

Abstract
Proteinuria is an early sign of kidney disease and has gained increasing attention over the past decade because of its close association with cardio-vascular and renal morbidity and mortality. Podocytes have emerged as the cell type that is critical in maintaining proper functioning of the kidney filter. A few genes have been identified that explain genetic glomerular failure and recent insights shed light on the pathogenesis of acquired proteinuric diseases. This review highlights the unique role of the cysteine protease cathepsin L as a regulatory rather than a digestive protease and its action on podocyte structure and function. We provide arguments why many glomerular diseases can be regarded as podocyte enzymatic disorders.
Keywords: Podocyte, Cathepsin L, Glomerular disease, Proteinuria, Proteases
Introduction
Proteinuria is a characteristic sign and a prognostic marker of many kidney diseases. The daily urinary excretion of more than 150 mg protein in adults or 100 mg/m2 in children [1] is considered abnormal. Several mechanisms can lead to increased urinary protein excretion including a diminished tubular reabsorption of low molecular weight (LMW) proteins that are normally filtered to the primary urine (tubular proteinuria) or the presence of an abnormally high concentration of LMW proteins in the blood whose filtration leads to oversaturation of the tubular reabsorption machinery (overflow proteinuria). However, by far the most common cause of proteinuria is glomerular damage leading to an increased loss of albumin and other intermediate and high molecular weight proteins into the urine.
The glomerulus is a highly specialized structure that normally ensures the selective ultrafiltration of plasma retaining essential proteins in the blood. Solutes and fluid leaving the bloodstream must pass three distinct layers of the glomerular capillary wall to reach the Bowman’s space, the site of primary urine collection [2]:
A fenestrated endothelium lining the inner surface of the glomerular capillaries
The glomerular basement membrane (GBM), a thin sheet of extracellular matrix
The inter-podocyte filtration slits that are covered by the slit diaphragm [3]
While the individual contribution of all layers is recognized, it is generally accepted that podocytes are most crucial in maintaining the integrity of the glomerular filter [4]. Podocytes are highly differentiated pericyte-like cells that reside on the external surface of the GBM and give rise to long major processes that extend along the capillaries to which they affix by numerous secondary processes more commonly termed foot processes (FP). The podocyte FPs are dynamic [5] and contain an actin-based contractile apparatus comparable to that of smooth muscle cells or pericytes [6]. The FP of neighboring podocytes regularly interdigitate, leaving between them narrow filtration slits that are bridged by the slit diaphragm, a Zipper-like structured, modified adherens junction [3]. Much of the evidence for a central role of podocytes in maintaining the integrity of the glomerular barrier comes from human genetic studies, which have identified several podocyte proteins that are crucial for the structural integrity of the slit diaphragm, including nephrin [7], podocin [8], TRPC6 [9, 10], PLCE1 [11], and α-actinin-4 [12]. Mutations in any of these genes lead to similar ultrastructural alterations characterized by blunting of podocyte membrane extensions as well as transformation of podocyte FPs into a band of cytoplasm (referred to as FPs effacement) [13]. While the aforementioned genetic studies have contributed invaluably to our understanding of the molecular structure of podocyte FPs and the slit diaphragm, familial mutations underlie only a small percentage of proteinuric kidney diseases. The majority of proteinuric diseases are acquired in the setting of intact gene function and thus must have other culprits. Since FP effacement is a reversible process and thus potentially amenable to therapy, the disease process needs to be defined in order to achieve better therapeutic options. Otherwise, FP effacement might result in progressive glomerular injury including podocyte depletion, scarring of the glomerulus, and eventually degeneration of the entire nephron leading to renal failure [14].
During recent years, it has become evident that podocytes are highly dynamic cells. The development of as well as recovery from FP effacement requires a high degree of podocyte motility implicating:
Changes in slit diaphragm structure and function
Reorganization of the podocyte actin cytoskeleton
Alterations in the adhesion of podocytes to the GBM
All these three aspects of podocyte dynamics are regulated through several enyzmatic pathways including signaling by phosphorylation and dephosphorylation as well as a recently discovered proteolytic regulatory cascade [5, 15, 16]. In the following, we will discuss the cathepsin L (CatL) proteolytic pathway in podocytes and its role in kidney health and disease.
The role of podocyte cathepsin L as a key enzyme in acquired proteinuria
CatL has long been known as a potent endoprotease primarily responsible for final protein breakdown within lysosomal compartments [17]. In addition, a secreted form of CatL has been shown to be involved in the degradation of extracellular matrix (ECM) in vivo [18] and in vitro [19]. Both the lysosomal and secreted forms of CatL have been implicated in cancer cell biology and metastasis [20]. The first evidence for a role of CatL in proteinuric kidney disease came from studies performed more than two decades ago, showing that the cathepsin inhibitor E-64 can reduce experimental proteinuria in a rat glomerulonephritis model [21]. The effects of E-64 were attributed to the inhibition of secreted CatL during matrix remodeling [21]. In 2004, the role of glomerular CatL was highlighted in a report on CatL mRNA and protein induction in proteinuric rats that received puromycin aminonucleoside (PAN) [5]. This study further demonstrated that the onset of experimental proteinuria is accompanied by an increased motility of podocytes, which was abrogated in CatL−/− podocytes [5]. As part of these studies, we noted that expression of a few intracellular podocyte proteins such as CD2AP declined, but only in the presence of CatL. In a subsequent study, we found that PAN and lipopolysaccharide (LPS, another proteinuric stimulus) specifically induce a short cytoplasmic variant of CatL devoid of the lysosomal targeting sequence [15] (Fig. 1). A shorter CatL variant has been previously shown to arise by translation from an alternative downstream AUG site and locates in the nucleus of fibroblasts where it can cleave the transcription factor CDP/Cux [22] or serve in Histone H3 processing during mouse embryonic stem cell differentiation [23]. This obviously broke with a dogma that CatL can only be active in the acidic pH of the lysosome. Whereas conventional CatL cleaves a variety of proteins very efficiently due to the denaturing conditions and low pH of the lysosome, short CatL exhibits a remarkable substrate specificity that allows a very specific enzymatic activity at cytosolic or nuclear pH [24]. So far, two substrates of cytosolic CatL have been described in podocytes: dynamin [15] and synaptopodin [16]. Both proteins contribute to the functional F-actin structure in normal podocyte FPs and allow FP effacement after their enzymatic processing by CatL.
Fig. 1.
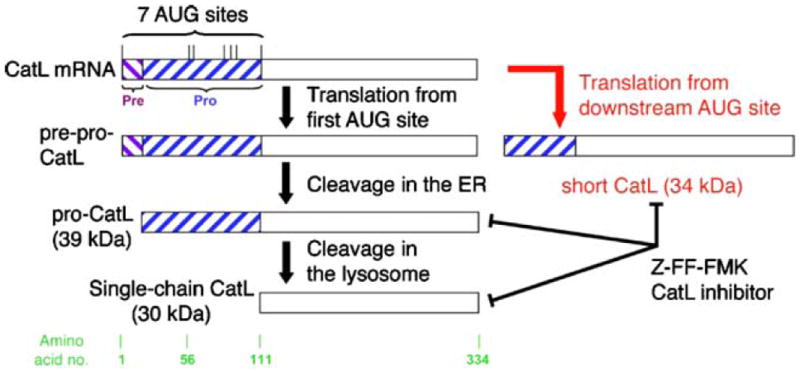
Schematic of CatL mRNA containing several AUG codons and resulting proteins. After translation from the first AUG, CatL is processed to yield a 30-kDa lysosomal form, called single-chain CatL (black arrows). However, alternative translation initiation from a downstream AUG produces a CatL isoform devoid of the lysosomal targeting sequence (short CatL), which localizes to the cytoplasm (red arrow). Adapted with permission from Sever et al. [15]
Evidence that supports relevance of cathepsin L to human glomerular disease
CatL was brought in context with human and murine renal disease as early as the 1980s. Active CatL was purified from normal human kidneys [25] and isolated rat glomeruli [26]. An extensive glomerular expression analysis performed by the group of Karl Tryggvason has also identified CatL as a glomerulus-enriched gene in the normal newborn and adult mouse glomerulus [27]. The first evidence for a pathogenic role of CatL in glomerular diseases came from a study that reported a marked reduction of proteinuria through administration of the cathepsin inhibitor E-64 in experimental anti-GBM disease. However, these studies did not differentiate between lysosomal, secreted and cytosolic CatL. Glomerular CatL was thought to be involved in extracellular matrix turnover, thereby affecting glomerular function. The aforementioned study by Sever et al. [15] was the first to identify cytosolic short CatL in podocytes and recognize its role in degrading intracellular cytoskeleton-associated proteins. The importance of podocyte CatL for glomerular pathology is highlighted by both in vitro and animal models of glomerular diseases as well as expression studies in human biopsies. CatL is significantly induced in at least two rodent models of proteinuria, i.e. the LPS mouse model (Fig. 2a) and the rat PAN model [5]. Stainings in cultured podocytes treated with LPS or PAN revealed a vast increase in CatL enzyme in the cytosol [15]. Enzymatic activity assays determined that cytosolic CatL is enzymatically active and can cleave its targets dynamin [15] and synaptopodin [16]. The significance of CatL induction is further underscored by the finding that CatL knockout mice are protected from LPS-induced FP effacement and proteinuria (Fig. 2). Human data stem from isolated glomeruli of explanted renal allografts with chronic allograft nephropathy [28] and microdissected glomeruli from kidney biopsies of patients with three types of glomerular disease, membranous nephropathy (MN), focal segmental glomerulosclerosis (FSGS), and diabetic nephropathy (DN) (Fig. 3a). All these cases revealed a two-fold or greater induction of CatL mRNA as measured by real-time RT-PCR [15]. Increased CatL protein is found in podocytes of patients with DN [15] (Fig. 3b, c).
Fig. 2.
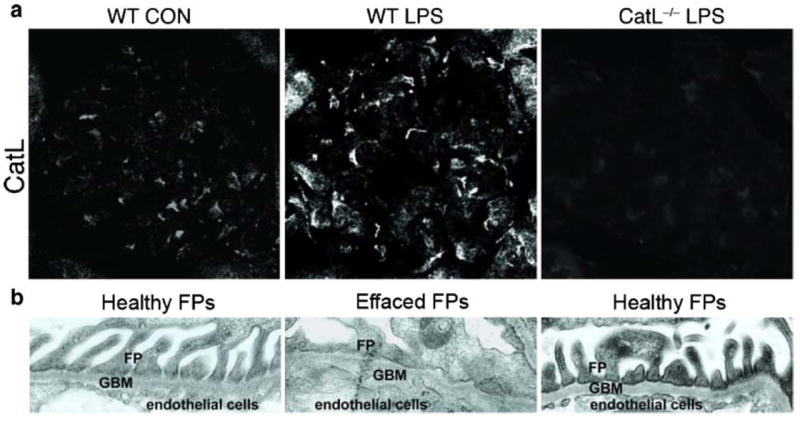
Cathepsin L (CatL) is essential for the development of proteinuria in the lipopolysaccharide (LPS) model. In wild-type (WT) mice, intraperitoneal injection of LPS leads to a T- and B-cell-independent transient form of proteinuria through the activation of podocyte TLR-4 and induction of B7-1 [39]. a Immunocytochemistry of mouse glomeruli using monoclonal anti-CatL antibody. WT mice receiving LPS (WT LPS) upregulate the expression of cytosolic CatL compared with control mice receiving PBS (WT CON). LPS was also injected into CatL−/− mice (CatL−/− LPS). Original magnification, ×400. b Electron micrographs of foot processes (FPs) showing effacement in LPS-treated WT, but not in CatL−/− mice. Adapted with permission from Sever et al. [15]
Fig. 3.
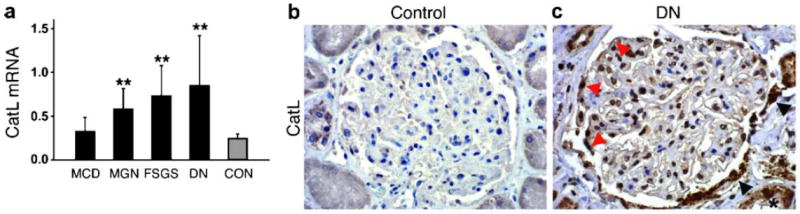
Cathepsin L (CatL) mRNA and protein expression are elevated in human proteinuric kidney diseases. a Quantitative rt-PCR of microdissected glomeruli from human biopsies of patients with acquired proteinuric diseases: minimal change disease (MCD; n=7), membranous glomerulonephritis (MGN; n=9), focal segmental glomerulosclerosis (FSGS; n=7), and diabetic nephropathy (DN; n=10). **P<0.01 for comparison with healthy controls (CON; n=8). b CatL labeling of normal human kidney. c CatL labeling of a kidney biopsy from a patient with diabetic nephropathy, mildly reduced renal function, and nephrotic range proteinuria. Adapted with permission from Sever et al. [15]
Cathepsin L proteolyzes dynamin and synaptopodin
The computer algorithm PEPS (prediction of endopeptidase proteolytic sites) has served to identify possible CatL substrates [24]. Since PEPS does not take into account the conditions of the environment, i.e. the pH of the compartment (lysosome vs cytosol), it is necessary to experimentally confirm the cleavage prediction using purified proteins [15, 16]. Using this algorithm, the first identified cleavage target in podocytes was the large GTPase dynamin. Dynamin is essential for the formation of clathrin-coated vesicles at the plasma membrane during endocytosis [29] and has also been implicated in the regulation of actin dynamics in certain cell types [30]. Dynamin is specifically cleaved in podocytes by CatL during LPS- or PAN-induced proteinuria in animal models and gene delivery of mutant dynamin forms resistant to cleavage by CatL protected mice from LPS-induced proteinuria [15]. Intact dynamin is required for proper podocyte structure and function [15]. Expression of dominant-negative dynamin mutants in podocytes caused proteinuria in vivo and led to a loss of actin stress fibers in vitro [15]. The role of dynamin in maintaining podocyte integrity does not depend on its function in endocytosis, but rather on its ability to stabilize F-actin organization in the FPs.
Synaptopodin is another major cleavage target of cytoplasmic CatL. Synaptopodin is the founding member of a unique class of proline-rich, actin-associated proteins that are expressed in highly dynamic cell compartments, such as the dendritic spine apparatus of neurons and podocyte FPs [31]. Synaptopodin binds to α-actinin and regulates the actin-bundling activity of α-actinin. Synaptopodin-deficient (synpo−/−) mice display impaired recovery from protamine sulfate-induced podocyte FP effacement and LPS-induced proteinuria. Similarly, synpo−/− podocytes show impaired actin filament reformation in vitro [32]. Synaptopodin is specifically proteolyzed at two cleavage sites by cytosolic CatL [16]. In vivo gene delivery or the podocyte-specific transgenic expression of a synaptopodin mutant that lacks these cleavage sites protected mice from LPS-induced proteinuria, suggesting that CatL-mediated cleavage of synaptopodin is required for the induction of FP effacement by LPS. Stabilized synaptopodin protein levels also help to maintain dynamin levels [16].
The good news about enzymes and substrates: treatment options seem logical
Why bother and try to clarify the disease process that underlies podocyte dysfunction, proteinuria and progression of renal disease? Since proteinuria is regarded as a silent but clearly contributable risk factor for kidney organ survival and for generalized health, novel efforts in the development of treatment options for proteinuric diseases need to be encouraged. The discovery of an enzymatic disease process within the podocytes harbors potential to either inhibit CatL or protect cleavage targets from being proteolyzed. The inhibition of CatL in general has been attempted in rats with glomerulonephritis and was partially successful [21]. We now know that the main deleterious action of CatL in podocytes stems from a novel CatL form that is active in the cytoplasm of podocytes (Fig. 4) and that is highly target-selective. It should be possible to develop CatL inhibitors that localize to the cytosol of podocytes and specifically inhibit the disease-causing CatL variant.
Fig. 4.
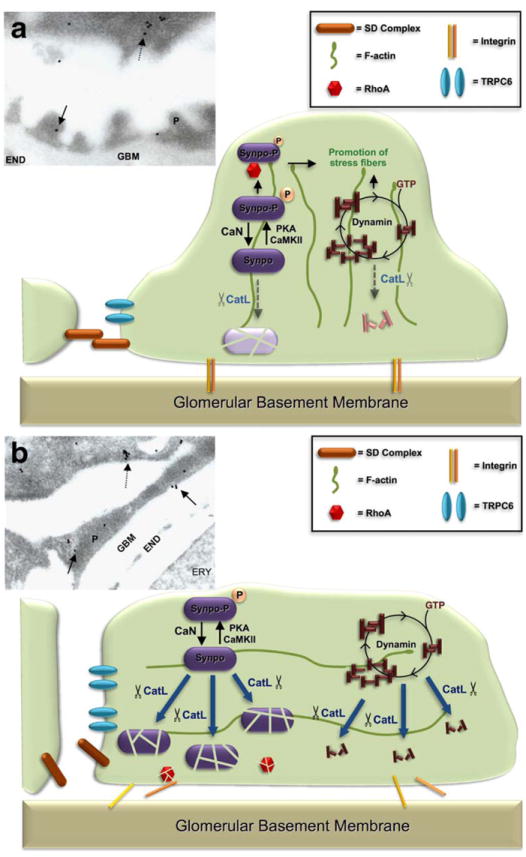
Cathepsin L (CatL) is induced in podocytes during FP effacement. a Schematic illustration of FP effacement and proteinuria as a podocyte enzymatic disease. Under normal conditions, synaptopodin and dynamin are involved in regulating podocyte F-actin. A small portion of CatL is in the cytosol and participates in a physiological turnover of synaptopodin and dynamin. The electron micrograph of control mice (insert), shows CatL expression located mainly in lysosomes of primary podocyte processes (dashed arrow). Only few gold labeling is found in FP (solid arrows). b The induction of cytosolic CatL causes proteolysis of synaptopodin and dynamin, thereby disrupting actin organization, causing podocyte FP effacement and proteinuria. The electron micrograph of LPS treated mice (insert) shows FP effacement and induction of CatL in lysosomes of primary processes (dashed arrow) and in effaced podocyte FPs (solid arrows); CaN calcineurin; PKA protein kinase A; CaMKII calcium-dependent protein kinase II; P podocyte; GBM glomerular basement membrane; END endothelial cells; ERY erythrocyte
This emerging concept of podocyte-directed therapy is further supported by our recent observation that the antiproteinuric activity of cyclosporine A (CsA) results from direct protection of the podocyte cytoskeleton from CatL-mediated injury [16]. CsA can induce remission of proteinuria in a number of diseases including MCD and FSGS [33, 34]. The well-characterized immunosuppressive action of CsA stems from the inhibition of the nuclear factor of activated T cells (NFAT) signaling by inhibiting the serine/threonine phosphatase calcineurin. Because T cell dysfunction is associated with some forms of proteinuria, including a subset of MCD in children, the antiproteinuric effect of CsA has been assumed to result from the inhibition of NFAT signaling in T cells. However, experimental support of this hypothesis is weak at best. Moreover, CsA can reduce proteinuria in human [35] and experimental [36] Alport syndrome, raising doubts about the above hypothesis. In the previously mentioned study by Faul et al. [16], a direct effect of CsA on podocytes has been demonstrated. Synaptopodin is phosphorylated at two amino acid residues by protein kinase A (PKA) and calcium/calmodulin-dependent protein kinase II (CaMKII) and dephosphorylated by calcineurin. Only in its phosphorylated form can synaptopodin bind to the chaperone-like protein 14-3-3 and is thereby protected from CatL-mediated cleavage [16]. Thus, inhibition of calcineurin during podocyte injury shifts the equilibrium between phosphorylated and dephosphorylated synaptopodin toward the phosphorylated form and thereby protects synaptopodin from CatL-mediated degradation. Since calcineurin is a calcium-sensitive phosphatase, one could easily envision a link to dysfunctional podocyte calcium channels, e.g. mutated TRPC6 [9, 10], or pathological TRPC6 overexpression [37], both known to cause glomerular disease [38].
Conclusions
Podocyte FP effacement can be caused by the translation of a novel CatL variant in the cytosol of podocyte FPs. CatL is induced in many proteinuric diseases. So far, two major cleavage targets have been described: dynamin and synaptopodin. Both proteins are critical regulators of podocyte cytoskeletal function. Additional targets are being investigated. The unraveling of these pathways not only greatly enhances our understanding of the pathophysiology of glomerular diseases, but also enables the development of specific therapies for proteinuric syndromes by directly targeting components of these enzymatic cascades in podocytes.
Acknowledgments
This work was supported by US National Institutes of Health (NIH) grant DK073495 (to J.R.).
Contributor Information
Andreas D. Kistler, Department of Nephrology, University Hospital, Zürich, Switzerland Miami Institute of Renal Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 NW 10th Street, Batchelor Bldg. #633A, Miami, FL 33136, USA.
Vasil Peev, Miami Institute of Renal Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 NW 10th Street, Batchelor Bldg. #633A, Miami, FL 33136, USA.
Anna-Lena Forst, Miami Institute of Renal Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 NW 10th Street, Batchelor Bldg. #633A, Miami, FL 33136, USA.
Shafic El Hindi, Miami Institute of Renal Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 NW 10th Street, Batchelor Bldg. #633A, Miami, FL 33136, USA.
Mehmet M. Altintas, Miami Institute of Renal Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 NW 10th Street, Batchelor Bldg. #633A, Miami, FL 33136, USA
Jochen Reiser, Email: jreiser@med.miami.edu, Miami Institute of Renal Medicine, Division of Nephrology and Hypertension, University of Miami Miller School of Medicine, 1580 NW 10th Street, Batchelor Bldg. #633A, Miami, FL 33136, USA.
References
- 1.Hogg RJ, Portman RJ, Milliner D, Lemley KV, Eddy A, Ingelfinger J. Evaluation and management of proteinuria and nephrotic syndrome in children: recommendations from a pediatric nephrology panel established at the National Kidney Foundation conference on proteinuria, albuminuria, risk, assessment, detection, and elimination (PARADE) Pediatrics. 2000;105:1242–1249. doi: 10.1542/peds.105.6.1242. [DOI] [PubMed] [Google Scholar]
- 2.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 3.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 4.Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. 2009;5:463–468. doi: 10.1038/nrneph.2009.108. [DOI] [PubMed] [Google Scholar]
- 5.Reiser J, Oh J, Shirato I, Asanuma K, Hug A, Mundel TM, Honey K, Ishidoh K, Kominami E, Kreidberg JA, Tomino Y, Mundel P. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem. 2004;279:34827–34832. doi: 10.1074/jbc.M401973200. [DOI] [PubMed] [Google Scholar]
- 6.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 8.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 9.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 10.Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Muller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’Toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nurnberg P, Hildebrandt F. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 13.Smoyer WE, Mundel P. Regulation of podocyte structure during the development of nephrotic syndrome. J Mol Med. 1998;76:172–183. doi: 10.1007/s001090050206. [DOI] [PubMed] [Google Scholar]
- 14.Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 15.Sever S, Altintas MM, Nankoe SR, Moller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, Cohen CD, Kretzler M, Kerjaschki D, Rudensky A, Nikolic B, Reiser J. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. 2007;117:2095–2104. doi: 10.1172/JCI32022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett AJ, Kirschke H. Cathepsin B, cathepsin H, and cathepsin L. Methods Enzymol. 1981;80(Pt C):535–561. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- 18.Ishidoh K, Kominami E. Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem Biophys Res Commun. 1995;217:624–631. doi: 10.1006/bbrc.1995.2820. [DOI] [PubMed] [Google Scholar]
- 19.Asanuma K, Shirato I, Ishidoh K, Kominami E, Tomino Y. Selective modulation of the secretion of proteinases and their inhibitors by growth factors in cultured differentiated podocytes. Kidney Int. 2002;62:822–831. doi: 10.1046/j.1523-1755.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- 20.Chauhan SS, Goldstein LJ, Gottesman MM. Expression of cathepsin L in human tumors. Cancer Res. 1991;51:1478–1481. [PubMed] [Google Scholar]
- 21.Baricos WH, O’Connor SE, Cortez SL, Wu LT, Shah SV. The cysteine proteinase inhibitor, E-64, reduces proteinuria in an experimental model of glomerulonephritis. Biochem Biophys Res Commun. 1988;155:1318–1323. doi: 10.1016/s0006-291x(88)81285-3. [DOI] [PubMed] [Google Scholar]
- 22.Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell. 2004;14:207–219. doi: 10.1016/s1097-2765(04)00209-6. [DOI] [PubMed] [Google Scholar]
- 23.Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, Allis CD. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell. 2008;135:284–294. doi: 10.1016/j.cell.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohmuller T, Wenzler D, Hagemann S, Kiess W, Peters C, Dandekar T, Reinheckel T. Toward computer-based cleavage site prediction of cysteine endopeptidases. Biol Chem. 2003;384:899–909. doi: 10.1515/BC.2003.101. [DOI] [PubMed] [Google Scholar]
- 25.Baricos WH, Zhou Y, Mason RW, Barrett AJ. Human kidney cathepsins B and L. Characterization and potential role in degradation of glomerular basement membrane. Biochem J. 1988;252:301–304. doi: 10.1042/bj2520301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baricos WH, Cortez SL, Le QC, Zhou YW, Dicarlo RM, O’Connor SE, Shah SV. Glomerular basement membrane degradation by endogenous cysteine proteinases in isolated rat glomeruli. Kidney Int. 1990;38:395–401. doi: 10.1038/ki.1990.218. [DOI] [PubMed] [Google Scholar]
- 27.He L, Sun Y, Patrakka J, Mostad P, Norlin J, Xiao Z, Andrae J, Tryggvason K, Samuelsson T, Betsholtz C, Takemoto M. Glomerulus-specific mRNA transcripts and proteins identified through kidney expressed sequence tag database analysis. Kidney Int. 2007;71:889–900. doi: 10.1038/sj.ki.5002158. [DOI] [PubMed] [Google Scholar]
- 28.Paczek L, Pazik J, Teschner M, Schaefer RM, Rowinski W, Szmidt J, Lao M, Abgarowicz K, Gradowska L, Morzycka-Michalik M, Heidland A. Human chronic kidney allograft rejection is accompanied by increased intraglomerular cathepsin B and L activity. Transpl Int. 1994;7(Suppl 1):S311–S313. doi: 10.1111/j.1432-2277.1994.tb01377.x. [DOI] [PubMed] [Google Scholar]
- 29.Kirchhausen T. Three ways to make a vesicle. Nat Rev Mol Cell Biol. 2000;1:187–198. doi: 10.1038/35043117. [DOI] [PubMed] [Google Scholar]
- 30.Schafer DA. Regulating actin dynamics at membranes: a focus on dynamin. Traffic. 2004;5:463–469. doi: 10.1111/j.1600-0854.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- 31.Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 33.Meyrier A. Treatment of focal segmental glomerulosclerosis. Expert Opin Pharmacother. 2005;6:1539–1549. doi: 10.1517/14656566.6.9.1539. [DOI] [PubMed] [Google Scholar]
- 34.Meyrier A. An update on the treatment options for focal segmental glomerulosclerosis. Expert Opin Pharmacother. 2009;10:615–628. doi: 10.1517/14656560902754029. [DOI] [PubMed] [Google Scholar]
- 35.Charbit M, Gubler MC, Dechaux M, Gagnadoux MF, Grunfeld JP, Niaudet P. Cyclosporin therapy in patients with Alport syndrome. Pediatr Nephrol. 2007;22:57–63. doi: 10.1007/s00467-006-0227-y. [DOI] [PubMed] [Google Scholar]
- 36.Chen D, Jefferson B, Harvey SJ, Zheng K, Gartley CJ, Jacobs RM, Thorner PS. Cyclosporine a slows the progressive renal disease of alport syndrome (X-linked hereditary nephritis): results from a canine model. J Am Soc Nephrol. 2003;14:690–698. doi: 10.1097/01.asn.0000046964.15831.16. [DOI] [PubMed] [Google Scholar]
- 37.Moller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 38.Moller CC, Flesche J, Reiser J. Sensitizing the Slit Diaphragm with TRPC6 Ion Channels. J Am Soc Nephrol. 2009;20:950–953. doi: 10.1681/ASN.2008030329. [DOI] [PubMed] [Google Scholar]
- 39.Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, Faul C, Kretzler M, Davidson A, Sugimoto H, Kalluri R, Sharpe AH, Kreidberg JA, Mundel P. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]