

Abstract
Small molecules that bind to normally unoccupied thyroxine (T4) binding sites within transthyretin (TTR) in the blood stabilize the tetrameric ground state of TTR relative to the dissociative transition state and dramatically slow tetramer dissociation, the rate-limiting step for the process of amyloid fibril formation linked to neurodegeneration and cell death. These so-called TTR kinetic stabilizers have been designed using structure-based principles and one of these has recently been shown to halt the progression of a human TTR amyloid disease in a clinical trial, providing the first pharmacologic evidence that the process of amyloid fibril formation is causative. Structure-based design has now progressed to the point where highly selective, high affinity TTR kinetic stabilizers that lack undesirable off-target activities can be produced with high frequency.
Transthyretin aggregation appears to cause the transthyretin amyloidoses
The TTR amyloid diseases (amyloidoses) appear to be caused by extracellular TTR tetramer dissociation, monomer misfolding and misassembly into a variety of aggregate morphologies [1,2,3•] (Figure 1). The field has hypothesized that oligomers formed during the process of amyloidogenesis lead to cellular toxicity [4,5]. The displacement of tissue by extracellular amyloid is also thought to exacerbate pathology [6]. The TTR amyloidoses include senile systemic amyloidosis (SSA), familial amyloid cardiomyopathy (FAC), familial amyloid polyneuropathy (FAP), and central nervous system selective amyloidoses (CNSA) [3•]. Those amyloidoses impacting the most patients include SSA (10–25% of the elderly population) [7] and FAC (3–4% of African Americans), leading to congestive heart failure [8] (Figure 2). Compelling genetic evidence is supportive of the amyloid hypothesis in the TTR amyloidoses, where the notion is that the process of amyloidogenesis is causatively linked to disease pathology [9•,10].
Figure 1.
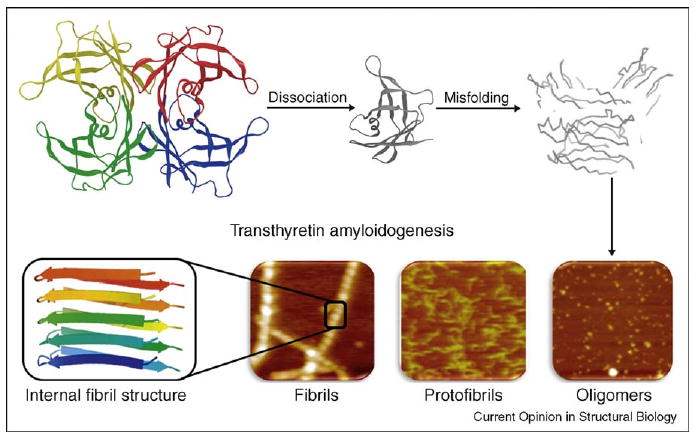
The transthyretin (TTR) amyloidogenesis cascade. For amyloidogenesis to occur, the TTR tetramer must dissociate into four folded monomers and undergo partial denaturation in order to subsequently misassemble into a spectrum of aggregate structures including cross-β-sheet amyloid fibrils.
Figure 2.
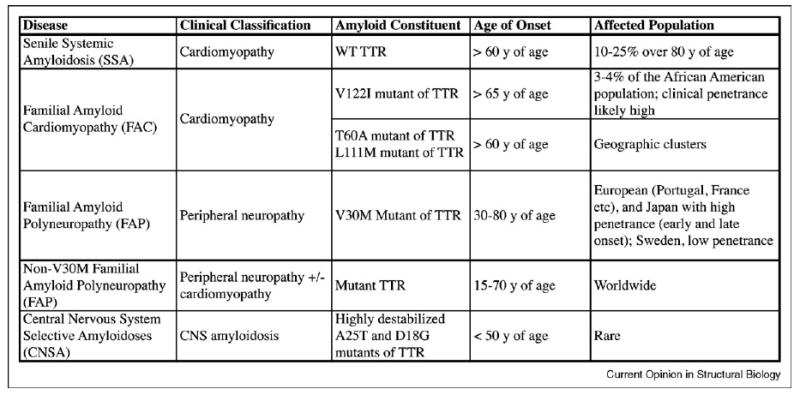
A table summarizing the transthyretin amyloidoses.
TTR is a homotetrameric protein, comprising 127-amino-acid, β-sheet-rich subunits [11,12], (Figure 3A). Amyloidogenesis occurs when the thermodynamically linked equilibrium between tetramer, natively folded monomer and partially denatured monomer is shifted toward the partially denatured TTR monomer that is amyloidogenic (Figure 1) [13]. The energetically weaker dimer–dimer interface of TTR (i, ii/iii, iv) creates the two T4 binding sites (Figure 3A and B) that are bisected by the crystallographic two-fold axis (C2) [14•]. In humans, the vast majority (>99.5%) of the TTR T4 binding sites in blood are unoccupied and available for small molecule binding [3•]. Occupancy of these sites by small molecule kinetic stabilizers has recently been shown to halt neurodegeneration in FAP patients (foldrx.com). TTR kinetic stabilizers differentially stabilize the tetramer over the dissociative transition state by selectively binding to the tetrameric state. This binding increases the free energy of activation for dissociation, dramatically slowing tetramer dissociation, which is rate limiting for TTR fibril formation (Figure 3C). This review focuses on recent developments, and outlines the current state of structure-based design of TTR ligands that kinetically stabilize TTR and prevent the process of amyloid fibril formation.
Figure 3.
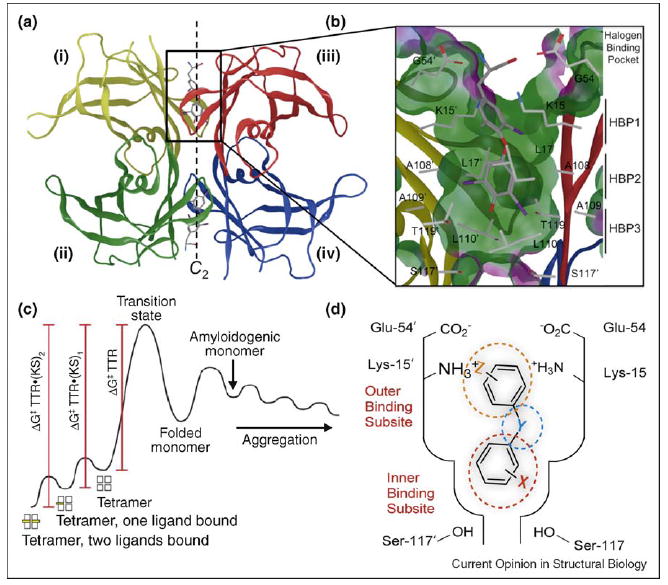
Structure of transthyretin•(T4)2 complex and the basis for TTR kinetic stabilization. (a) Crystal structure of TTR with T4 bound to each T4 binding site (PDB accession code 2ROX [16]). (b) Close-up view of one T4 binding site with T4 bound. Primed amino acids refer to those comprising symmetry-related halogen binding pockets (HBPs) of TTR monomers. (b) Kinetic stabilization of TTR by small molecule binding to the natively folded tetramer stabilizes the ground state and increases the tetramer dissociation barrier, preventing amyloidogenesis and pathology. (d) Schematic depiction of one of the two T4 binding pockets within TTR occupied by a small molecule TTR kinetic stabilizer, where Y represents a linker of variable chemical structure (e.g. NH, O, CH=CH, C(O)NH, etc.) joining the two aryl rings, which typically bear a combination of alkyl, carboxyl, halide, trifluoromethyl, or hydroxyl substituents (X and Z). The inner and outer binding subsites are indicated.
Transthyretin's thyroxine binding sites
Each T4 binding site is characterized by a series of subsites [15•] (Figure 3D), an outer binding subsite, an inner binding subsite, and an intervening interface that are all composed of pairs of symmetric hydrophobic depressions referred to as halogen binding pockets (HBPs), wherein the iodine atoms of T4 reside (Figure 3B) [16]. HBPs 1 and 1′ in the outer binding subsite comprise the side chains of Ala108/108′, Thr106/106′, Met13/13′ and Lys15/15′ with the pocket lined by methyl and methylene groups of Lys15/15′, Ala108/108′ and Thr106/106′, whereas HBPs 2 and 2′, are made up of the side chains from Leu110/110′, Ala109/109′, Lys15/15′ and Leu17/17′. Finally, HBPs 3 and 3′ are composed of the methyl and methylene groups of Ser117/117′, Thr119/119′, Ala109/109′ and Leu110/110′ (Figure 3B) [16]. T4 and most ligands bind to TTR with negative cooperativity, apparently resulting from conformational changes within the tetramer upon binding to the first T4 site [3•], although this has yet to be definitively confirmed crystallographically. Comparative studies of TTR from human and rat in complex with T4 suggest that conformational changes and hydrogen bonding to Lys15/15′ in the outer subsite and Ser117/117′ in the inner subsite affect both binding orientation and ligand affinity [17].
Small molecule TTR ligands and the basis for kinetic stabilization
Over one thousand aromatic small molecules exhibiting structural complementarity to the T4 binding sites within TTR have been synthesized [18–25,26••,27••,28••,29,30] and are typically composed of two aromatic rings occupying the inner and outer T4 binding subsites (Figure 3D). The aromatic rings composing TTR kinetic stabilizers can either be linked directly (e.g. biphenyls) [19,20] or tethered through short hydrophobic linkers [26••,27••] (Figure 3D). Most of the kinetic stabilizers were conceived of by structure-based drug design principles, taking advantage of > 100 high resolution TTR•(small molecule)2 crystal structures [11,16,19,21,22,26••,27••,28••,31–54] (PDB accession codes and compounds are listed in Appendix A, Table S1).
Ligands that bind to the T4 sites do so utilizing two dominant forces, the hydrophobic effect and electrostatic interactions. Proper choice of the aryl substitution pattern and hydrophobic substituents enable maximal occupancy of the HBPs, thus facilitating ligand interactions with neighboring monomeric subunits through hydrophobic interactions. These bridging hydrophobic interactions help impose kinetic stabilization on the weaker dimer–dimer interface of TTR by stabilizing the tetramer over the dissociative transition state, thereby raising the barrier for tetramer dissociation [9•], Figure 3C. Bridging hydrogen bonds between the ligand substructure in the inner binding subsite and the Thr116/116′ or Ser117/117′ residues of TTR further differentially stabilize the native non-amyloidogenic tetramer. Lysine and glutamic acid residues (Lys15/15′, Glu54/54′) on the periphery of the outer T4 binding subsite electrostatically complement polar substituents on the aryl rings further stabilizing the native state as also observed in the binding of T4 whose ammonium groups salt bridge with Glu54/54′ [16] (Figure 3B and D). These tetramer stabilizing forces combine to make the dissociation barrier insurmountable under physiological conditions (Figure 3C) [3•,9•,47].
Structure-based design of TTR kinetic stabilizers
Kinetic stabilizers for the treatment of the TTR amyloidoses must be both highly potent and selective. The ligands must not interact with the thyroid hormone receptor (THR), a major concern given the similarity of some kinetic stabilizers with triiodothyronine (T3, the primary thyroid hormone) and T4 (the prohormone). Nonsteroidal anti-inflammatory drug (NSAID) activity is also contraindicated for treating TTR amyloidosis patients; thus, TTR kinetic stabilizers should exhibit minimal NSAID activity [55]. While sometimes unpredictable, these activities can usually be minimized by maximizing TTR amyloid inhibition potency, TTR binding affinity and binding selectivity to TTR in human plasma, which we have accomplished using a combination of structure-based drug design and assays to evaluate the above mentioned criteria [3•].
A wide variety of TTR kinetic stabilizers have been identified including naturally derived flavonoid and xanthone derivatives [56–58], as well as synthetic compounds belonging to five families including: bisaryloxime ethers, biphenyls, 1-aryl-4,6-biscarboxydibenzofurans, 2-phenylbenzoxazoles and biphenylamines [18–25, 26••,27••,28••,29,30]. Kinetic stabilizers have also been identified through halogenation of NSAIDs, such as salicylic acid [52], diflunisal [21,48,59,60], and analogues of flufenamic [53,60–62]. More recently, additional ligands, such as isatin [63] and β-aminoxypropionic acid linked aryl or fluorenyl derivatives have been identified [54,56]. As of 2005, the primary candidates for the most potent and selective kinetic stabilizers were biphenyls, 2-phenylbenzoxazoles and dibenzofurans, since some bisaryloxime ethers have lower than desirable chemical stability [18]. This relative lack of selective, structurally diverse TTR kinetic stabilizers is largely due to the fact that, until recently, no systematic optimization had been attempted on the three structural elements composing a typical TTR amyloidogenesis inhibitor [26••,27••,28••].
Optimization of the ideal aryl substituents and their substitution pattern on aromatic rings X and Z, as well as the linker substructure joining the rings (Figure 3D), revealed numerous high affinity solutions for occupancy of TTR's thyroxine binding sites [26••,27••,28••]. On the basis of these new SAR data, it is now possible to make accurate predictions of which structures will exhibit TTR kinetic stabilizer potency, TTR plasma binding selectivity and the desired binding orientation [64••]. Detailed herein is a summary of the emerging trends from this structure-based, TTR kinetic stabilizer design, with an exhaustive tabulated list of recently synthesized TTR kinetic stabilizers, complete with so-called efficacy scores (Appendix A, equation S2), integrating TTR amyloidogenesis inhibitor potency and TTR binding selectivity in plasma (Appendix A, Tables S3, S5 and S7).
Optimal aryl-X ring substructures
To optimize the aryl-X ring, a library of 2-arylbenzoxazoles was synthesized in which the benzoxazole ring was purposefully unmodified and the 2-aryl ring was systematically substituted (Figure 4A). The library members were evaluated for TTR amyloid inhibition potency, plasma TTR binding selectivity, as well as undesired COX-1 and THR binding activity (Appendix A, Table S4). Substituents at the 3 and 4 positions and, especially, the 3, 4 and 5 positions afforded the most potent and selective TTR amyloidogenesis inhibitors (Appendix A, Table S3). The most potent inhibitors had a 4-hydroxyl substituent and hydrophobic substituents at the 3 and 5 positions [27••,64••,65]. The results showed that hydrogen and fluorine substituents at the 3 and 5 positions tended to afford poorer TTR kinetic stabilizers than their chloro, bromo, iodo, and methyl counterparts, probably due to less efficient interactions with the HBPs (Figure 4A). Halogens are known to contribute more to ligand binding affinity than would be expected on the basis of their hydrophobicity alone. This enhanced binding is probably due to formation of a charge-transfer complex between the halogen and a neighboring oxygen atom [65,66]. 3,5-substituents are generally superior to 3-substituents only. Substitution at both the 3 and the 5 position enables simultaneous interactions with HBPs on adjacent TTR subunits, whereas mono-meta-substitution enables occupancy of only one HBP on one subunit, less efficiently stabilizing the weaker dimer–dimer interface [27••]. Recent studies concerning the iodination of salicylic acid show that 3,5-diiodo salicylic acid binds in the inner T4 binding subsite, interacting with HBP 3 and 3′ as would be predicted by these results. However, these data demonstrate that a second ring is needed to occupy the outer T4 binding subsite for high binding affinity, also consistent with our observations [52].
Figure 4.
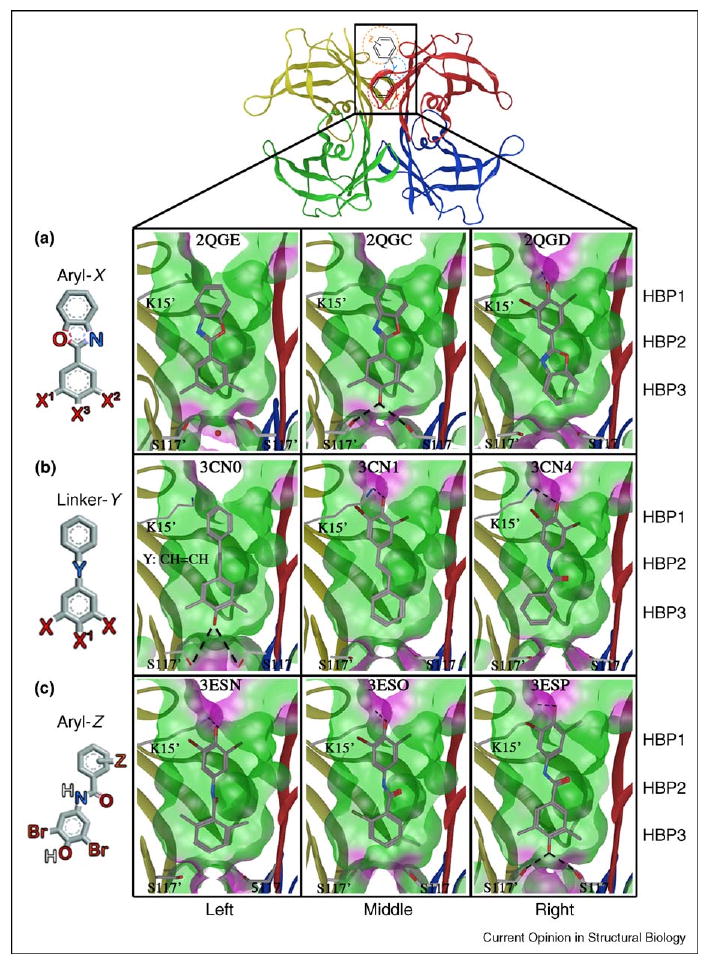
Crystal structures of WT–TTR in complex with kinetic stabilizers from substructure X, Y and Z optimization studies. Ribbon diagram depiction of WT–TTR highlighting one of the two identical T4 binding sites containing a schematic representation of a typical TTR kinetic stabilizer. (a) Aryl-X substructure optimization, PDB accession codes 2QGE, 2QGC and 2QGD, as indicated. (b) Linker-Y substructure optimization, PDB accession codes 3CN0, 3CN1 and 3CN4, as indicated. (c) Aryl-Z substructure optimization, PDB accession codes 3ESN, 3ESO and 3ESP, as indicated. All structures have ‘Connolly analytical’ surface representation of the pocket (green = hydrophobic, purple = polar), defined as all residues within 8 Å of ligand. Lysine 15′ and serines 117′ and 117 are shown with corresponding bonds with ligand. Figure is generated using the program MOE (2008.10), Chemical Computing Group, Montreal, Canada.
The electrostatic interactions made by the 4-hydroxyl group also appear to be important in maximally stabilizing the tetrameric, non-amyloidogenic state of TTR. In the case of the 2-(3,5-dimethyl-hydroxyphenyl)-benzoxazole, the phenol forms hydrogen bonds that bridge Ser117/117′ hydroxyls deep in the inner binding subsite (Figure 4A, middle panel), mimicking the structural water oxygen placement seen in the apo TTR structure 2QGB or in 2QGE where the ligand is devoid of a 4-hydroxyl group [27••] (Figure 4A, left panel). Electron-donating 3,5-dimethyl substituents result in a higher 4-hydroxyl pKa (calculated to be 9.21), enabling it to be protonated, thus favoring hydrogen bonding with the Ser117/117′ hydroxyls in the so-called ‘forward’ binding mode. By contrast, in the isostructural 2-(3,5-dibromo-4-hydroxyphenyl)-benzoxazole, the substituted ring is placed in the outer binding subsite in ‘reverse’ binding mode with the putative phenolate substituent engaged in an electrostatic interaction with the ε-NH3+ of Lys15/15′ (Figure 4A, right panel), apparently owing to the lower pKa of the phenol (calculated to be 5.35) [27••]. Thus, the 3,5-X2-4-hydroxyphenyl ring binding orientation in the context of 2-arylbenzoxazoles is predicted by the pKa of the phenol. These amyloidogenesis inhibitors display little, if any, inhibition of COX-1 activity, and with the exception of the thyroxine-like 3,5-I2-4-OH substituted 2-aryl ring, the majority of the most potent TTR amyloidogenesis inhibitors do not significantly interact with the THR either (Appendix A, Table S4).
Optimal linker substructures
We next evaluated possible substructures to link the aryls occupying the inner and outer TTR T4 binding subsites, including CH=CH, −CH2CH2−, polar 1–3 atom linkers and fused five-membered ring heteroaromatic linkers. One aromatic ring is always unsubstituted in analogues made for this purpose, whilst the other ring comprises a 3,5-X2 or a 3,5-X2-4-hydroxyphenyl substructure, where X is Br or Me (Figure 4B and Appendix A, Table S5). Analysis of the numerous TTR•(ligand)2 structures in the Protein Data Bank (PDB) reveals that the two aryl rings are typically bound out-of-plane with respect to each other, thus optimal linkers will probably have to permit this [18–22,26••,27••,28••,47]. Co-consideration of TTR amyloid inhibition potency and plasma TTR binding selectivity revealed that the direct linkage of two aryl rings (i.e. biphenyls or 2-arylbenzoxazole), or linkage through a nonpolar substructure such as an E-olefin or a −CH2CH2− bridge, afforded the most potent and selective TTR amyloidogenesis inhibitors [26••] (Appendix A, Table S5). Irrespective of whether a compound binds in the ‘forward’ or ‘reverse’ orientation, the linker is positioned within the hydrophobic vicinity of HBPs 2 and 2′ (Figure 4B, left and middle panels). Stilbenes composed of a trans-CH=CH linker exhibit excellent structural complementarity to the hydrophobic micro-environments in the vicinity of HBPs 2 and 2′ (Figure 4B, left and middle panels), unlike polar amide linkers (Figure 4B, right panel). The CH=CH linker positioned proximal to HBPs 2 and 2′ interacts with Leu17/17′, Ala108/108′, Leu110/110′ and Val121/121′ side chains as seen in the TTR• (Resveratrol)2 structure (Figure S9) [22]. Of the 40 compounds tested, only four displayed appreciable inhibition of COX-1 activity (Appendix A, Table S6). All four were composed of a common 3,5-dimethyl-4-hydroxyphenyl substructure but contained distinct linkers, suggesting the linker contribution to COX-1 activity is negligible.
Optimal aryl-Z ring substructures
Evaluation of aryl-Z ring candidates was carried out using a library composed of a fixed aryl-X substructure (N-(3,5-dibromo-4-hydroxyphenyl) as well as a fixed and suboptimal amide linker (Figure 4C) [28••]. Regardless of the diverse range of aryl-Z substituents evaluated, most were capable of increasing inhibitor potency and plasma TTR binding selectivity (Appendix A, Table S7). Independent of the substituent, 2,6-substituents are generally more potent and selective than 2,5-substituents that are more potent and selective than 2-substituents [3,18– 25,26••,27••,28••,29,30,47]. This effect is probably due to more efficient hydrophobic interactions with HBPs from adjacent TTR subunits in the case of bis-alkyl and halide substituted aromatics (Figure 4C). Of 56 compounds synthesized, only 5 were observed to displace >10% of T3 from the THR (Appendix A, Table S8). The random distribution of substitution patterns in thyroid-displacing TTR kinetic stabilizers suggests that this undesirable characteristic could be prevented, perhaps by employing an optimal linker.
Substructure combination strategy is effective
Using SAR data from the aryl-X, aryl-Z and linker-Y substructure optimization studies [26••,27••,28••], we envisioned that a library rich in highly potent and selective TTR kinetic stabilizers could be generated by simply combining the most highly ranked substructures.
As a demonstration, we utilized the highly ranked 3,5-dibromo-4-hydroxyphenyl aryl-X ring (and isosteric variants replacing the OH functional group with a NH2), a variety of highly ranked aryl-Z rings and two hydrophobic linkers (trans−CH=CH− and −CH2CH2−) that previously yielded potent TTR kinetic stabilizers [22,26••] (Figure 5).
Figure 5.
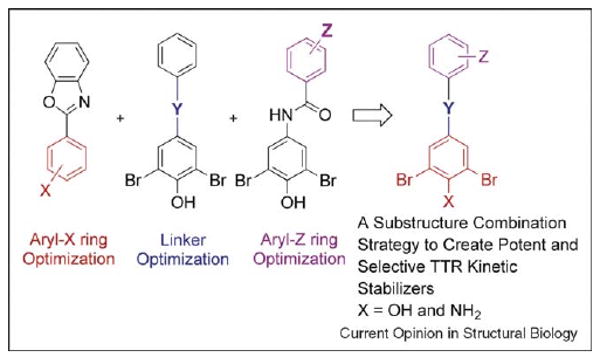
Overview of the substructure combination strategy. Potent and selective TTR kinetic stabilizers were created by combining highly ranked aryl-X and aryl-Z rings with appropriate linker-Y.
Of the 88 3,5-dibromo-4-hydroxyphenyl-based stilbene and dihydrostilbene compounds evaluated, 80 were excellent amyloidogenesis inhibitors allowing < 10% WT–TTR fibril formation at a concentration of 7.2 μM (minimal concentration required to occupy both T4 sites) and between 15 and 30% fibril formation at a concentration of 3.6 μM. These potent compounds were further evaluated for their binding selectivity for TTR in human plasma. Notably, 37 of these exhibited exceptional selectivity to plasma TTR with >1.5 equivalents bound per tetramer (the maximum being 2). Nonetheless, it was disappointing that more compounds than would have been predicted displaced T3 at the THR, with 70% of the promising stilbene compounds displacing >20% of T3. In the case of the stilbenes that displace T3 from the THR, replacing the CH=CH linker with a dihydrostilbene (CH2−CH2) linker yielded a library that was largely free of T3 THR displacement (24% displacing >20% of T 3) [64••].
The 4-amino group was found to be equivalent to the 4-hydroxyl group in terms of potency and exhibited excellent TTR binding selectivity in plasma. Notably, none of the library members composed of a 4-NH2 displaced T3 from the THR, suggesting that further aryl-X optimization may be an effective strategy to reduce THR binding [64••]. While the 3,5–Br2–4–OH ring prefers to occupy the outer binding subsite in the ‘reverse’ binding mode [26••,27••,28••], the 3,5-dibromo-4-aminophenyl ring strongly prefers to occupy the inner binding subsite in the ‘forward’ binding mode, and identifies a key piece of new data in that it is desirable to have more substructures that bind with subsite selectivity.
The kinetic stabilizer efficacy scores (integrating TTR amyloid inhibition efficacy and TTR binding selectivity data in human plasma) reveal that the substructure combination strategy is very effective for efficiently producing TTR kinetic stabilizers, in that nearly 70% of the stilbenes examined and 93% of the dihydrostilbenes examined exhibit exceptional TTR kinetic-stabilizer activity [64••].
Conclusions
The systematic optimization of the three substructures comprising a typical TTR kinetic stabilizer has enabled rank ordering of these substructural components and these data now enable us to confidently predict the structures of diverse, potent and highly selective TTR kinetic stabilizers that inhibit TTR amyloidogenesis in vivo. From the high-resolution X-ray crystallographic data, we now know why some aryls prefer the inner binding subsite and others prefer the outer binding subsite. We also now appreciate that those that maximize HBP occupancy and electrostatic interactions with one or more TTR subunits are the most potent kinetic stabilizers [26••,27••,28••]. The substructure subsite preferences revealed by X-ray crystallography now inform us regarding which aryl-X, aryl-Y and linker substructures should be combined to yield potent and selective TTR kinetic stabilizers.
Supplementary Material
Acknowledgments
We are grateful to the NIH (DK46335 to JWK and AI42266 to IAW), as well as the Skaggs Institute for Chemical Biology and the Lita Annenberg Hazen Foundation for long standing financial support. The progress outlined within would not have been possible without the hard work, determination, and creativity of numerous co-workers cited within, including those who co-authored this review.
Footnotes
This review comes from a themed issue on Folding and binding
Edited by Laura Itzhaki and Peter Wolynes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.sbi.2009.12.009.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 2.Kelly JW, Colon W, Lai Z, Lashuel HA, McCulloch J, McCutchen SL, Miroy GJ, Peterson SA. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv Protein Chem. 1997;50:161–181. doi: 10.1016/s0065-3233(08)60321-6. [DOI] [PubMed] [Google Scholar]
- 3•.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]; Review summarizing why ligand binding causes stabilization of the native state over the dissociative transition state raising the kinetic barrier for dissociation and, thus preventing amyloidogenesis.
- 4.Walsh DM, Selkoe DJ. Abeta oligomers– a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 5.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 6.Pepys MB. Amyloidosis. In: Little me. Brown and Company, editor. Immunological Diseases. Vol. 1. 1988. pp. 631–674. [Google Scholar]
- 7.Cornwell GC, Sletten K, Johansson B, Westermark P. Evidence that the amyloid fibril protein in senile systemic amyloidosis is derived from normal prealbumin. Biochem Biophys Res Commun. 1988;154:648–653. doi: 10.1016/0006-291x(88)90188-x. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 9•.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]; A comparison of pharmacologic and genetic evidence demonstrating that kinetic stabilization of TTR should prevent amyloid disease. This manuscript describes a series of transthyretin amyloidosis inhibitors that functioned by increasing the kinetic barrier for dissociation. The trans-suppressor mutation, T119 M, which is known to ameliorate familial amyloid disease, was also shown to function through kinetic stabilization.
- 10.Coelho T, Carvalho M, Saraiva MJ, Alves I, Almeida MR, Costa PP. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR Met 30 and TTR Met 119. J Rheumatol. 1993;20:179. [Google Scholar]
- 11.Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 12.Blake CC, Geisow MJ, Swan ID, Rerat C, Rerat B. Structure of human plasma prealbumin at 2–5 Å resolution. A preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J Mol Biol. 1974;88:1–12. doi: 10.1016/0022-2836(74)90291-5. [DOI] [PubMed] [Google Scholar]
- 13.Hurshman Babbes AR, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry. 2008;47:6969–6984. doi: 10.1021/bi800636q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.Hornberg A, Eneqvist T, Olofsson A, Lundgren E, Sauer-Eriksson AE. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J Mol Biol. 2000;302:649–669. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]; Structural comparisons of three wild-type, three non-amyloidogenic mutants, seven amyloidogenic mutants and nine complexes of TTR.
- 15•.Sacchettini JC, Kelly JW. Therapeutic strategies for human amyloid diseases. Nat Rev Drug Discov. 2002;1:267–275. doi: 10.1038/nrd769. [DOI] [PubMed] [Google Scholar]; A review of both small-molecule and macromolecular approaches for intervention in human amyloid diseases.
- 16.Wojtczak A, Cody V, Luft JR, Pangborn W. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-l-thyronine at 2.2 Å resolution. Acta Crystallogr D Biol Crystallogr. 1996;52:758–765. doi: 10.1107/S0907444996003046. [DOI] [PubMed] [Google Scholar]
- 17.Cody V. Mechanisms of molecular recognition: crystal structure analysis of human and rat transthyretin inhibitor complexes. Clin Chem Lab Med. 2002;40:1237–1243. doi: 10.1515/CCLM.2002.214. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SM, Petrassi HM, Palaninathan SK, Mohamedmohaideen NN, Purkey HE, Nichols C, Chiang KP, Walkup T, Sacchettini JC, Sharpless KB, et al. Bisaryloxime ethers as potent inhibitors of transthyretin amyloid fibril formation. J Med Chem. 2005;48:1576–1587. doi: 10.1021/jm049274d. [DOI] [PubMed] [Google Scholar]
- 19.Purkey HE, Palaninathan SK, Kent KC, Smith C, Safe SH, Sacchettini JC, Kelly JW. Hydroxylated polychlorinated biphenyls selectively bind transthyretin in blood and inhibit amyloidogenesis: rationalizing rodent PCB toxicity. Chem Biol. 2004;11:1719–1728. doi: 10.1016/j.chembiol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN, Deechongkit S, Chiang KP, Dendle MT, Sacchettini JC, et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew Chem Int Ed Engl. 2003;42:2758–2761. doi: 10.1002/anie.200351179. [DOI] [PubMed] [Google Scholar]
- 21.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47:355–374. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 22.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 23.Oza VB, Petrassi HM, Purkey HE, Kelly JW. Synthesis and evaluation of biaryl amines and benzylamines as inhibitors of TTR amyloid fibril formation. Book of Abstracts, 217th ACS National Meeting; Anaheim, California. 1999. ORGN-447. [Google Scholar]
- 24.Oza VB, Petrassi HM, Purkey HE, Kelly JW. Synthesis and evaluation of anthranilic acid-based transthyretin amyloid fibril inhibitors. Bioorg Med Chem Lett. 1999;9:1–6. doi: 10.1016/s0960-894x(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 25.Oza VB, Smith C, Raman P, Koepf EK, Lashuel HA, Petrassi HM, Chiang KP, Powers ET, Sachettinni J, Kelly JW. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J Med Chem. 2002;45:321–332. doi: 10.1021/jm010257n. [DOI] [PubMed] [Google Scholar]
- 26••.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2008;51:6348–6358. doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first of three studies aimed at systematically optimizing the three substructures comprising typical TTR kinetic stabilizers, the two aryl rings and the linker joining them. This paper outlines biochemical and structural data on a library of 2-arylbenzoxazoles bearing thyroid hormone-like aryl substituents on the 2-aryl ring to optimize the aryl-X substructure.
- 27••.Johnson SM, Connelly S, Wilson IA, Kelly JW. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem. 2008;51:260–270. doi: 10.1021/jm0708735. [DOI] [PubMed] [Google Scholar]; The second of three studies optimizing the linker substructure comprising typical TTR kinetic stabilizers, whereby 40 molecules composed of 10 unique linker substructures were evaluated both biochemically and structurally to determine how these linkages influence inhibitor potency and selectivity.
- 28••.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward optimization of the second aryl substructure common to transthyretin amyloidogenesis. Inhibitors using biochemical and structural studies. J Med Chem. 2009;52:1115–1125. doi: 10.1021/jm801347s. [DOI] [PMC free article] [PubMed] [Google Scholar]; The last paper of a 3-part series focused on optimizing the second aryl ring of a typical TTR kinetic stabilizer. Both biochemical and structural evaluation of a library of compounds employing a suboptimal linker and an optimal aryl-X was utilized to rank order the aryl-Z substructures.
- 29.Petrassi HM, Johnson SM, Purkey HE, Chiang KP, Walkup T, Jiang X, Powers ET, Kelly JW. Potent and selective structure-based dibenzofuran inhibitors of transthyretin amyloidogenesis: kinetic stabilization of the native state. J Am Chem Soc. 2005;127:6662–6671. doi: 10.1021/ja044351f. [DOI] [PubMed] [Google Scholar]
- 30.Petrassi HM, Klabunde T, Sacchettini J, Kelly JW. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J Am Chem Soc. 2000;122:2178–2192. [Google Scholar]
- 31.Wojtczak A, Luft J, Cody V. Mechanism of molecular recognition. Structural aspects of 3,3′-diiodo-l-thyroxine binding to human serum transthyretin. J Biol Chem. 1992;267:353–357. [PubMed] [Google Scholar]
- 32.Ciszak E, Cody V, Luft JR. Crystal structure determination at 2.3 A resolution of human transthyretin-3′,5′-dibromo-2′,4,4′,6-tetrahydroxyaurone complex. Proc Natl Acad Sci. 1992;89:6644–6648. doi: 10.1073/pnas.89.14.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamilton JA, Steinrauf LK, Braden BC, Liepnieks J, Benson MD, Holmgren G, Sandgren O, Steen L. The X-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30 to Met variant to 1.7-Å resolution. J Biol Chem. 1993;268:2416–2424. [PubMed] [Google Scholar]
- 34.Wojtczak A, Luft JR, Cody V. Structural aspects of inotropic ipyridine binding. Crystal structure determination to 1.9 Å of the serum transthyretin–milrinone complex. J Biol Chem. 1993;268:6202–6206. doi: 10.2210/pdb1tlm/pdb. [DOI] [PubMed] [Google Scholar]
- 35.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 36.Zanotti G, D'Acunto MR, Malpeli G, Folli C, Berni R. Crystal structure of the transthyretin—retinoic–acid complex. Eur J Biochem. 1995;234:563–569. doi: 10.1111/j.1432-1033.1995.563_b.x. [DOI] [PubMed] [Google Scholar]
- 37.Sunde M, Richardson SJ, Chang L, Pettersson TM, Schreiber G, Blake CC. The crystal structure of transthyretin from chicken. Eur J Biochem. 1996;236:491–499. doi: 10.1111/j.1432-1033.1996.00491.x. [DOI] [PubMed] [Google Scholar]
- 38.Peterson SA, Klabunde T, Lashuel H, Purkey H, Sacchettini JC, Kelly JW. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci U S A. 1998;95:12956–12960. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naylor HM, Newcomer ME. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry. 1999;38:2647–2653. doi: 10.1021/bi982291i. [DOI] [PubMed] [Google Scholar]
- 40.Sebastiao MP, Merlini G, Saraiva MJ, Damas AM. The molecular interaction of 4′-iodo-4′-deoxydoxorubicin with Leu-55Pro transthyretin ‘amyloid-like’ oligomer leading to disaggregation. Biochem J. 2000;351:273–279. doi: 10.1042/0264-6021:3510273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wojtczak A, Cody V, Luft JR, Pangborn W. Structure of rat transthyretin (rTTR) complex with thyroxine at 2.5 A resolution: first non-biased insight into thyroxine binding reveals different hormone orientation in two binding sites. Acta Crystallogr D Biol Crystallogr. 2001;57:1061–1070. doi: 10.1107/s0907444901007235. [DOI] [PubMed] [Google Scholar]
- 42.Wojtczak A, Neumann P, Cody V. Structure of a new polymorphic monoclinic form of human transthyretin at 3 Å resolution reveals a mixed complex between unliganded and T4-bound tetramers of TTR. Acta Crystallogr D Biol Crystallogr. 2001;57:957–967. doi: 10.1107/s0907444901006047. [DOI] [PubMed] [Google Scholar]
- 43.Muziol T, Cody V, Wojtczak A. Comparison of binding interactions of dibromoflavonoids with transthyretin. Acta Biochim Pol. 2001;48:885–892. [PubMed] [Google Scholar]
- 44.Eneqvist T, Lundberg E, Karlsson A, Huang S, Santos CR, Power DM, Sauer-Eriksson AE. High resolution crystal structures of piscine transthyretin reveal different binding modes for triiodothyronine and thyroxine. J Biol Chem. 2004;279:26411–26416. doi: 10.1074/jbc.M313553200. [DOI] [PubMed] [Google Scholar]
- 45.Morais-de-Sa E, Pereira PJ, Saraiva MJ, Damas AM. The crystal structure of transthyretin in complex with diethylstilbestrol: a promising template for the design of amyloid inhibitors. J Biol Chem. 2004;279:53483–53490. doi: 10.1074/jbc.M408053200. [DOI] [PubMed] [Google Scholar]
- 46.Neto-Silva RM, Macedo-Ribeiro S, Pereira PJ, Coll M, Saraiva MJ, Damas AM. X-ray crystallographic studies of two transthyretin variants: further insights into amyloidogenesis. Acta Crystallogr D Biol Crystallogr. 2005;61:333–339. doi: 10.1107/S0907444904034316. [DOI] [PubMed] [Google Scholar]
- 47.Wiseman RL, Johnson SM, Kelker MS, Foss T, Wilson IA, Kelly JW. Kinetic stabilization of an oligomeric protein by a single ligand binding event. J Am Chem Soc. 2005;127:5540–5551. doi: 10.1021/ja042929f. [DOI] [PubMed] [Google Scholar]
- 48.Gales L, Macedo-Ribeiro S, Arsequell G, Valencia G, Saraiva MJ, Damas AM. Human transthyretin in complex with iododiflunisal–structural features associated with a potent amyloid inhibitor. Biochem J. 2005;388:615–621. doi: 10.1042/BJ20042035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neumann P, Cody V, Wojtczak A. Ligand binding at the transthyretin dimer–dimer interface: structure of the transthyretin–T4Ac complex at 2.2 Å resolution. Acta Crystallogr D Biol Crystallogr. 2005;61:1313–1319. doi: 10.1107/S0907444905022523. [DOI] [PubMed] [Google Scholar]
- 50.Morais-de-Sa E, Neto-Silva RM, Pereira PJ, Saraiva MJ, Damas AM. The binding of 2,4-dinitrophenol to wild-type and amyloidogenic transthyretin. Acta Crystallogr D Biol Crystallogr. 2006;62:512–519. doi: 10.1107/S0907444906006962. [DOI] [PubMed] [Google Scholar]
- 51.Razavi H, Powers ET, Purkey HE, Adamski-Werner SL, Chiang KP, Dendle MT, Kelly JW. Design, synthesis, and evaluation of oxazole transthyretin amyloidogenesis inhibitors. Bioorg Med Chem Lett. 2005;15:1075–1078. doi: 10.1016/j.bmcl.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 52.Gales L, Almeida MR, Arsequell G, Valencia G, Saraiva MJ, Damas AM. Iodination of salicylic acid improves its binding to transthyretin. Biochim Biophys Acta, Proteins Proteomics. 2008;1784:512–517. doi: 10.1016/j.bbapap.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 53.Mairal T, Nieto J, Pinto M, Almeida MR, Gales L, Ballesteros A, Barluenga J, Perez JJ, Vazquez JT, Centeno NB, et al. Iodine atoms: a new molecular feature for the design of potent transthyretin fibrillogenesis inhibitors. PLoS One. 2009;4:e4124. doi: 10.1371/journal.pone.0004124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palaninathan SK, Mohamedmohaideen NN, Orlandini E, Ortore G, Nencetti S, Lapucci A, Rossello A, Freundlich JS, Sacchettini JC. Novel transthyretin amyloid fibril formation inhibitors: synthesis, biological evaluation, and X-ray structural analysis. PLoS One. 2009;4:e6290. doi: 10.1371/journal.pone.0006290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Epstein M. Non-steroidal anti-inflammatory drugs and the continuum of renal dysfunction. J Hypertens Suppl. 2002;20:S17–23. [PubMed] [Google Scholar]
- 56.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 57.Lueprasitsakul W, Alex S, Fang SL, Pino S, Irmscher K, Kohrle J, Braverman LE. Flavonoid administration immediately displaces thyroxine (T4) from serum transthyretin, increases serum free T4, and decreases serum Thyrotropin in the rat. Endocrinology. 1990;126:2890–2895. doi: 10.1210/endo-126-6-2890. [DOI] [PubMed] [Google Scholar]
- 58.Maia F, Almeida Mdo R, Gales L, Kijjoa A, Pinto MM, Saraiva MJ, Damas AM. The binding of xanthone derivatives to transthyretin. Biochem Pharmacol. 2005;70:1861–1869. doi: 10.1016/j.bcp.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 59.Almeida MR, Macedo B, Cardoso I, Alves I, Valencia G, Arsequell G, Planas A, Saraiva MJ. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem J. 2004;381:351–356. doi: 10.1042/BJ20040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–552. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 61.Dolado I, Nieto J, Saraiva MJ, Arsequell G, Valencia G, Planas A. Kinetic assay for high-throughput screening of in vitro transthyretin amyloid fibrillogenesis inhibitors. J Comb Chem. 2005;7:246–252. doi: 10.1021/cc049849s. [DOI] [PubMed] [Google Scholar]
- 62.Baures PW, Oza VB, Peterson SA, Kelly JW. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the nonsteroidal antiinflammatory drug flufenamic acid. Bioorg Med Chem. 1999;7:1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez A, Quirante J, Nieto J, Almeida MR, Saraiva MJ, Planas A, Arsequell G, Valencia G. Isatin derivatives, a novel class of transthyretin fibrillogenesis inhibitors. Bioorg Med Chem Lett. 2009;19:5270–5273. doi: 10.1016/j.bmcl.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 64••.Choi S, Reixach N, Connelly S, Johnson SM, Wilson IA, Kelly JW. A substructure combination strategy to create potent and selective transthyretin kinetic stabilizers that prevent amyloidogenesis and cytotoxicity. J Am Chem Soc. 2010;132:1359–1370. doi: 10.1021/ja908562q. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent report into the use of a substructure combination strategy using SAR data from refs. [26••,27••,28••] on optimized substructures to generate a library of potent and selective TTR kinetic stabilizers.
- 65.Andrea TA, Cavalieri RR, Goldfine ID, Jorgensen EC. Binding of thyroid hormones and analogs to the human plasma protein prealbumin. Biochemistry. 1980;19:55–63. doi: 10.1021/bi00542a009. [DOI] [PubMed] [Google Scholar]
- 66.Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci USA. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.