

Abstract
Rhabdoid tumors of early infancy are highly aggressive with consequent poor prognosis. Most cases show inactivation of the SMARCB1 (also known as INI1 and hSNF5) tumor suppressor, a core member of the ATP-dependent SWI/SNF chromatin-remodeling complex. Familial cases, described as rhabdoid tumor predisposition syndrome (RTPS), have been linked to heterozygous SMARCB1 germline mutations. We identified inactivation of another member of the SWI/SNF chromatin-remodeling complex, its ATPase subunit SMARCA4 (also known as BRG1), due to a SMARCA4/BRG1 germline mutation and loss of heterozygosity by uniparental disomy in the tumor cells of two sisters with rhabdoid tumors lacking SMARCB1 mutations. SMARCA4 is thus a second member of the SWI/SNF complex involved in cancer predisposition. Its general involvement in other tumor entities remains to be established.
Main Text
Rhabdoid tumors (RT) are highly malignant, aggressive, embryonal neoplasms of early infancy and childhood (median age at onset: 11 mo) that may originate from virtually any tissue.1 In the CNS, these tumors are termed AT/RT (atypical teratoid, rhabdoid tumor), whereas for the kidney, the term RTK (rhabdoid tumor of kidney) has been applied. The prognosis of RT is dismal, and despite recent advances using aggressive, multimodality treatment schedules,2 the long-term outcome remains doubtful.
The vast majority of RT demonstrate biallelic somatic inactivation of the SMARCB1 (aliases INI1, hSNF5, BAF47 [MIM ∗601607]) tumor suppressor within tumor cells.3–5 Different mechanisms contribute to SMARCB1 inactivation, including gross chromosomal aberrations or loss of heterozygosity (LOH) of the chromosomal region 22q11.2 containing the SMARCB1 gene, as well as a range of private point mutations. Remarkably, heterozygous germline mutations can be observed in up to 20% of patients with RT,6 including familial cases described as having RT-predisposition syndrome (RTPS [MIM #609322]). Such germline mutations predict a fatal course in almost any case.6,7 These findings, along with the development of rhabdoid-like tumors in SMARCB1−/+ mice, have qualified SMARCB1 as a bona fide tumor suppressor for RT.
SMARCB1 encodes for a core member of the ATP-dependent SWI/SNF chromatin-remodeling complex, a master regulator of gene expression involved in cancer.8,9 Theoretically, other members of this complex may also be implicated in RTPS. Nevertheless, until now, no germline mutation in any of the respective gene loci has been demonstrated to be involved in RTPS.
We have recently seen two sisters with RT lacking germline or somatic inactivation of the SMARCB1 gene.10 Patient III-2 was diagnosed at 8 mo of age with symptoms of increased irritability and vomiting. Neuroimaging revealed a mass originating from the right cerebello-pontine angle, involving the brain stem (Figure 1A). Neurosurgical biopsy was performed. However, no further treatment was initiated, because the patient deteriorated rapidly after the neurosurgical procedure and died of disease progression only weeks later. Extensive immunohistochemical analysis by several independent neuropathologists revealed the diagnosis of an INI1-expressing AT/RT.
Figure 1.

Synopsis of Clinical and Molecular Data of the Investigated Family
(A) T1-weighted imaging with contrast demonstrates in axial, coronal, and sagittal planes (from left) a cerebellopontine angle tumor with a heterogeneous enhancement pattern and infiltration of the brainstem indicative of a highly malignant neoplasm in patient III-2.
(B) MRI and CT imaging of patient III-3 demonstrates a large right flank mass originating from the kidney with metastases to the lungs (yellow arrows) and mediastinum.
(C) Pedigree of the investigated family and segregation of the SMARCA4 nonsense mutation. The unaffected sibling, individual III-1, was not available for investigation.
(D) Domain structure of normal SMARCA4 and of the derived p.R1189X truncation mutant. The truncated protein would lack the C-terminal part of the ATPase subunit, the AT hook, and the Bromo domain.
Patient III-3 presented at 7 mo of age with increased abdominal circumference and pain. Imaging studies were suggestive of a stage IV Wilms tumor with metastases to the lungs and mediastinum (Figure 1B). Because of the family history, a biopsy was performed, yielding the diagnosis of an INI1-positive RTK. Local disease responded well to an individual polychemotherapy regimen enabling a tumor nephrectomy at 12 mo. However, mediastinal metastases persisted, and the child died of distant and local disease before radiotherapy could be initiated, at 18 mo of age.
The family history is unremarkable except for the maternal grandfather's death of lung carcinoma and metastasis to the brain at the age of 66 yrs (Figure 1C). The father and an unaffected brother of the two children with RTs are well, without any sign of tumors or schwannomatosis at the time of the writing of this manuscript. In contrast to the more than 60 SMARCB1-negative RT cases in our German registry, SMARCB1 expression was preserved in both cases.
As recently reported,10 extensive molecular studies of the SMARCB1 locus in this family showed that in concordance with the SMARCB1 expression, SMARCB1 mutations were not detected in tumor cells or leukocytes. Moreover, fluorescence in situ hybridization (FISH) and array comparative genomic hybridization (CGH) analyses could not identify chromosomal changes at the SMARCB1 locus, but they also lacked evidence for chromosomal imbalances affecting other regions of the tumor genome. Finally, SMARCB1 haplotyping ruled out SMARCB1 as the causative gene for the RTs in this family.10 These findings strongly pointed to the existence of a yet-unidentified second gene locus involved in RTPS in this particular family.
To identify the genetic cause underlying RTPS in this family, we sequenced the exons and intron-exon borders of four candidate genes from the core unit of the SWI/SNF complex—SMARCA4 (MIM ∗603254), SMARCA2 or BRM (MIM ∗600014), SMARCC1 or BAF155 (MIM ∗601732), and SMARCC2 or BAF170 (MIM ∗601734)—in germline DNA derived from one of the affected siblings (III-3). Whereas analyses of the latter three genes revealed no deviations from the published sequence except allelic variants of known SNPs, mutation analysis of SMARCA4 identified the heterozygous nonsense mutation c.3565C>T (p.Arg1189X), suggesting either a severely truncated translation product or nonsense-mediated decay of mRNA as possible consequences (Figure 1D and Figure 2A). The same mutation was also detected in the germline of the healthy father but not in the mother. No germline DNA of patient III-2 was available, but her AT/RT carried the same mutation in a homozygous state, suggesting LOH at the SMARCA4 locus in the tumor (Figure 2A). The same holds true for the RTK of patient III-3, leading us to investigate the mechanism of LOH in both RTs (PCR primers and conditions are given in Table S1, available online).
Figure 2.
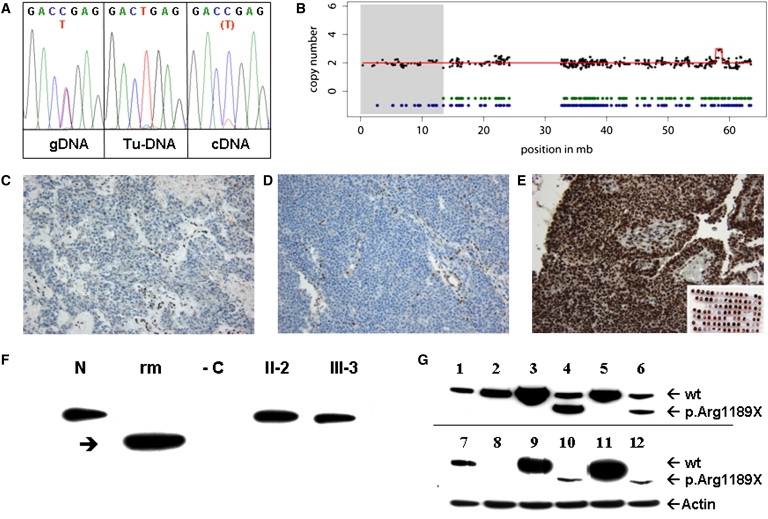
SMARCA4 Immunohistochemistry and Molecular Studies
(A) Identification of the nonsense mutation p.R1189X (c.3565C>T) in germline DNA, detection of loss of heterozygosity in tumor DNA (Tu-DNA), and low concentration mutant cDNA obtained by RT PCR from patient III-3's immortalized B cells compared to the heterozygous state seen in genomic germline DNA (gDNA).
(B) Copy-neutral loss of heterozygosity in 19p containing the SMARCA4 locus in the RT of patient III-2. 100K SNP array (GeneChip Human Mapping 100K Set, Affymetrix; Santa Clara, CA, USA) mapping was performed on DNA derived from the RT of patient III-2, as well as from germline DNA of the father (II-1, for comparison). Analysis of chromosome 19 is displayed with black dots (raw copy-number data smoothed by a median filter) and a red line showing copy-number estimations derived from the Copy Number Analyser for GeneChip (2 = balanced) of the RT of patient III-2 (the gain in 19q derived from a known copy-number variant). Heterozygous calls are indicated by green dots (RT of III-2) and blue dots (germline DNA of the father, II-1). In contrast to II-1, the RT of III-2 shows loss of heterozygosity in 19p (gray shaded area, chr19: 341,341–13,425,865 bp), containing the SMARCA4 gene at chr19:10,932,598–11,033,958 (based on the May 2004 UCSC Genome Browser; hg17).
(C–E) Immunohistochemistry. Tumor cells from both case individuals (C: patient III-2, AT/RT; D: patient III-3, RTK) lack SMARCA4 staining; immunoreactivity remains restricted to the vasculature. In contrast, 14 sporadic AT/RT (E) (representative staining result) as well as a panel of embryonal tumors (inset) display nuclear SMARCA4 staining.
(F) Lack of expression of truncated mutant SMARCA4 in immortalized B cell lines of patient III-3 in comparison to B cells of her mother (II-2) and of a normal control (N). Arrow points to truncated recombinant mutant SMARCA4 (rm) overexpressed in the SMARCA4-negative cell line NCI H1299. −C: nontransfected NCI H1299 as negative control.
(G) Expression of two naturally occurring splice variants of SMARCA4 (one without exon 27 and one including exon 27), both harboring the p.R1189X mutation, in comparison to recombinant WT SMARCA4 in different cell lines. 1 and 7: nontransfected HELA cells constitutively expressing SMARCA4, 2–6: 293-EBNA cells constitutively expressing SMARCA4 (2: mock transfection, 3: WT SMARCA4 long splice variant, 4: mutant SMARCA4 long splice variant, 5: WT SMARCA4 short splice variant, 6: mutant SMARCA4 short splice variant). 8–12: NCI-H1299 cells lacking constitutive SMARCA4 expression (8: mock transfection, 9: WT SMARCA4 long splice variant, 10: mutant SMARCA4 long splice variant, 11: WT SMARCA4 short splice variant, 12: mutant SMARCA4 short splice variant). The truncated mutant p.R1189X fragment is expressed under a strong cytomegalovirus promoter and is clearly separated from WT SMARCA4 by electrophoresis. Size differences between the long and the short SMARCA4 splice variants are not visible because of limited electrophoretic resolution.
In line with previous 2.6 K array CGH analyses10 that did not reveal any chromosomal imbalances in the RT of both sisters, FISH did not identify a chromosomal aberration of the SMARCA4 locus in any of the tumors (BAC clones are listed in Table S2). In contrast, SNP array analysis (GeneChip Human Mapping 100K Set; mapped to hg17, NCBI build 35) identified a long stretch (57 SNPs) of copy-neutral homozygosity in 19p13 (chr19: 341,341–13,425,865 bp) in the RT of patient III-2, suggesting partial uniparental disomy of the paternal allele to be the cause of LOH in the tumors (Figure 2B). Remarkably, with the exception of one other region in chromosome 6q21 (chr6:105,998,201–107,639,853 bp), this was the only long stretch of homozygosity in the RT. This, as well as the absence of chromosomal imbalances (except known copy-number variations), lends further support to SMARCA4 being the causative gene in this form of RTPS.
After deparaffinization and boiling at pH 9 for antigen retrieval, immunohistochemistry was performed on 2 μm sections via the Avidin Biotin Complex (ABC) method on an automated staining system (TechMate, DAKO; Glostrup, Denmark). For SMARCA4/BRG1 staining, a rabbit antiserum against BRG1 (catalog number 07-478, 1:2000, Upstate; Lake Placid, NY, USA) was employed. The specificity of this antiserum for BRG1 has been documented previously.11 Tumor cells from both cases lacked SMARCA4 immunoreactivity (Figures 2C and 2D). In contrast, strong nuclear SMARCA4 staining was observed in all 14 cases of a series of randomly collected AT/RT, as well as in a panel of other embryonal tumors, consisting of 40 medulloblastomas, seven primitive neuroectodermal tumors, one neuroblastoma, and one ependymoblastoma (Figure 2E). Because the antibody used for immunohistochemistry detects amino acids 214–279 of human SMARCA4/BRG1, located N-terminal of the truncating mutation in the family, the complete absence of detectable protein suggests nonsense-mediated decay of the mutant transcript. This is supported by the observation that in lymphoblastoid cells of patient III-3, the mutant transcript was detected by RT-PCR only at a much lower level than the wild-type (WT) allele (see cDNA in Figure 2A) (Primers and RT-PCR conditions can be provided upon request.)
We also performed an immunoblot of SMARCA4 from immortalized B cells of patient II-2 and her mother. Cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS), followed by cell lysis in lysis buffer (250 mM NaCl, 50 mM Tris/HCl pH 8, 0.1% NP40, complete proteinase inhibitor) and freezing-thawing. The lysate was centrifuged, and aliquots of the supernatant, each containing 100 μg protein, were applied to a NUPAGE Novex Bis-Tris Gradient Gel, 4%–12% (Invitrogen). Electrophoresis was followed by immunoblotting onto Hybond ECL membrane (GE Healthcare, Freiburg, Germany) with the use of a monoclonal BRG1 (G-7) antibody (Santa Cruz Biotechnology; Santa Cruz, CA, USA) as first antibody and a goat-anti-mouse HRP-conjugated antibody (DAKO Cytomation, Hamburg, Germany) for detection of the bands by luminescence generated from lumilight substrate (Roche Diagnostics, Mannheim, Germany). However, we identified only a band correlating with the expected size of the WT protein but not with a mutant truncated translation product (Figure 2F).
Finally, aiming at further characterizing the mutant SMARCA4 protein, we carried out recombinant expression studies of WT and mutant SMARCA4. A SMARCA4 cDNA ((#TC118209, OriGene (Rockville, USA) was subcloned into the vector pIRESneo2 (C). As this cDNA is a short SMARCA4 transcript lacking sequence of exon 27 (nucleotide c.3775 - 3893 / p.1259 - 1290) due to alternative splicing we additionally obtained the full-length cDNA by in vitro mutagenesis of this clone using the Quick Change mutagenesis kit (Stratagene, La Jolla, USA). The same method was applied to introduce the patients' mutation using forward and reverse primers of 40 bp in length (detailed description of these methods can be provided upon request).
Two different cell lines, 293-EBNA (Invitrogen, Karlsruhe, Germany) and NCI-H1299 (CRL-5803, ATCC, USA), the latter lacking constitutive SMARCA4 expression, were transiently transfected with WT and mutant pIRESneo2-SMARCA4, respectively, by means of lipofectamine 2000 (Invitrogen) as described previously.12 They were cultured for 72 hr in high-glucose Dulbecco's modified Eagle's medium/10% fetal bovine serum and were prepared for immunoblotting as described above. After overexpression of mutant SMARCA4 cDNA in either 293 EBNA or NCI-H1299 cells, we indeed identified an aberrant band correlating with the expected size of a mutant truncated protein. No difference in expression of the mutant transcript was seen between the two splicing variants with and without exon 27, suggesting that they do not influence the consequences of the mutation (Figures 2F and 2G). These expression studies indicate that the mutated SMARCA4 allele can in principle be translated in a truncated protein, further corroborating that lack of detectable expression in the tumor cells of the patients is due to nonsense-mediated decay.
Our findings provide strong evidence that SMARCA4/BRG1 is a second gene, besides SMARCB1, of the SWI/SNF chromatin-remodeling complex involved in RTPS. SMARCA4 has been proposed as a bona fide tumor suppressor (i) because somatic mutations of SMARCA4 have previously been identified in epithelial cancer cell lines such as lung cancer, pancreatic cancer, breast cancer, and prostate cancer,13–15 though only in a few primary tumors,14 and (ii) because SMARCA4−/+ mice develop epithelial tumors at a low rate.16 However, RTs have never been observed in SMARCA4−/+ mice, and a causative role of a single reported germline in-frame 24 bp duplication of SMARCA4 could not be established in a patient with lung carcinoma because the respective allele was lost in the tumor.14 Nevertheless, the association of SMARCA4 to RTPS in the family presented herein is strongly supported by the following observations:
-
(1)
The RT of both siblings lacked expression of the SMARCA4 protein.
-
(2)
Both affected sisters carried the same SMARCA4 nonsense mutation.
-
(3)
RT-PCR on the patient's lymphoblastoid cells and expression studies indicated nonsense-mediated decay as the molecular mechanism for the lack of SMARCA4 expression in the tumors.
-
(4)
Copy-neutral LOH encompassing the SMARCA4 locus in 19p13 was identified as a “second hit” in the tumor cells on the background of a balanced genome.
-
(5)
The classical RT suppressor gene SMARCB1 and three additional genes coding for core members of the SWI/SNF chromatin-remodeling complex did not show chromosomal or molecular alterations.
Remarkably, the patients' father is an as-yet-unaffected carrier of the same SMARCA4 mutation. This could be due to incomplete penetrance, as well as to a “parent of origin” effect of the mutation. Unfortunately, this could not be further studied because the family rejected testing of additional members. Nevertheless, incomplete penetrance is not truly surprising in RTPS, because it has also been observed in three of nine published families with RTPS due to SMARCB1 mutations.17–19 Similarly, incomplete penetrance also exists in SMARCB1−/+ heterozygous mice, in which only 6%–15% of animals develop RTs.20,21
These observations of RTPS linked to germline SMARCB1 and, as shown here, SMARCA4 mutations, suggest that RT, similar to other tumors of infancy and early childhood such as retinoblastoma, neuroblastoma, and nephroblastoma, is a developmental disorder that arises in children in only a limited time frame.22 This is also supported by the fact that the manifestation of RT occurs at a very early age, with a median of 5.5 mo in children with SMARCB1 germline mutations and a median of 13 mo in children without SMARCB1 germline mutations,6 but is very rare in older children or adults.
SMARCA4 plays a central role in the ATP-dependent chromatin-remodeling complex by carrying its ATPase activity. Therefore, its possible involvement in RTPS is a priori obvious. But until now, no other RT patients had been diagnosed with SMARCA4 mutations. Furthermore, in a recent study it was shown that SMARCA4 loss is antagonistic to oncogenesis caused by SMARCB1 loss and that presence of SMARCA4 is essential for tumor formation caused by SMARCB1 loss in conditional SMARCB1−/− mice.23 SMARCA4 loss in our patients with RTs seems to challenge these observations. However, the situation is different in our patients because SMARCB1 is present. This could indicate compensatory mechanisms for SMARCA4 loss in the oncogenic process. One such mechanism could be the replacement of SMARCA4 by SMARCA2/BRM, another member of the complex that also carries ATPase activity. However, the two proteins seem to be mutually exclusive in the complex, suggesting that they do not act redundantly.8 Additionally, we obviously cannot rule out a genetic or epigenetic hit additional to SMARCA4 inactivation that is not detectable with the applied strategies.
Finally, our finding that SMARCA4 seems to be not only dispensable but also rather causative in the manifestation of RT may also point to a yet-unknown role of SMARCA4 in oncogenesis apart from its ATPase activity in the chromatin-remodeling complex.
Besides the role of SMARCA4 as a, to our knowledge, previously unreported RTPS locus that would affect only a small number of children, SMARCA4 germline mutations might also be involved in the manifestation of other cancers in adults. Incomplete penetrance concerning RT, as evident in the reported family, does not exclude the manifestation of other cancers later in life for mutation carriers. The lack of respective data compromises appropriate genetic counseling. Larger systematic studies screening for SMARCA4 germline mutations in patients with other cancers lacking SMARCA4 expression are therefore desirable.
Acknowledgments
This work was supported by the “Foerdergemeinschaft Kinderkrebszentrum Hamburg e.V.” (R.Sc.), the “KinderKrebsInitiative Buchholz/Holm-Seppensen” (R.Si.), and the “Wasowicz-Stiftung im Stifterverband für die Deutsche Wissenschaft” (M.C.F.). Claudia Becher, Birgit Lechtape, Magret Ratjen, and Barbara Riesmeier provided expert technical assistance. Our study is in accordance with our institutional and national ethical standards on human experimentation. Informed consent for the molecular genetic and clinical studies was obtained from both parents by M.C.F.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
University of California-Santa Cruz (UCSC) Genome Browser, http://genome.ucsc.edu
University Medical Center Hamburg-Eppendorf SMARCB1 Database, http://www.uke.de/kliniken/haematologie/index_60091.php
References
- 1.Roberts C.W., Biegel J.A. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol. Ther. 2009;8:412–416. doi: 10.4161/cbt.8.5.8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chi S.N., Zimmerman M.A., Yao X., Cohen K.J., Burger P., Biegel J.A., Rorke-Adams L.B., Fisher M.J., Janss A., Mazewski C. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J. Clin. Oncol. 2009;27:385–389. doi: 10.1200/JCO.2008.18.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson E.M., Sievert A.J., Gai X., Hakonarson H., Judkins A.R., Tooke L., Perin J.C., Xie H., Shaikh T.H., Biegel J.A. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin. Cancer Res. 2009;15:1923–1930. doi: 10.1158/1078-0432.CCR-08-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sévenet N., Sheridan E., Amram D., Schneider P., Handgretinger R., Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 1999;65:1342–1348. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Versteege I., Sévenet N., Lange J., Rousseau-Merck M.F., Ambros P., Handgretinger R., Aurias A., Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 6.Kordes U., Gesk S., Frühwald M.C., Graf N., Leuschner I., Hasselblatt M., Jeibmann A., Oyen F., Peters O., Pietsch T. Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer. 2010;49:176–181. doi: 10.1002/gcc.20729. [DOI] [PubMed] [Google Scholar]
- 7.Savla J., Chen T.T., Schneider N.R., Timmons C.F., Delattre O., Tomlinson G.E. Mutations of the hSNF5/INI1 gene in renal rhabdoid tumors with second primary brain tumors. J. Natl. Cancer Inst. 2000;92:648–650. doi: 10.1093/jnci/92.8.648. [DOI] [PubMed] [Google Scholar]
- 8.Roberts C.W., Orkin S.H. The SWI/SNF complex—chromatin and cancer. Nat. Rev. Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 9.Reisman D., Glaros S., Thompson E.A. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 10.Frühwald M.C., Hasselblatt M., Wirth S., Köhler G., Schneppenheim R., Martin T., Subero J.I., Siebert R., Kordes U., Jürgens H., Vormoor J. Non-linkage of familial rhabdoid tumors to SMARCB1 implies a second locus for the rhabdoid tumor predisposition syndrome. Pediatr. Blood Cancer. 2006;47:273–278. doi: 10.1002/pbc.20526. [DOI] [PubMed] [Google Scholar]
- 11.Wurster A.L., Pazin M.J. BRG1-mediated chromatin remodeling regulates differentiation and gene expression of T helper cells. Mol. Cell. Biol. 2008;28:7274–7285. doi: 10.1128/MCB.00835-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hassenpflug W.A., Budde U., Obser T., Angerhaus D., Drewke E., Schneppenheim S., Schneppenheim R. Impact of mutations in the von Willebrand factor A2 domain on ADAMTS13-dependent proteolysis. Blood. 2006;107:2339–2345. doi: 10.1182/blood-2005-04-1758. [DOI] [PubMed] [Google Scholar]
- 13.Wong A.K., Shanahan F., Chen Y., Lian L., Ha P., Hendricks K., Ghaffari S., Iliev D., Penn B., Woodland A.M. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–6177. [PubMed] [Google Scholar]
- 14.Medina P.P., Carretero J., Fraga M.F., Esteller M., Sidransky D., Sanchez-Cespedes M. Genetic and epigenetic screening for gene alterations of the chromatin-remodeling factor, SMARCA4/BRG1, in lung tumors. Genes Chromosomes Cancer. 2004;41:170–177. doi: 10.1002/gcc.20068. [DOI] [PubMed] [Google Scholar]
- 15.Medina P.P., Romero O.A., Kohno T., Montuenga L.M., Pio R., Yokota J., Sanchez-Cespedes M. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum. Mutat. 2008;29:617–622. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 16.Bultman S., Gebuhr T., Yee D., La Mantia C., Nicholson J., Gilliam A., Randazzo F., Metzger D., Chambon P., Crabtree G., Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- 17.Ammerlaan A.C., Ararou A., Houben M.P., Baas F., Tijssen C.C., Teepen J.L., Wesseling P., Hulsebos T.J. Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br. J. Cancer. 2008;98:474–479. doi: 10.1038/sj.bjc.6604156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janson K., Nedzi L.A., David O., Schorin M., Walsh J.W., Bhattacharjee M., Pridjian G., Tan L., Judkins A.R., Biegel J.A. Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr. Blood Cancer. 2006;47:279–284. doi: 10.1002/pbc.20622. [DOI] [PubMed] [Google Scholar]
- 19.Taylor M.D., Gokgoz N., Andrulis I.L., Mainprize T.G., Drake J.M., Rutka J.T. Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am. J. Hum. Genet. 2000;66:1403–1406. doi: 10.1086/302833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guidi C.J., Sands A.T., Zambrowicz B.P., Turner T.K., Demers D.A., Webster W., Smith T.W., Imbalzano A.N., Jones S.N. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol. 2001;21:3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts C.W., Galusha S.A., McMenamin M.E., Fletcher C.D., Orkin S.H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA. 2000;97:13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scotting P.J., Walker D.A., Perilongo G. Childhood solid tumours: a developmental disorder. Nat. Rev. Cancer. 2005;5:481–488. doi: 10.1038/nrc1633. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Sansam C.G., Thom C.S., Metzger D., Evans J.A., Nguyen P.T., Roberts C.W. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69:8094–8101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.