

Abstract
Background and purpose:
Cilostazol is a specific inhibitor of 3′-5′-cyclic adenosine monophosphate (cAMP) phosphodiesterase, which is widely used to treat ischemic symptoms of peripheral vascular disease. Although cilostazol has been shown to exhibit vasodilator properties as well as antiplatelet and anti-inflammatory effects, its cellular mechanism in microglia is unknown. In the present study, we assessed the anti-inflammatory effect of cilostazol on the production of pro-inflammatory mediators in lipopolysaccharide (LPS)-stimulated murine BV2 microglia.
Experimental approach:
We examined the effects of cilostazol on LPS-induced nuclear factor-kappaB (NF-κB) activation and phosphorylation of mitogen-activated protein kinases (MAPKs).
Key results:
Cilostazol suppressed production of nitric oxide (NO), prostaglandin E2 (PGE2) and the proinflammatory cytokines, interleukin-1 (IL-1), tumour necrosis factor-α, and monocyte chemoattractant protein-1 (MCP-1), in a concentration-dependent manner. Inhibitory effects of cilostazol were not affected by treatment with an adenylate cyclase inhibitor, SQ 22536, indicating that these actions of cilostazol were cAMP-independent. Cilostazol significantly inhibited the DNA binding and transcriptional activity of NF-κB. Moreover, cilostazol blocked signalling upstream of NF-κB activation by inhibiting extracellular signal-regulated kinases 1 and 2 (ERK1/2) and c-Jun N-terminal kinase (JNK), but without affecting the activity of p38 MAPK.
Conclusion and implications:
Our results demonstrate that suppression of the NF-κB, ERK, JNK signalling pathways may inhibit LPS-induced NO and PGE2 production. Therefore, cilostazol may have therapeutic potential for neurodegenerative diseases by inhibiting pro-inflammatory mediators and cytokine production in activated microglia.
Keywords: cilostazol, inducible nitric oxide synthase, cyclooxygenase-2, nuclear factor-κB, monocyte chemoattractant protein-1, cAMP
Introduction
Cilostazol is a drug that was first introduced to increase the intracellular level of 3′-5′-cyclic adenosine monophosphate (cAMP) by blocking its hydrolysis by phosphodiesterase 3 (PDE3) (Kimura et al., 1985). It is an inhibitor of platelet aggregation and a vasodilator, useful for the treatment of ischemic symptoms of peripheral vascular disease (Kimura et al., 1985; Yasuda et al., 1985; Dawson et al., 1998). Besides its antiplatelet and vasodilator properties, cilostazol delays the onset of atherosclerosis (da Rosa et al., 2006), protects vascular endothelium, inhibits vascular smooth muscle cell proliferation in vitro and has been shown to suppress neointimal formation in the balloon-injured rat carotid artery (Takahashi et al., 1992; Ishizaka et al., 1999). In addition, cilostazol exerts neuroprotective effects by reducing cerebral hypoperfusion and focal cerebral ischemic damage in rodents (Honda et al., 2006; Watanabe et al., 2006; Lee et al., 2008). This compound was also shown to prevent lipopolysaccharide (LPS)-stimulated nitric oxide (NO) and cytokine production in microglia (Yoshikawa et al., 1999; Yoshikawa et al., 2002), although its functional mechanisms as well as its role in prostaglandin E2 (PGE2) production in these cells are not completely understood.
Microglia are specialized macrophages, widely distributed in the brain. Microglia comprise approximately 10–20% of the total glial cells in the adult central nervous system (CNS) (Fanarraga et al., 2009). Microglia may play a dual role, participating in host defence of the brain as well as acting as phagocytes to engulf tissue debris and dead cells. Microglia can also augment neuroinflammation by secreting various neurotoxic and pro-inflammatory mediators in chronic brain diseases, causing neuronal death and demyelination (Glezer et al., 2007). In response to brain injury or neuroinflammatory stimuli, microglia may overproduce proinflammatory and/or cytotoxic factors, including NO, PGE2, monocyte chemoattractant protein-1 (MCP-1), interleukin-1 (IL-1), IL-6 and tumour necrosis factor-α (TNF-α). These factors are characteristic of various neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), trauma, multiple sclerosis (MS) and cerebral ischemia (McGeer and McGeer, 1995; Gonzalez-Scarano and Baltuch, 1999). Reduction of proinflammatory mediators in microglia could attenuate the severity of these disorders (Liu and Hong, 2003; Eikelenboom and van Gool, 2004). These results indicate that activated microglia are a major cellular source of proinflammatory and/or cytotoxic factors that cause neuronal damage in the CNS. Therefore, controlling microglial activation has been considered to be an important therapeutic strategy for the treatment of many neuroinflammatory diseases.
Lipopolysaccharide, a bacterial endotoxin, initiates a number of major cellular effects that play critical roles in the pathogenesis of inflammatory responses and has been employed to induce microglial activation during infection by Gram-negative bacteria. LPS stimulation of the microglia is therefore a useful model for the study of mechanisms underlying neuronal injury by various pro-inflammatory and neurotoxic factors released by activated microglia (Kim et al., 2004a,b;). Several studies have reported that excessive production of these proinflammatory factors has emerged as a critical determinant of the cytotoxicity associated with inflammation in pathologies such as AD, PD, trauma, cerebral ischemia and MS (Misko et al., 1995; Pasinetti and Aisen, 1998; Tomimoto et al., 2000). Based on this information, pharmacological inhibition of these inflammatory mediators is an important target for potential treatment of several neurodegenerative disorders associated with inflammatory processes.
In the present study, we investigated the anti-inflammatory effects and mechanisms of action of cilostazol on LPS-activated production of pro-inflammatory and cytotoxic factors in murine BV2 microglia. Our findings suggest that cilostazol may be a therapeutic candidate for the treatment of various neurodegenerative diseases.
Methods
Cell culture
The murine BV2 cell line was maintained in DMEM supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin at 37°C in a humidified incubator with 5% CO2. Confluent cultures were passaged by trypsinization. For the experiments, cells were washed twice with warm DMEM (without phenol red) and treated in serum-free medium for at least 4 h before treatments. In all experiments, cells were treated with cilostazol for the indicated times before addition of LPS (1 µg·mL−1).
Cell viability assay
Cell viability was measured based on the formation of blue formazan metabolized from colourless MTT by mitochondrial dehydrogenases, which are active only in live cells. BV2 cells were plated into 24-well plates at a density of 2 × 105 cells per well for 24 h, and then washed. Cells incubated with various concentrations of cilostazol were treated with LPS (Escherichia coli 026:B6) for 24 h, and then incubated in 0.5 mg·mL−1 MTT solution. Three hours later, the supernatant was removed and formation of formazan was measured at 540 nm using a microplate reader (Dynatech MR-7000; Dynatech Laboratories).
Intracellular cAMP measurements
The cAMP levels were measured using a Parameter™ ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's protocols. The absorbance at 450 nm was determined using a microplate reader.
NO production
Concentrations of NO in the culture supernatants were determined by measuring nitrite, a major stable product of NO, using the Griess reagent (1% sulfanilamide/0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5% H3PO4). Cells (5 × 105 cells·mL−1) were stimulated in 24-well plates for 24 h, and then 100 µL of each culture medium was mixed with an equal volume of Griess reagent. Nitrite levels were determined using an enzyme-linked immunosorbent assay (ELISA) plate reader at 540 nm (Dynatech MR-7000; Dynatech Laboratories), and nitrite concentrations were calculated by reference to a standard curve generated by known concentrations of sodium nitrite.
RNA isolation and RT-PCR
BV2 cells were plated overnight in 12-well culture plates at a density of 2 × 105 cells per well, and then the cells were further incubated in serum-free medium for at least 4 h before treatments. Total RNA was isolated using TRIzol reagent (Invitrogen, CA, USA). The total RNA (1.0 µg) obtained from cells was reverse-transcribed using M-MLV reverse transcriptase (Promega, Madison, WI, USA) to produce cDNAs. The inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), IL-1β, TNF-α and MCP-1 genes were amplified from the cDNA by PCR. PCR primers were as follows: mouse iNOS (5′-ATGTCCGAAGCAAACATCAC-3′ and 5′-TAATGTCCAGGAAGTAGGTG-3′), COX-2 (5′-CAGCAAATCCTTGCTGTTCC-3′ and 5′-TGGGCAAAGAATGCAAACATC-3′), IL-1β (5′-ATGGCAACTGTTCCTGAACTCAACT-3′ and 5′-TTTCCTTTCTTAGATATGGACAGGAC-3′), TNF-αα(5′-ATGAGCACAGAAAGCATGATC-3′ and 5′-TACAGGCTTGTCACTCGAATT-3′) and MCP-1 (5′-CCTGCTGTTCACAGTTGCC-3′ and 5′-TGAGGTGGTTGTGGAAAAGG-3′). After amplification, PCR reactions were electrophoresed in an agarose gel.
Western blot analysis
BV2 cells were plated overnight in 6-well culture plates at a density of 4 × 105 cells per well, then the cells were further incubated in serum-free medium for at least 4 h before treatments. Cells were washed three times with PBS and lysed in lysis buffer (1% Triton X-100, 1% deoxycholate, 0.1% NaN3) containing protease inhibitor cocktail tablets (Roche Diagnostics, Mannheim, Germany). Equal amounts of protein were separated on 10% SDS-polyacrylamide minigels. Proteins were transferred to Immobilon PVDF membranes (Millipore) and subsequently blocked in 5% bovine serum albumin (BSA)-Tris-buffered saline Tween (TBST, 100 mM Tris, pH 8.0, 150 mM NaCl and 0.1% Tween 20) for 1 h at room temperature. Specific antibodies against inducible NO synthase (iNOS), COX-2, p65 (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), extracellular signal-regulated kinase (ERK), phosphorylated (p)-ERK 1/2, p38, p-p38, c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK) and p-JNK/SAPK (1:1000; Cell Signaling Technology, Beverly, MA, USA) were diluted in 5% BSA-TBST. After incubation with the appropriate primary antibody, membranes were incubated for 1 h at room temperature with a secondary antibody conjugated to horseradish peroxidase (1:10 000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Following three washes in TBST, immunoreactive bands were visualized using the ECL detection system (Pierce, Rockford, IL, USA). In a parallel experiment, nuclear protein was prepared using nuclear extraction reagents (Pierce) according to the manufacturer's protocol.
Cytokine assays
Cytokine levels were determined by ELISA. ELISA kits from R&D Systems were used to measure IL-1β, TNF-α and MCP-1 levels, and a kit from Cayman Chemical (Ann Arbor. MI, USA) was used to measure PGE2 levels. The absorbance at 450 nm was determined using a microplate reader.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared using NE-PER nuclear extraction reagent (Pierce). An oligonucleotide containing the immunoglobulin κ-chain binding site (κB, 5′-CCGGTTAACAGAGGGGGCTTTCCGAG-3′) was synthesized as a probe for the gel retardation assay, and the probe was labelled with biotin (Pierce). The binding reactions contained 10 µg of nuclear extract protein, buffer (10 mM Tris, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM dithiothreitol, 0.05% Nonidet P-40 and 2.5% glycerol), 50 ng of poly (dI-dC) and 20 fM of biotinylated DNA. The reactions were incubated for 20 min at room temperature in a final volume of 20 µL. In all experiments, DNA-binding specificity was verified using 100-fold excess of unlabelled κB to the reaction mixture. The reaction mixture was analysed by electrophoresis in a 5% polyacrylamide gel in 0.5× Tris-borate buffer (Tris-HCl 89 mM, boric acid 89 mM, EDTA 2 mM, pH 8.0). The reactions were transferred to nylon membranes and the biotinylated DNA was detected using a LightShift chemiluminescent EMSA kit (Pierce).
Confocal laser scanning microscopy study
Nuclear factor-kappaB p65 nuclear localization was detected by indirect immunofluorescence assays using confocal microscopy. BV2 microglia were cultured directly on glass coverslips in 24-well plates for 24 h. After stimulation with 1 µg·mL−1 of LPS and/or 30 µg·mL−1 of cilostazol, cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.2% Triton X-100 in PBS and blocked with 1.5% normal donkey serum (Sigma). Polyclonal antibodies against NF-κB p65 (1 µg per well) were applied for 1 h, followed by a 1 h incubation with FITC-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). After washing with PBS, the coverslips were mounted with Fluoromount-G (Southern Biotechnology Associates Inc., Birmingham, AL, USA), and the fluorescence was visualized using a Zeiss LSM 510 laser scanning confocal device.
Statistical analyses
Data values represent means ± SEM Statistical significance was determined by analysis of variance followed by Scheffe's test. A value of P < 0.05 was accepted as statistically significant.
Materials
Cilostazol, forskolin, MTT, Griess reagents and LPS (E. coli 026:B6) were purchased from Sigma (St. Louis, MO, USA). Dibutyryl-cAMP and SQ 22536 were obtained from BIOMOL Research Laboratories Inc. (Plymouth Meeting, PA, USA). Drug and molecular target nomenclature conforms to Alexander et al. (2008).
Results
Effect of cilostazol on NO and PGE2 production in LPS-stimulated BV2
Initially, we wished to evaluate the effect of cilostazol on NO and PGE2 production in LPS-stimulated BV2 microglia. To determine NO production, we measured nitrite released into the culture medium using the Griess reagent. BV2 microglia were treated with various concentrations of cilostazol (0, 10, 20 or 30 µM) for 6 h before addition of LPS (1 µg·mL−1). Incubation with LPS alone markedly increased (about 15-fold) NO production from the cells, compared with that generated under control conditions (Figure 1A). However, pretreatment with cilostazol prevented this increase in levels of NO production in LPS-stimulated BV2 microglia in a concentration-dependent manner (Figure 1A). Significant repression was observed with 10 µM of cilostazol.
Figure 1.
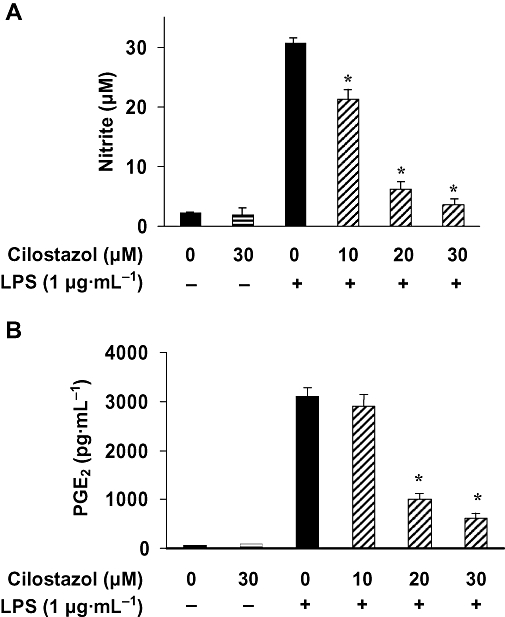
Inhibition of NO and PGE2 production by cilostazol in LPS-stimulated BV2 microglia. BV2 microglia were pretreated with various concentrations of cilostazol (10, 20 and 30 µM) for 6 h before incubation with LPS (1 µg·mL−1) for 24 h. Nitrite content was measured using the Griess reaction (A) and the PGE2 concentration in culture media was measured using a commercial ELISA kit (B). Each value indicates the mean ± SEM, and is representative of results obtained from four independent experiments. *P < 0.05 indicates a significant difference from cells treated with LPS in the absence of cilostazol. ELISA, enzyme-linked immunosorbent assay; LPS, lipopolysaccharide; NO, nitric oxide; PGE2, prostaglandin E2.
Prostaglandin E2 represents the most important inflammatory product of COX-2 activity; therefore, we quantified the PGE2 levels present in the supernatant. To assess whether cilostazol could inhibit production of LPS-induced PGE2 in BV2 microglia, cells were pretreated with cilostazol for 6 h and then stimulated with 1 µg·mL−1 of LPS. After incubation for 24 h, the cell culture medium was harvested, and the production of PGE2 was measured using ELISA. As shown in Figure 1B, the amount of PGE2 present in the culture medium increased baut 50-fold after 24 h of exposure to LPS alone. Pretreatment with cilostazol (10–30 µg·mL−1) decreased PGE2 synthesis, concentration dependently.
The cytotoxic effects of cilostazol in BV2 microglia were evaluated in the absence or presence of LPS using MTT assays. The concentrations (10–30 µM) used to inhibit NO and PGE2 production did not affect cell viability (Figure 2). Thus, concentrations of 10–30 µM cilostazol were used in subsequent experiments.
Figure 2.
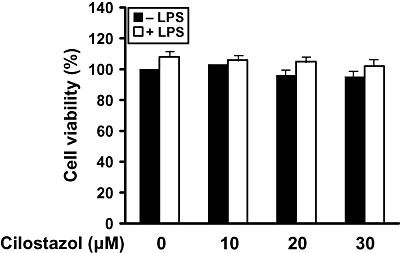
Effect of cilostazol on the viability of BV2 microglia. Cells were treated with the indicated concentrations of cilostazol (10, 20 and 30 µM) for 6 h prior to LPS (1 µg·mL−1) treatment for 24 h. Cell viability was assessed by MTT reduction assays, and the results are expressed as the percentage of surviving cells over control cells (no addition of cilostazol). Each value indicates the mean ± SEM and is representative of results obtained from three independent experiments. LPS, lipopolysaccharide.
Effect of cilostazol on LPS-induced expression of protein and mRNA for iNOS and COX-2
As cilostazol was found to inhibit NO and PGE2 production, we examined the relationship between cilostazol levels and the expression of iNOS and COX-2. The inhibitory effects of cilostazol on the expression of iNOS and COX-2 protein and mRNA were determined by Western blot analysis and RT-PCR respectively. Levels of iNOS and COX-2 proteins were markedly up-regulated after 24 h of LPS (1 µg·mL−1) treatment, and cilostazol significantly attenuated iNOS and COX-2 protein expression in LPS-stimulated BV2 microglia in a concentration-dependent manner (Figure 3A). The effects of cilostazol on iNOS and COX-2 mRNA expression were also evaluated (Figure 3B). RT-PCR analysis showed that the reduction in iNOS and COX-2 mRNAs correlated with the reduction in the corresponding protein levels.
Figure 3.
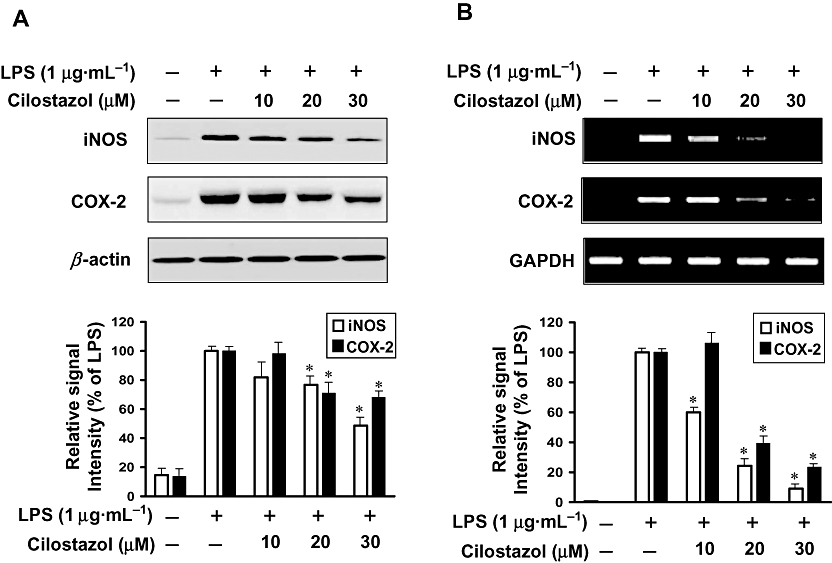
Inhibition of iNOS and COX-2 protein (A) and mRNA (B) expression by cilostazol in LPS-stimulated BV2 microglia. (A) BV2 microglia were pretreated with cilostazol (10, 20 and 30 µM) 6 h prior to incubation with LPS (1 µg·mL−1) for 24 h. Cell lysates were then prepared and Western blots were performed using an antibodies specific for murine iNOS or COX-2. (B) After LPS treatment for 6 h, total RNA was prepared for RT-PCR analysis of iNOS and COX-2 gene expression in LPS-stimulated BV2 microglia. β-actin and GAPDH were used as internal controls for Western blot analysis and RT-PCR assays respectively. The experiment was repeated three times and similar results were obtained. Each value indicates the mean ± SEM and is representative of results obtained from five independent experiments. *P < 0.05 indicates a significant difference from cells treated with LPS in the absence of cilostazol. COX-2, cyclooxygenase-2; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide.
Effect of cilostazol on cAMP production in LPS-stimulated BV2 microglia
Cilostazol is a potent inhibitor of PDE3 and thus increases the intracellular levels of cAMP. To evaluate a possible involvement of cAMP in the intracellular signalling pathway related to the inhibitory effect of cilostazol, we examined the effects of cilostazol on intracellular cAMP levels in BV2 microglia. Intracellular cAMP levels were slightly increased by LPS alone. Regardless of LPS presence, cilostazol increased cAMP levels (Figure 4A). We next examined the effect of forskolin (a direct activator of adenylate cyclase), dibutyryl-cAMP (a cell-permeable cAMP analogue) on the expression of iNOS and COX-2. Neither forskolin and dibutyryl-cAMP also had any effect on LPS induced inflammatory responses (Figure 4B). In addition, in order to verify the cAMP-dependent or -independent action of cilostazol, we examined the effects of SQ 22536 (an adenylate cyclase inhibitor), irreversible and selective cAMP antagonist, on the expression of iNOS and COX-2 production. This study found that SQ 22536 did not reverse the inhibitory effect of cilostazol on the expression of iNOS and COX-2 (Figure 4C). These results suggested that the intracellular cAMP signalling pathway could not be involved in the inhibitory effect of cilostazol on LPS-induced iNOS and COX-2 expression in the BV2 microglia.
Figure 4.
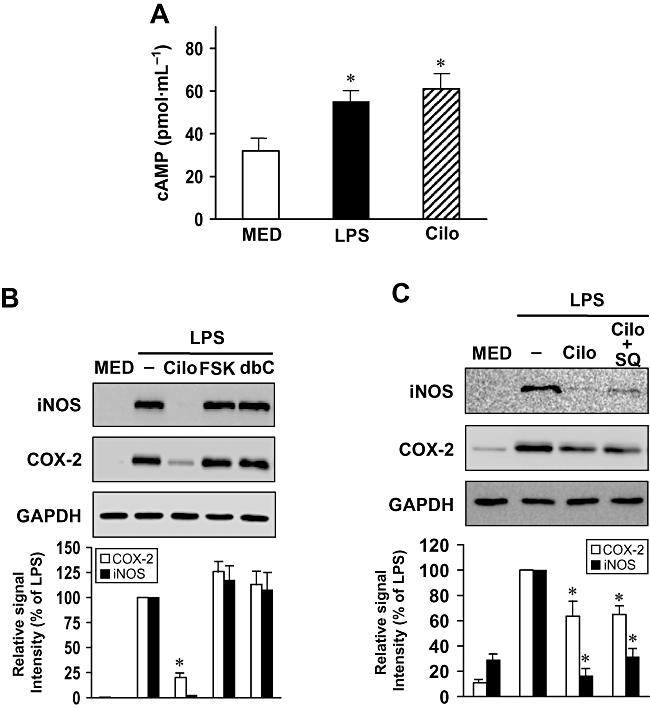
Effect of cilostazol induced-intracellular cAMP on iNOS and COX-2 expression in LPS-stimulated BV2 microglia. (A) Intracellular cAMP levels. Cells were treated with LPS and cilostazol (Cilo) for 10 min. Each value indicates the mean ± SEM, and is representative of results obtained from three independent experiments. *P < 0.05 indicates a significant difference from untreated cells (MED). (B) Effect of cAMP-elevating agents on iNOS and COX-2 expression. Cells were pretreated with cilostazol (Cilo, 30 µM, 6 h), forskolin (FSK, 10 µM, 30 min) and dibutyryl-cAMP (dbC, 100 µM, 30 min) and then stimulated with LPS (1 µg·mL−1) for another 24 h. (C) Effect of cilostazol on iNOS and COX-2 expression by SQ 22536, cAMP antagonist (SQ) in LPS-stimulated microglia. Cells were pretreated with SQ (30 µM) for 10 min, followed by cilostazol (30 µM) for 6 h, and then stimulated with LPS (1 µg·mL−1) for another 24 h. Cell lysates were prepared for the determination of protein levels of iNOS and COX-2. Each value indicates the mean ± SEM and is representative of results obtained from three independent experiments. *P < 0.05 indicates a significant difference from cells treated with LPS in the absence of cilostazol. cAMP, 3′-5′-cyclic adenosine monophosphate; COX-2, cyclooxygenase-2; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide.
Effects of cilostazol on LPS-induced TNF-α and IL-1β production
Next, we analysed the effects of cilostazol on proinflammatory cytokines such as TNF-α and IL-1β. BV2 microglia were incubated with cilostazol (10, 20 or 30 µM) in the presence or absence of LPS (1 µg·mL−1) for 24 h, and cytokine levels in the culture media were measured by ELISA. As shown in Figure 5A, TNF-α and IL-1β levels were increased in the culture media of LPS-stimulated BV2 microglia, and these increases were significantly decreased in a concentration-dependent manner by treatment with cilostazol. In a parallel experiment, RT-PCR was performed to determine whether cilostazol inhibits the expression of these cytokines at the transcriptional level. As shown in Figure 5B, treatment of BV2 microglia with different concentrations of cilostazol 6 h before LPS treatment resulted in a dose-dependent decrease in IL-1β and TNF-α mRNA. Thus, the results indicate that cilostazol inhibited the expression of these cytokines, which are involved in the inflammatory process.
Figure 5.
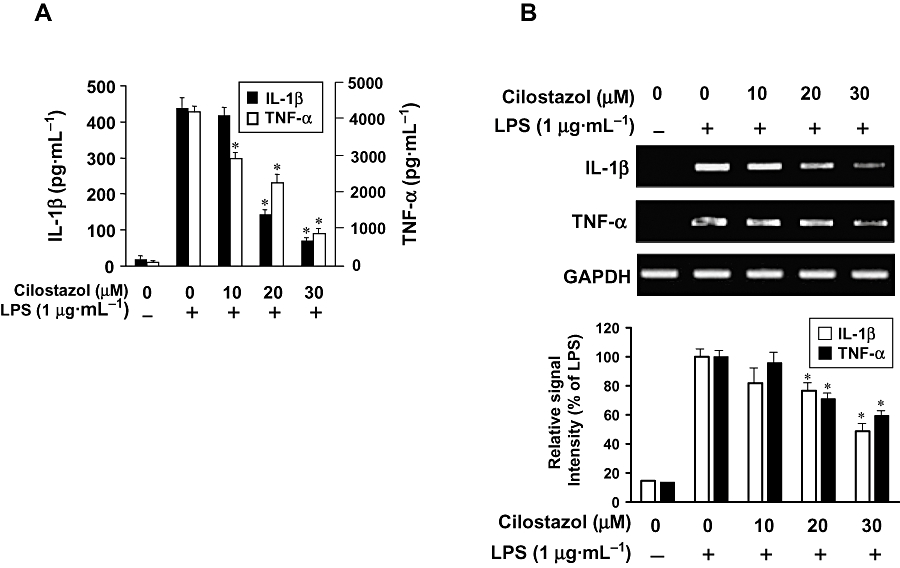
Effect of cilostazol on proinflammatory cytokines in LPS-stimulated BV2 microglia. Cells were pretreated with the indicated concentrations of cilostazol for 6 h before LPS treatment (1 µg·mL−1), and total RNA and the supernatants were isolated at 6 h or 24 h after LPS treatment respectively. (A) After incubation for 24 h, the levels of IL-1β and TNF-α present in the supernatants were measured. (B) After incubation for 6 h, the levels of IL-1β and TNF-α mRNA were determined by RT-PCR. Each value indicates the mean ± SEM and is representative of results obtained from three independent experiments. *P < 0.05 indicates a significant difference from cells treated with LPS in the absence of cilostazol. IL, interleukin; LPS, lipopolysaccharide; TNF, tumour necrosis factor.
Effects of cilostazol on LPS-stimulated MCP-1 protein and mRNA expression
We investigated whether cilostazol suppresses LPS-stimulated production of MCP-1 by RT-PCR and ELISA (Figure 6). The expression of mRNA for MCP-1 was found to be significantly increased in microglial cells treated with LPS (Figure 6A). This induction was significantly inhibited by addition of cilostazol to BV2 microglia cultures. In line with this finding, MCP-1 protein secretion, as revealed by ELISA, was increased in BV2 microglia cultures treated with LPS, and cilostazol significantly inhibited LPS-stimulated MCP-1 release from activated BV2 microglia (Figure 6B).
Figure 6.
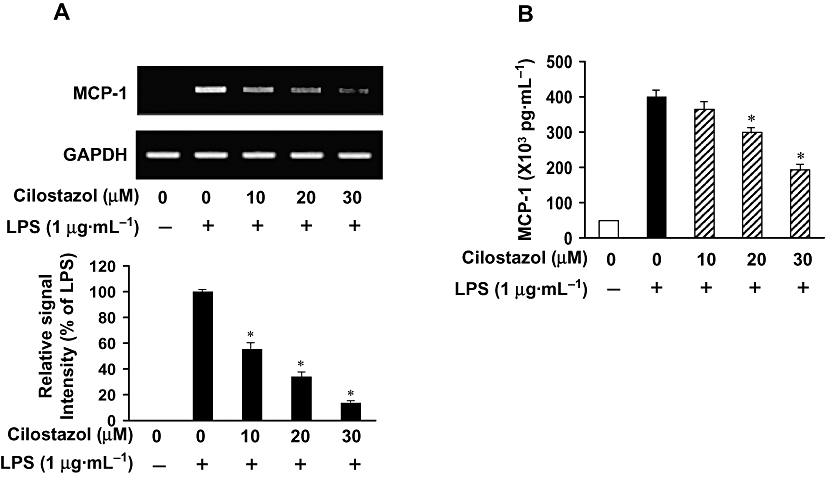
Effect of cilostazol on MCP-1 production in LPS-stimulated BV2 microglia. BV2 microglia were pretreated with cilostazol (10, 20 and 30 µM) 6 h prior to incubation with LPS (1 µg·mL−1), and total RNA and the supernatants were isolated at 6 h and 24 h after LPS treatment respectively. (A) After LPS treatment for 6 h, total RNA was prepared for RT-PCR analysis of MCP-1 mRNA gene expression in LPS-stimulated BV2 microglia. Results are representative of those obtained from three independent experiments, and the densitometric data below the RT-PCR results are presented as fold changes as compared with their respective controls. (B) Extracellular levels of MCP-1 were measured in culture media using commercial ELISA kits. Experiments were repeated three times and similar results were obtained. *P < 0.05 indicates a significant difference from cells treated with LPS in the absence of cilostazol. ELISA, enzyme-linked immunosorbent assay; LPS, lipopolysaccharide; MCP-1, monocyte chemoattractant protein-1.
Effect of cilostazol on NF-κB activity
Activation of NF-κB is necessary for induction of the iNOS and COX-2 genes. To further characterize the mechanism by which cilostazol inhibits proinflammatory and/or cytotoxic factor expression, the effect of cilostazol on the DNA-binding activity of NF-κB was determined by EMSA (Figure 7A). LPS treatment caused a significant increase in the DNA-binding activity of NF-κB. In contrast, treatment with cilostazol markedly reduced the LPS-induced DNA-binding activity of NF-κB.
Figure 7.
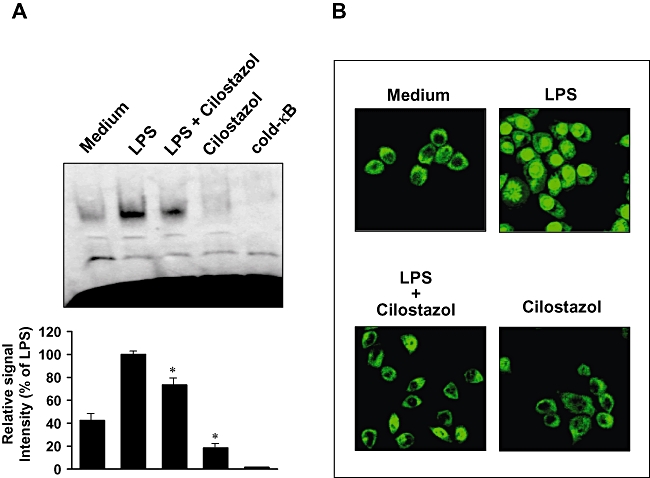
Effect of cilostazol on NF-κB activity in LPS-stimulated BV2 microglia. (A) Nuclear extracts (4 µg) were prepared and analysed for the DNA binding activity of NF-κB by EMSA. Binding specificity was determined using the unlabelled probe (100-fold in excess; shown as ‘cold’) to compete with the labelled oligonucleotide. BV2 microglial cells were pretreated with vehicle or the indicated concentrations of cilostazol for 6 h before stimulation with LPS (1 µg·mL−1) for another 1 h. The results shown are representative of three independent experiments. *P < 0.05 indicates a significant difference from LPS alone group. (B) BV2 microglia cells were pretreated with 30 µM cilostazol for 6 h prior to stimulation with LPS (1 µg·mL−1) for 1 h. p65 protein localization was determined using an anti-p65 antibody and a FITC-labelled anti-rabbit IgG antibody, and cells were visualized using laser confocal scanning microscopy. A representative result from three to five independent experiments is shown. EMSA, electrophoretic mobility shift assay; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB.
Effect of cilostazol on the nuclear translocation of NF-κB
We also investigated the effect of cilostazol on LPS-induced NF-κB p65 nuclear translocation, as translocation of NF-κB to the nucleus has been shown to be required for NF-κB-dependent transcription following LPS stimulation. The cellular localization of NF-κB p65 was investigated by immunofluorescence staining and visualization with confocal microscopy (Figure 7B). Confocal images revealed that NF-κB p65 was normally sequestered in the cytoplasm (Figure 7B, shown as Medium), and that nuclear accumulation of NF-κB p65 was strongly induced after stimulation of BV2 microglia with LPS (Figure 7B, shown as LPS). The LPS-induced translocation of NF-κB p65 was completely abolished by pretreating the cells with cilostazol (Figure 7B, LPS+cilostazol). Nuclear translocation of NF-κB p65 was not induced in the cells after pretreatment with cilostazol alone in the absence of LPS stimulation (Figure 7B, cilostazol panel). Taken together, these results indicate that the inhibition of NF-κB activation by cilostazol may be the mechanism responsible for suppression of NO, PGE2 and pro-inflammatory cytokines in BV2 microglia.
Effect of cilostazol on the phosphorylation of MAP kinases in LPS-stimulated BV2 microglia
Experiments were designed to elucidate the signalling cascades that control the expression of iNOS and COX-2 genes in BV2 microglial cells in response to LPS stimulation. Previously published evidence described the role of MAP kinases in the regulation of cell growth and differentiation and in the control of cellular responses to cytokines and stresses. They play a critical role in the activation of NF-κB. Moreover, MAP kinases are known to be important for the expression of iNOS and COX-2. MAP kinases thus act as a specific target in inflammatory responses. To investigate whether the inhibition of inflammation by cilostazol is regulated by the MAP kinase pathway, we examined the effect of cilostazol on LPS-induced phosphorylation of ERK-1/2, JNK and p38 kinase in BV2 microglia by Western blot analysis. We first demonstrated that ERK-1/2, JNK and p38 kinase were phosphorylated after stimulation of BV2 cells with LPS, and then examined the effect of cilostazol on LPS-induced activation of MAP kinases. As shown in Figure 8, cilostazol (20 or 30 µM) markedly inhibited ERK-1/2 and JNK activation, while phosphorylation of p38 kinase was not affected. The amounts of total ERK-1/2 and JNK were unaffected by either LPS or cilostazol treatment. These results suggest that the ERK-1/2 and JNK pathways are of major relevance during LPS-mediated expression of iNOS and COX-2.
Figure 8.
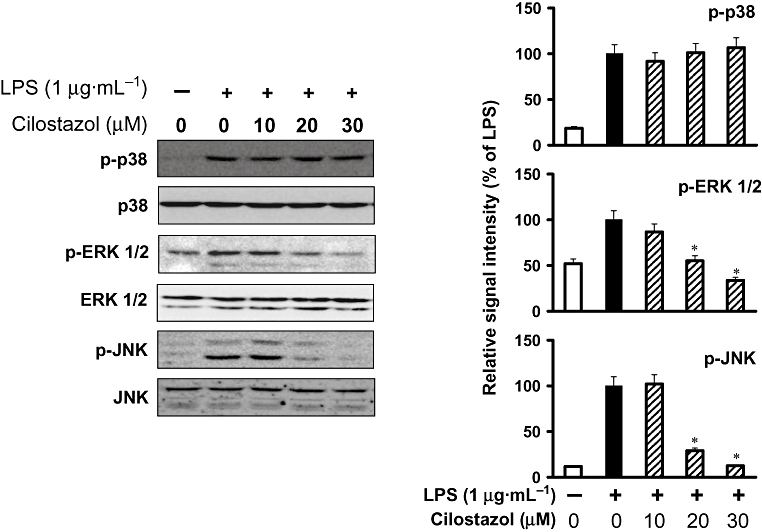
Effect of cilostazol on LPS-induced phosphorylation of ERK-1/2, SAPK/JNK and p38 MAP kinase in BV-2 microglia. BV-2 microglia were treated with vehicle or the indicated concentrations of cilostazol for 6 h before being incubated with LPS (1 µg·mL−1) for 30 min. Cell extracts were then prepared and subjected to Western blotting with antibodies specific for phosphorylated forms of ERK-1/2, SAPK/JNK and p38. The results presented are representative of three independent experiments. *P < 0.05 indicates a significant difference from cells treated with LPS in the absence of cilostazol. ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; MAP, mitogen-activated protein; SAPK, stress-activated protein kinase.
Discussion
Inflammation in the brain caused by activated microglia is a prominent pathological feature associated with the neurodegeneration caused by several diseases, including PD, AD, MS and amyotrophic lateral sclerosis (McGeer and McGeer, 1995; McGeer et al., 2001; Liu and Hong, 2003). Activation of microglia is observed in brain injuries and neurodegenerative diseases, and is also induced after exposure to LPS, interferon (IFN)-γ or β-amyloid (Zielasek and Hartung, 1996). Under pathological conditions, activated microglia release a variety of neurotoxic compounds and pro-inflammatory mediators, including NO, PGE2, reactive oxygen species, complement factors and pro-inflammatory cytokines (IL-1, IL-6 and TNF-α). These neurotoxic and pro-inflammatory factors are thought to be responsible for the pathological conditions associated with neurodegenerative diseases.
Cilostazol is a selective inhibitor of PDE3 and increases the intracellular cAMP level. Previous reports indicate that activation of cAMP contributes to the anti-inflammatory effects of cilostazol, which inhibits the mitogen-activated protein kinase (MAPK) activation and NF-κB signalling pathways (Tsai et al., 2008). In the present study, microglial cAMP levels were slightly elevated by cilostazol (Figure 4A). We next examined the effect of forskolin, dibutyryl-cAMP on the expression of iNOS and COX-2. Forskolin and dibutyryl-cAMP had no effect on LPS induced inflammation response (Figure 4B). In addition, in order to verify the cAMP-dependent or -independent action of cilostazol, we examined the effects of SQ 22536, an irreversible and selective cAMP antagonist, on the expression of iNOS and COX-2 production. This study found that SQ 22536 did not reverse the inhibitory effect of cilostazol on the expression of iNOS and COX-2 (Figure 4C). Thus, we suggested that the inhibitory effect of cilostazol on iNOS and COX-2 inhibition was likely to be independent of intracellular cAMP signalling pathway in BV2 microglia.
A previous study reported that cilostazol affects cytokine and NO production in microglia (Yoshikawa et al., 1999). However, the molecular mechanisms underlying its anti-inflammatory activity remain unclear. In addition, the inhibitory effects of cilostazol on the release of PGE2 in LPS-stimulated BV2 microglia were not previously known. In the present study, we investigated the pharmacological effects and mechanisms of action of cilostazol on the production of pro-inflammatory mediators in BV2 microglia stimulated with LPS. We found that cilostazol inhibited the expression of iNOS, COX-2, MCP-1 and cytokines (IL-1β and TNF-α), which are NF-κB-regulated inflammation-associated genes in LPS-activated microglia. This study suggests that cilostazol modulated LPS-induced inflammatory responses, and may be a potential therapeutic candidate for the treatment of neurodegenerative conditions.
Prostaglandin E2, NO and pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α are known to be important mediators in the process of inflammation. These proinflammatory mediators are thought to be responsible for some of the harmful effects of brain injuries and diseases, including ischemia, AD and neural death (Meda et al., 1995). Microglia activation induced by CNS injury or infection is associated with neurodegenerative disorders and the release of NO and PGE2, and subsequent release of pro-inflammatory cytokines (Gonzalez-Scarano and Baltuch, 1999). Many reports have shown that LPS strongly activates microglia and induces the COX-2 gene, leading to the synthesis of PGE2 (Bauer et al., 1997; Fiebich et al., 2003). In addition, NO has been shown to be an important regulatory molecule in diverse physiological functions, including vasodilation, neural communication and host defence (Mitchell et al., 1995; MacMicking et al., 1997). In mammalian cells, NO is synthesized by three different isoforms of NOS: endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS). Importantly, iNOS is induced by bacterial LPS and certain inflammatory cytokines in a variety of cells. It is well known that inflammatory mediators, including iNOS and COX-2, are responsible for the symptoms of many neuroinflammatory diseases such as AD, PD and HIV dementia (Minghetti and Levi, 1998; Gonzalez-Scarano and Baltuch, 1999). We demonstrated that inhibition of NO and PGE2 production is due to changes in iNOS and COX-2 expression at the mRNA as well as the protein level as shown by RT-PCR and Western blot. In the brain, microglia appear to rapidly express COX-2 and iNOS in response to pathological stimuli. Abnormalities in the production or function of cytokines such as TNF-α and IL-1β have been reported to play a key role in many inflammatory lesions (De Nardin, 2001). The neuroinflammatory response is represented by activated microglia producing elevated levels of pro-inflammatory cytokines, including TNF-α and IL-1β (Boka et al., 1994; Hunot et al., 1999). TNF-α is primarily produced by monocytes, macrophages and T cells, and has various pro-inflammatory effects on many cell types. Microglia are the major producers of TNF-α in the brain and may play a role in certain pathological conditions in the brain (Sawada et al., 1989). TNF-α overexpression is implicated in the pathogenesis of several human CNS disorders, including AD, MS, PD and cerebral ischemia (Meda et al., 1995; Botchkina et al., 1997; Akassoglou et al., 1998; Sriram et al., 2002). IL-1β is a potent pro-inflammatory cytokine that acts through the IL-1 receptors found on numerous cell types, including neurons and microglia. Moreover, IL-1β is proposed to be an important mediator of neuroimmune interactions that participate directly in neurodegeneration (Rothwell et al., 1997). It may interact with other molecules that are either released or induced in response to damage, or it may affect only compromised neurons. Thus, inhibition of cytokine production or function serves as a key mechanism in the control of neurodegeneration.
Central nervous system inflammation caused by injury is amplified by the recruitment of activated microglia to sites of injury. The migration of microglia is regulated by chemokines, which are also important for neurotoxic and neuroprotective activity. Chemokines are well-known regulators of peripheral immune cell recruitment and trafficking under both physiological and pathological conditions, can cause secondary damage during inflammation and can affect neurodegeneration. MCP-1 is a particularly important chemokine that is primarily responsible for the initiation and progression of inflammatory responses by promoting migration and recruitment of inflammatory cells (Huang et al., 2000). Previous reports indicated that MCP-1 regulates the migration of activated microglial cells to sites of inflammation in the CNS (McManus et al., 2000). MCP-1 expression is up-regulated in a number of CNS pathologies, including AD, MS, HIV dementia, ischemia and brain injury (Ishizuka et al., 1997; Ivacko et al., 1997; Kelder et al., 1998; McManus et al., 1998; Muessel et al., 2000). Therefore, understanding the neurotoxic effect of microglial cells and modulation of chemokines may result in the development of treatments specific for neurodegenerative diseases. In the present study, cilostazol inhibited the LPS-stimulated mRNA expression and protein release of MCP-1 in activated microglia.
Nuclear factor-kappaB is known as a pleiotropic regulator of various genes involved in the production of many pro-inflammatory cytokines and enzymes related to the inflammatory process. NF-κB is a central regulator of microglial responses to activating stimuli, including LPS and cytokines (O'Neill and Kaltschmidt, 1997). NF-κB, as a consequence of its key role in several pathologic conditions, is a major drug target in a variety of diseases. NF-κB activation is critical for the expression of various cytokines, iNOS and COX-2 in microglia in response to LPS (Baldwin, 1996; Moon et al., 2007b). We showed that cilostazol prevents LPS-induced p65 nuclear translocation and NF-κB binding to a consensus sequence. Therefore, inhibition of NF-κB signalling pathways in microglia by cilostazol might result in the down-regulation of pro-inflammatory mediators, thereby resulting in an anti-inflammatory effect.
Various intracellular signalling pathways are involved in inflammatory mediator expression. MAPKs are a group of signalling molecules that also appear to play key roles in inflammatory processes. Previous studies have shown that activation of MAPKs have a significant effect on the regulation of COX-2, iNOS, MCP-1 and pro-inflammatory cytokine gene expression by controlling the activation of NF-κB in microglia (Moon et al., 2007a; Zhou et al., 2007; Jung et al., 2009). Early regulation occurs in the cytoplasm with the activation of the inhibitor of NF-κB (IκB) and subsequent IκB phosphorylation (Brown et al., 1995; O'Connell et al., 1998). This phosphorylation results in IκB degradation, thus allowing NF-κB translocation to the nucleus. MAPK/ERK kinase kinase 1 (MEKK1) directly interacts with IκB kinase (IKK) and phosphorylates it (Nemoto et al., 1998). So, it appeared that MAPKs can regulate NF-κB-dependent transcription by activating IKK. It is possible that neuroprotective mechanisms are associated with inhibition of MAP kinases or inflammatory mediator production in activated BV2 microglia. Therefore, we investigated the effect of cilostazol on LPS-stimulated phosphorylation of ERK-1/2, JNK and p38 kinase in BV2 microglia. We found that phosphorylation of ERK-1/2 and JNK in response to LPS was decreased by cilostazol treatment; however, no significant effects of cilostazol were observed on the LPS-induced phosphorylation of p38 kinase. This finding is consistent with the results reported by Pocivavsek et al. (2009). They showed that the p38 pathway was not essential for iNOS expression. On the other hand, our previous report showed that activation of p38 is involved in LPS-induced iNOS expression (Jung et al., 2009). Our results implicate ERK-1/2 and JNK as major mediators of iNOS induction and inflammatory signalling in BV2 microglia. The reason for this discrepancy is mainly due to variation in individual MAPK gene expression with cell types and stimulators. We next examined whether or not cilostazol suppressed the activation of ERK and JNK. In nonstimulated microglia, neither MAPK/ERK kinase 1/2 (MEK1/2; a major upstream kinase responsible for the activation of ERK) nor SAPK/ERK1/MAPK kinase kinase 4 (SEK1/MKK4; a major upstream kinase responsible for the activation of JNK) was phosphorylated. Stimulation with LPS resulted in phosphorylation of MEK1/2 and SEK1/MKK4. However, cilostazol led to a significant suppression of MEK1/2 and SEK1/MKK4 phosphorylation in LPS-stimulated BV2 microglia (Figure S1). Thus, cilostazol was found to suppress the phosphorylation of MEK1/2 and SEK1/MKK4, thereby in turn suppressing the activation of downstream kinases ERK and JNK respectively. Depression of the MAPK pathway appeared to be caused by the inhibition of a more upstream kinase (MAPK kinase) by cilostazol. Taken together, these results suggest that MEK1/2-ERK-1/2 and SEK1/MKK4-JNK, but not p38 kinase, are involved in the inhibitory effect of cilostazol on LPS-induced iNOS and COX-2 expression and NF-κB activation.
In summary, our results show that cilostazol treatment of BV2 microglia results in decreased levels of pro-inflammatory mediators following LPS stimulation, and also reveal new information about the underlying molecular mechanisms. Cilostazol significantly attenuated the release of iNOS, PGE2, TNF-α, IL-1β, and MCP-1 in a concentration-dependent manner; moreover, it acts at the level of transcription. The inhibitory effect of cilostazol was mediated by inhibition of NF-κB, ERK-1/2 and JNK activation in activated microglia. These data indicate that cilostazol targets ERK-1/2, JNK and NF-κB in microglia and inhibits iNOS, COX-2, MCP-1 and pro-inflammatory cytokine expression. Given the fact that microglial activation contributes to the pathogenesis of neurodegenerative human brain diseases, cilostazol may be considered as a potential therapeutic agent for various inflammatory and neurodegenerative diseases.
Acknowledgments
This study was supported by research funds from Chosun University 2009.
Glossary
Abbreviations:
- COX-2
cyclooxygenase-2
- IKK
IκB kinase
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- IκB
inhibitor of NF-κB
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemoattractant protein-1
- MEKK1
MAPK/ERK kinase kinase 1
- NF-κB
nuclear factor-κB
- PGE2
prostaglandin E2
- SEK1/MKK4
SAPK/ERK1/MAPK kinase kinase 4
- TNF-α
tumour necrosis factor-α
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of cilostazol on LPS-induced phosphorylation of MEK1/2 and SEK1/MKK4 in BV-2 microglia. BV-2 microglia were treated with vehicle or the inducated concentrations of cilostazol for 6 h and then stimulated with LPS (1 μg·mL−1) for different time nitervals (10 min: phosphorylation of MEK1/2 (A); 15 min: phosphorylation of SEK1 (B) (protein expression). Cell extracts were then prepared and subjected to western blotting with antibodies specific for phosphorylated forms of MEK1/2 and SEK1/MKK4. The results presented are representative of three independent experiments. *P < 0.05 indicates a significant difference from the value obtained for cells treated with LPS in the absence of cilostazol.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, et al. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol. 1998;153:801–813. doi: 10.1016/S0002-9440(10)65622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AS., Jr. The NF-kappaB and I-kappaB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bauer MK, Lieb K, Schulze-Osthoff K, Berger M, Gebicke-Haerter PJ, Bauer J, et al. Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem. 1997;243:726–731. doi: 10.1111/j.1432-1033.1997.00726.x. [DOI] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- Botchkina GI, Meistrell ME, III, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- Dawson DL, Cutler BS, Meissner MH, Strandness DE., Jr. Cilostazol has beneficial effects in treatment of intermittent claudication. Circulation. 1998;98:678–686. doi: 10.1161/01.cir.98.7.678. [DOI] [PubMed] [Google Scholar]
- De Nardin E. The role of inflammatory and immunological mediators in periodontitis and cardiovascular disease. Ann Periodontol. 2001;6:30–40. doi: 10.1902/annals.2001.6.1.30. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, van Gool WA. Neuroinflammatory perspectives on the two faces of Alzheimer's disease. J Neural Transm. 2004;111:281–294. doi: 10.1007/s00702-003-0055-1. [DOI] [PubMed] [Google Scholar]
- Fanarraga ML, Villegas JC, Carranza G, Castaño R, Zabala JC. Tubulin cofactor B regulates microtubule densities during microglia transition to the reactive states. Exp Cell Res. 2009;315:535–541. doi: 10.1016/j.yexcr.2008.10.045. [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Lieb K, Kammerer N, Hüll M. Synergistic inhibitory effect of ascorbic acid and acetylsalicylic acid on prostaglandin E2 release in primary rat microglia. J Neurochem. 2003;86:173–178. doi: 10.1046/j.1471-4159.2003.01822.x. [DOI] [PubMed] [Google Scholar]
- Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147:867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Honda F, Imai H, Ishikawa M, Kubota C, Shimizu T, Fukunaga M, et al. Cilostazol attenuates gray and white matter damage in a rodent model of focal cerebral ischemia. Stroke. 2006;37:223–228. doi: 10.1161/01.STR.0000196977.76702.6d. [DOI] [PubMed] [Google Scholar]
- Huang D, Han Y, Rani MR, Glabinski A, Trebst C, Sorensen T, et al. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol Rev. 2000;177:52–67. doi: 10.1034/j.1600-065x.2000.17709.x. [DOI] [PubMed] [Google Scholar]
- Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, et al. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaka N, Taguchi J, Kimura Y, Ikari Y, Aizawa T, Togo M, et al. Effects of a single local administration of cilostazol on neointimal formation in balloon-injured rat carotid artery. Atherosclerosis. 1999;142:41–46. doi: 10.1016/s0021-9150(98)00147-6. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci. 1997;51:135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Ivacko J, Szaflarski J, Malinak C, Flory C, Warren JS, Silverstein FS. Hypoxic–ischemic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. J Cereb Blood Flow Metab. 1997;17:759–770. doi: 10.1097/00004647-199707000-00006. [DOI] [PubMed] [Google Scholar]
- Jung WK, Ahn YW, Lee SH, Choi YH, Kim SK, Yea SS, et al. Ecklonia cava ethanolic extracts inhibit lipopolysaccharide-induced cyclooxygenase-2 and inducible nitric oxide synthase expression in BV2 microglia via the MAP kinase and NF-kappaB pathways. Food Chem Toxicol. 2009;47:410–417. doi: 10.1016/j.fct.2008.11.041. [DOI] [PubMed] [Google Scholar]
- Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. β-Chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998;44:831–835. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- Kim HS, Whang SY, Woo MS, Park JS, Kim WK, Han IO. Sodium butyrate suppresses interferon-gamma-, but not lipopolysaccharide-mediated induction of nitric oxide and tumor necrosis factor-alpha in microglia. J Neuroimmunol. 2004a;151:85–93. doi: 10.1016/j.jneuroim.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Kim WK, Jang PG, Woo MS, Han IO, Piao HZ, Lee K, et al. A new anti-inflammatory agent KL-1037 represses proinflammatory cytokine and inducible nitric oxide synthase (iNOS) gene expression in activated microglia. Neuropharmacology. 2004b;47:243–252. doi: 10.1016/j.neuropharm.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Tani T, Kanbe T, Watanabe K. Effect of cilostazol on platelet aggregation and experimental thrombosis. Arzneimittelforschung. 1985;35:1144–1149. [PubMed] [Google Scholar]
- Lee JH, Park SY, Shin HK, Kim CD, Lee WS, Hong KW. Protective effects of cilostazol against transient focal cerebral ischemia and chronic cerebral hypoperfusion injury. CNS Neurosci Ther. 2008;14:143–152. doi: 10.1111/j.1527-3458.2008.00042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. The inflammatory response system of brain: implication for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson's disease. Adv Neurol. 2001;86:83–89. [PubMed] [Google Scholar]
- McManus CM, Berman JW, Brett FM, Stauton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–29. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- McManus CM, Liu JS, Hahn MT, Hua LL, Brosnan CF, Berman JW, et al. Differential induction of chemokines in human microglia by type I and II interferons. Glia. 2000;29:273–280. [PubMed] [Google Scholar]
- MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Ann Rev Immunol. 1997;15:323–330. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr., Baron P, Villalba M, et al. Activation of microglial cells by β-amyloid protein and IFN-γ. Nature. 1995;373:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Misko TP, Trotter JL, Cross AH. Mediation of inflammation by encephalitogenic cells: interferon-γ induction of nitric oxide synthase and cyclooxygenase 2. J Neuroimmunol. 1995;61:195–204. doi: 10.1016/0165-5728(95)00091-f. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Larkin S, Williams TJ. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem Pharmacol. 1995;50:1535–1542. doi: 10.1016/0006-2952(95)00212-x. [DOI] [PubMed] [Google Scholar]
- Moon DO, Choi YH, Kim ND, Park YM, Kim GY. Anti-inflammatory effects of beta-lapachone in lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol. 2007a;7:506–514. doi: 10.1016/j.intimp.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Moon DO, Park SY, Lee KJ, Heo MS, Kim KC, Kim MO, et al. Bee venom and melittin reduce proinflammatory mediators in lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol. 2007b;7:1092–1101. doi: 10.1016/j.intimp.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Muessel MJ, Berman NEJ, Klein RM. Early and specific expression of monocyte chemoattractant protein-1 in the thalamus induced by cortical injury. Brain Res. 2000;870:211–221. doi: 10.1016/s0006-8993(00)02450-1. [DOI] [PubMed] [Google Scholar]
- Nemoto S, DiDonato JA, Lin A. Coordinate regulation of IκB kinases by mitogen-activated protein kinase kinase kinase 1 and NF-κB-inducing kinase. Mol Cell Biol. 1998;18:7336–7343. doi: 10.1128/mcb.18.12.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MA, Bennett BL, Mercurio F, Manning AM, Mackman N. Role of IKK1 and IKK2 in lipopolysaccharide signaling in human monocytic cells. J Biol Chem. 1998;273:30410–30414. doi: 10.1074/jbc.273.46.30410. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Aisen PS. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience. 1998;87:319–324. doi: 10.1016/s0306-4522(98)00218-8. [DOI] [PubMed] [Google Scholar]
- Pocivavsek A, Burns MP, Rebeck GW. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia. 2009;57:444–453. doi: 10.1002/glia.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rosa MP, Baroni GV, Portal VL. Cilostazol, a phosphodiesterase III inhibitor: future perspectives in atherosclerosis. Arq Bras Cardiol. 2006;87:222–226. doi: 10.1590/s0066-782x2006001800035. [DOI] [PubMed] [Google Scholar]
- Rothwell N, Allan S, Toulmond S. The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. 1997;100:2648–2652. doi: 10.1172/JCI119808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989;491:394–397. doi: 10.1016/0006-8993(89)90078-4. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson's disease. FASEB J. 2002;16:1474–1476. doi: 10.1096/fj.02-0216fje. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Oida K, Fujiwara R, Maeda H, Hayashi S, Takai H, et al. Effect of cilostazol, a cyclic AMP phosphodiesterase inhibitor, on the proliferation of rat aortie smooth muscle cells in culture. J Cardiovasc Pharmacol. 1992;20:900–906. doi: 10.1097/00005344-199212000-00009. [DOI] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Wakita H, Lin JX, Budka H. Cyclooxygenase-2 is induced in microglia during chronic cerebral ischemia in humans. Acta Neuropathol. 2000;99:26–30. doi: 10.1007/pl00007402. [DOI] [PubMed] [Google Scholar]
- Tsai CS, Lin FY, Chen YH, Yang TL, Wang HJ, Huang GS, et al. Cilostazol attenuates MCP-1 and MMP-9 expression in vivo in LPS-administrated balloon-injured rabbit aorta and in vitro in LPS-treated monocytic THP-1 cells. J Cell Biochem. 2008;103:54–66. doi: 10.1002/jcb.21388. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Zhang N, Liu M, Tanaka R, Mizuno Y, Urabe T. Cilostazol protects against brain white matter damage and cognitive impairment in a rat model of chronic cerebral hypoperfusion. Stroke. 2006;37:1539–1545. doi: 10.1161/01.STR.0000221783.08037.a9. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Sakuma M, Tanabe T. Hemodynamic effect of cilostazol on increasing peripheral blood flow in arteriosclerosis obliterans. Arzneimittelforschung. 1985;35:1198–1200. [PubMed] [Google Scholar]
- Yoshikawa M, Suzumura A, Tamaru T, Takayanagi T, Sawada M. Effects of phosphodiesterase inhibitors on cytokine production by microglia. Mult Scler. 1999;5:126–133. doi: 10.1177/135245859900500210. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M, Suzumura A, Ito A, Tamaru T, Takayanagi T. Effect of phosphodiesterase inhibitors on nitric oxide production by glial cells. Tohoku J Exp Med. 2002;196:167–177. doi: 10.1620/tjem.196.167. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Ling EA, Dheen ST. Dexamethasone suppresses monocyte chemoattractant protein-1 production via mitogen activated protein kinase phosphatase-1 dependent inhibition of Jun N-terminal kinase and p38 mitogen-activated protein kinase in activated rat microglia. J Neurochem. 2007;102:667–678. doi: 10.1111/j.1471-4159.2007.04535.x. [DOI] [PubMed] [Google Scholar]
- Zielasek J, Hartung HP. Molecular mechanisms of microglial activation. Adv Neuroimmunol. 1996;6:191–202. doi: 10.1016/0960-5428(96)00017-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.