE that contributes to the caspase activity threshold in living cells. The Journal of cell biology. 2007;179:1467–1480. doi: 10.1083/jcb.200706027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–895. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. A conserved tetrapeptide motif: potentiating apoptosis through IAP-binding. Cell death and differentiation. 2002;9:93–95. doi: 10.1038/sj.cdd.4400957. [DOI] [PubMed] [Google Scholar]
- Shi Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci. 2004;13:1979–1987. doi: 10.1110/ps.04789804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Erlich RL, Hunter M, Bernstein E, Schneider D, Maniatis T. Immune activation of NF-kappaB and JNK requires Drosophila TAK1. J Biol Chem. 2003;278:48928–48934. doi: 10.1074/jbc.M304802200. [DOI] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Stöven S, Pandey N, Hultmark D, Maniatis T. A Drosophila IkappaB kinase complex required for Relish cleavage and antibacterial immunity. Genes Dev. 2000;14:2461–2471. doi: 10.1101/gad.817800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöven S, Silverman N, Junell A, Hedengren-Olcott M, Erturk D, Engstrom Y, Maniatis T, Hultmark D. Caspase-mediated processing of the Drosophila NF-{kappa}B factor Relish. Proc Natl Acad Sci U S A. 2003 doi: 10.1073/pnas.1035902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, Kong L, Xu L, Hilliard B, Hu S, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133:415–426. doi: 10.1016/j.cell.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Kawai T, Kumar H, Sato S, Yonehara S, Akira S. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol. 2006;176:4520–4524. doi: 10.4049/jimmunol.176.8.4520. [DOI] [PubMed] [Google Scholar]
- Tenev T, Zachariou A, Wilson R, Ditzel M, Meier P. IAPs are functionally non-equivalent and regulate effector caspases through distinct mechanisms. Nature cell biology. 2005;7:70–77. doi: 10.1038/ncb1204. [DOI] [PubMed] [Google Scholar]
- Thevenon D, Elodie E, Avet-Rochex A, Gottar M, Bergeret E, Tricoire H, Benaud C, Baudier J, Taillebourg E, Fauvarque M. The Drosophila Ubiquitin Specific Protease dUSP36/Scny targets IMD to prevent constitutive immune signalling. Cell Host and Microbes. 2009 doi: 10.1016/j.chom.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. Ubiquitin fusion technique and its descendants. Methods in enzymology. 2000;327:578–593. doi: 10.1016/s0076-6879(00)27303-5. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–297. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Coulson EJ, Vaux DL. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome biology. 2001;2:REVIEWS3009. doi: 10.1186/gb-2001-2-7-reviews3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes Dev. 2001;15:1900–1912. doi: 10.1101/gad.203301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Skaug B, Zeng W, Chen ZJ. A Ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol Cell. 2009;36:302–314. doi: 10.1016/j.molcel.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Silverman N, Hong M, Liao DS, Chung Y, Chen ZJ, Maniatis T. The role of ubiquitnation in Drosophila innate immunity. J Biol Chem. 2005 doi: 10.1074/jbc.M506655200. [DOI] [PubMed] [Google Scholar]
- Zhuang ZH, Sun L, Kong L, Hu JH, Yu MC, Reinach P, Zang JW, Ge BX. Drosophila TAB2 is required for the immune activation of JNK and NF-kappaB. Cell Signal. 2006;18:964–970. doi: 10.1016/j.cellsig.2005.08.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.