

Abstract
Autoreactive CD4+ T cells are involved in the pathogenesis of many autoimmune diseases, but the antigens that stimulate their responses have been difficult to identify and in most cases are not well defined. In the nonobese diabetic (NOD) mouse model of type 1 diabetes (T1D), we have identified a peptide WE14 from chromogranin A (ChgA) as the antigen for highly diabetogenic CD4+ T cell clones. Truncation and extension analysis showed that WE14 binds to the NOD mouse MHCII molecule, I-Ag7, in an atypical manner, occupying only the C-terminal half of the I-Ag7 peptide-binding groove. This finding extends the list of T cell antigens in T1D and supports the idea that autoreactive T cells respond to unusually presented self-peptides.
The self-antigen targets in many autoimmune diseases for both humans and mice have been identified by the presence of serum autoantibodies. Examples are DNA and chromatin in systemic lupus erythematosis (SLE), immunoglobulin in rheumatoid arthritis (RA), and insulin in type I diabetes (T1D). Most autoimmune diseases also involve autoreactive CD4+ T cells that not only are required for autoantibody production, but also can be pathogenic as in T1D. In some instances, such as insulin, epitopes for autoreactive CD4+ T cells have been found in the same proteins targeted by autoantibodies. In general, however, identification of the relevant T cell autoantigen epitopes has been much more difficult and the targets of autoreactive T cells have remained largely undefined.
The pathogenic CD4+ T cells that arise in the non-obese diabetic (NOD) mouse model of T1D provide a striking example. Over the years numerous laboratories have identified CD4+ T cell clones in NOD mice that are reactive with pancreatic antigens in vitro and that also cause or accelerate diabetes in various types of in vivo experiments. Some of these clones have turned out to be specific for insulin epitopes, but the antigenic targets of other highly pathogenic CD4+ T cell clones are unknown. The most extensively studied of these are the BDC clones, which include BDC-2.5, that were isolated from the spleens and lymph nodes of diabetic NOD mice1,2. The BDC clones are not responsive to insulin, but respond in vitro to pancreatic islet cells or cell extracts from beta cell adenomas presented in the context of the major histocompatibility complex (MHC) antigen I-Ag7 (refs.3,4). Polymorphisms unique to this class II MHC allele are very strongly associated with disease5,6. The highly pathogenic nature of these clones has been demonstrated by adoptive transfer studies into young NOD mice in which they greatly accelerate development of T1D2,7. T cells from BDC T cell receptor (TCR) transgenic mice are similarly aggressive in vivo8,9. In addition, introduction of the genes encoding BDC-specific TCRs into T cell deficient NOD.scid mice (retrogenic mice) rapidly induces T1D10. Previous studies suggested that the majority of these clones react to a common pancreatic antigen3,4, but despite numerous efforts, this antigen had never been identified.
Here we identify chromogranin A (ChgA) as the source of the antigen for BDC-2.5 and two other clones, based on mass spectrometric analysis of biochemically purified antigenic fractions from an islet beta cell tumor and on the demonstration that this antigen was missing from the pancreatic islet cells from Chga−/− mice. Peptide antigen mimotopes for these T cells, identified here, and in previous studies11,12, shared a common amino acid motif in the predicted p5 to p9 portion of the peptides [WX(R/K)M(D/E)], which suggested that the ChgA amino acid (aa) sequence 354–362 (EDKRWSRMD) was the probable antigenic epitope. Surprisingly, peptides containing this sequence did not activate the T cells, but the clones were activated by an overlapping peptide, WE14 (aa 359–372, WSRMDQLAKELTAE), a natural cleavage product of ChgA13. This finding was quite unexpected, because despite the presence of the antigen motif, the stimulating WE14 peptide lacked the N-terminal amino acids that would occupy positions p1 to p4 of the I-Ag7 peptide-binding groove and are normally important for stable MHC class II binding. Binding studies suggest that the nine C-terminal amino acids of WE14 make up for this loss by interacting with I-Ag7 at a site outside of the normal binding groove.
RESULTS
Proteomic identification of chromogranin A (ChgA)
Our first approach to identification of candidate antigens for the BDC clones was through biochemical separation and proteomic analysis of antigens in a partially purified protein preparation from the secretory granules of a beta cell adenoma tumor14. We had shown previously that the relevant antigen was present in these cells and that several of the BDC clones reacted with the same granule fraction3,4. We further purified the antigen through the combination of size exclusion (SEC) and ion exchange chromatography (IEX), tracking the antigen via the response of the prototypic T cell clone, BDC-2.5 (Fig. 1a,b). The final highly purified fractions also stimulated two other T cell clones, BDC-10.1 and BDC-5.10.3 (data not shown). The relative degree of purification obtained with each step of separation is provided in Table 1 and is illustrated by a representative silver-stained SDS-PAGE gel (Fig. 1c) that reveals the protein content of the combined antigen-containing SEC fractions (lane 2) and peak antigenic fractions from IEX (lanes 3–7).
Figure 1.
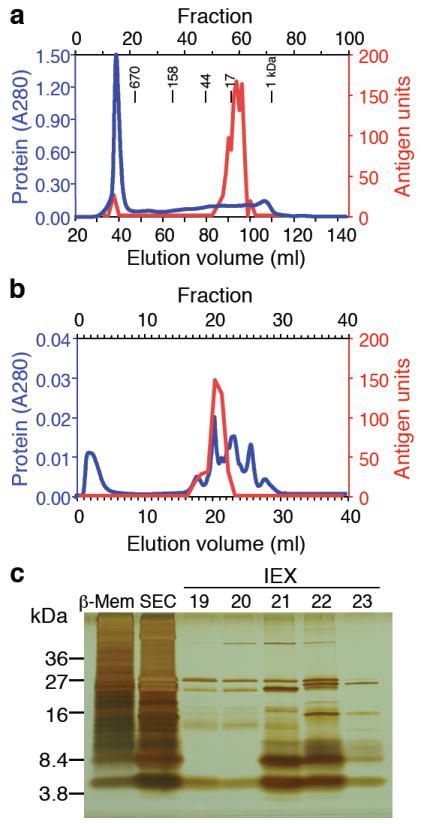
Purification of the antigen for the T cell clone BDC-2.5. (a) Size exclusion chromatography (SEC) of 13.8 mg β-cell membrane lysate and (b), anion exchange (IEX) chromatography of pooled antigenic SE fractions 60-62. The protein content for each chromatographic fractionation was monitored by its absorption at 280 nm (blue lines). The fractions obtained were tested for the presence of antigen with the T cell clone BDC-2.5 (red lines). One antigen unit (A.U.) induces the production of 10 ng/ml IFN-γ under standard antigen assay conditions. (c) Tricine-Tris gel electrophoresis of 4 A.U. β-cell membrane lysate (β-Mem) and 4 A.U. pooled antigenic SEC fractions 60-62. Remaining lanes contain 4 A.U. of the peak antigenic IEX fraction 21 and identical sample sizes (<4 A.U.) of the adjacent IEX fractions 19, 20, 22 and 23. Data from chromatograms and gel electrophoresis are representative of at least 3 independent experiments. The purification table (Table 1) correlates to the data obtained from the chromatograms and SDS-PAGE in this figure.
Table 1.
| Procedure | Activity [applied] (A.U.) | Activity [yield] (A.U.) | Yield (%) | Total protein (mg) | Specific activity (A.U./mg) | Purification factor |
|---|---|---|---|---|---|---|
| Crude cell extract | 32,778 | 100 | 89.43 | 367 | 1.0 | |
| Differential centrifugation | 32,778 | 2,964 | 9 | 17.63 | 168 | 0.5 |
| Size exclusion (fractions 60-62) | 2,325 | 699 | 30 | 0.33 | 2,095 | 5.7 |
| Ion exchange (fractions 21-22) | 373 | 275 | 74 | 0.05 | 5,870 | 16.0 |
To identify the proteins present in the IEX fractions containing the antigenic activity, tryptic digests from each fraction were analyzed by mass spectrometry. Resulting peptides were sequenced and matched to proteins via a search of the Swissprot protein database using the database search program SpectrumMill (Agilent Technologies). A total of 21 proteins were identified in fractions 19–23 using this technique and spectral intensities (Fig. 2a) indicate relative abundance of individual proteins identified in each fraction. A comparison of spectral intensities with corresponding antigenicity in each fraction resulted in a list of potential antigen candidates including secretogranins 1 and 2, insulin-like growth factor II, and ChgA; representative mass spectra and matching sequence are shown for 2 peptides (Fig. 2b,c). Nearly identical results were obtained in 5 independent experiments, with the exception of insulin-like growth factor, which was only identified in one experiment. Overall, 6 ChgA peptides mapping to the more C-terminal portion of the protein (aa 233–463) were confidently identified in highly antigenic fractions with 4 peptides being reproducibly detected in 3 experiments (Fig. 2d). The predicted molecular weight of this potentially truncated ChgA protein (aa 233–463) is approximately 26 kDa, which is consistent with results from SEC. Based on these results and the close correlation of antigenicity with the distribution of ChgA in the fractions, we decided to further investigate ChgA as a candidate antigen.
Figure 2.
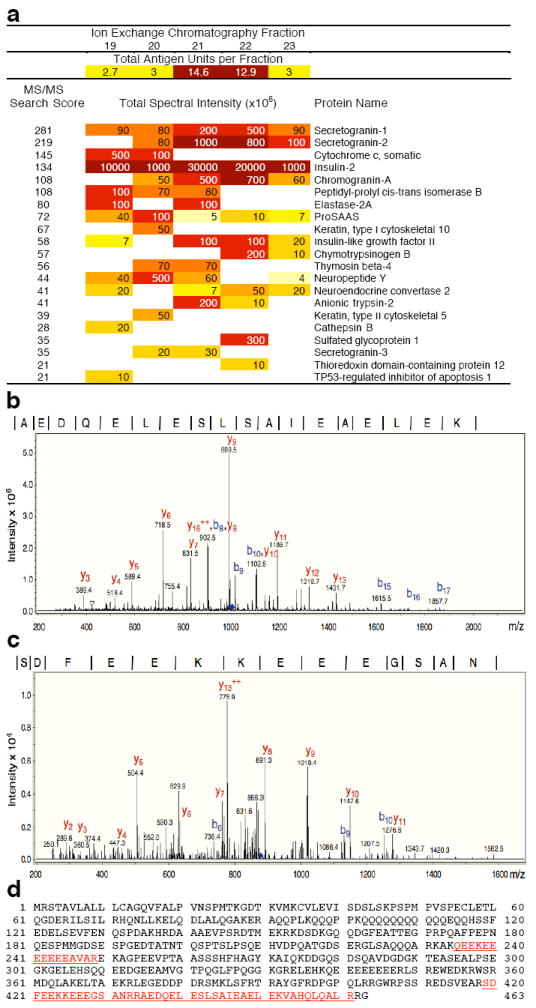
Mass spectrometric analysis of IEX fractions. Proteins in highly purified antigenic IEX fraction 21 and adjacent fractions (Fig. 1b) that displayed lower antigenic activity (fractions 19, 20, 22 and 23) were digested with trypsin and after separation by HPLC, were analyzed using an ion trap mass spectrometer. Resulting spectra were searched against a protein sequence database. (a) Proteins identified in each fraction following database searching. The summarized numeric spectral intensity of the individual peptides identified is an indicator for the relative abundance of a specific protein in a fraction. Darker colors indicate higher intensity. MS/MS Search scores (far left column) greater than 20 are considered significant. (b,c) Ion trap mass spectra matching to two ChgA peptides: AEDQELESLSAIEAELEK (b) and SDFEEKKEEEGSAN (c) are shown (data are representative of 5 independent runs). Peptide amino acid sequence is shown at the top; vertical lines in the sequence correspond to b-ion series (progressing from the N-terminus to the C-terminus) and the complementary y-ion series (progressing from the C-terminus to the N-terminus). (d) Complete ChgA amino acid sequence is shown and the four peptides that were detected and matched to ChgA are underlined.
Islets lacking ChgA do not stimulate the T cell clones
To determine whether ChgA was the source of antigen for the BDC-2.5 T cell clone, and the similar BDC-10.1 and BDC-5.10.3 clones, we compared antigen abundance in pancreatic islet cells from Chga−/− versus Chga+/+ mice15. While the Chga−/− mice are apparently healthy and normal in most respects, they do exhibit some irregularities in terms of islet numbers, size and insulin secretion16. Therefore, we included the PD-12.4.4 insulin reactive clone17 as a control for any global deficiencies in the Chga−/− mice in islet beta cell or granule formation. The T cell clones were cultured with I-Ag7 antigen-presenting cells (APCs) and various numbers of islet cells from the Chga−/− versus Chga+/+ mice as a source of antigen; the beta cell tumor antigen preparation was used as a positive control. We used the production of interferon-γ (IFN-γ) as a measure of T cell activation. All four T cell clones responded well to the control antigen (Fig. 3a). The PD-12.4.4 insulin reactive clone responded equally well to islet cells from either Chga+/+ or Chga−/− mice, suggesting equivalent insulin abundance in individual islet beta cells from either source (Fig. 3b,c). The three BDC clones also responded well to Chga +/+ islet cells, but not at all to islet cells obtained from Chga−/− mice (Fig. 3b,c). These results confirm ChgA as the source of the antigen for these BDC T cell clones.
Figure 3.
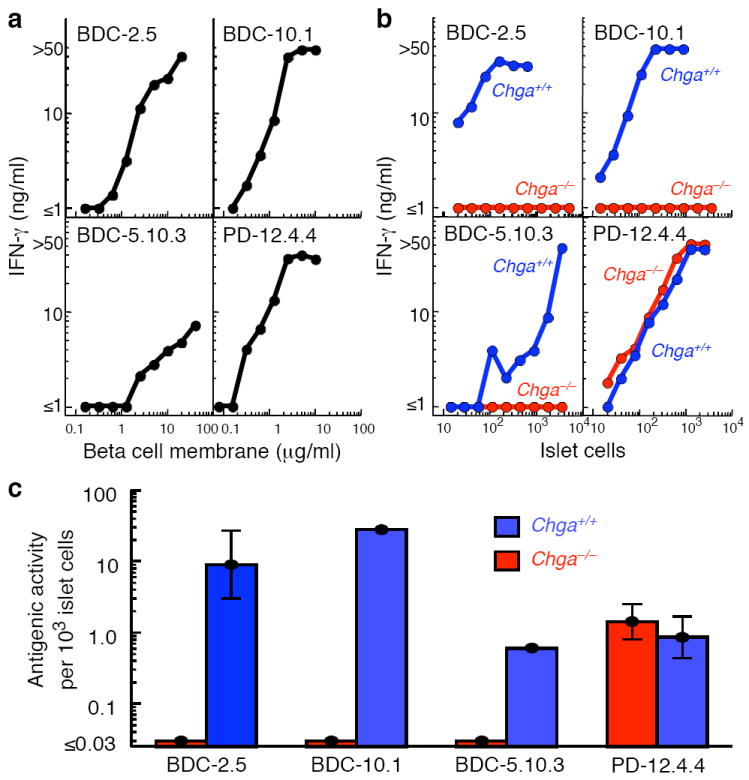
Absence of the T cell antigen in islets from Chga−/− mice. Examples of the IFN-γ response (ng/ml) of the BDC-2.5, BDC-10.1, BDC-5.10.3 and (insulin-reactive) PD-12.4.4 T cell clones to (a) various concentrations of beta cell tumor membrane proteins or (b) various numbers of islet cells obtained from Chga−/− mice (red) or control Chga+/+ mice (blue). (c) Summary of all experiments performed as in (b). The average concentration of antigen in the islet cells from Chga−/− (red bars) or Chga+/+ (blue bars) mice is expressed as antigen units per 103 islet cells, with one unit of antigen defined as the amount required to induce the production of 10 ng/ml of IFN-γ. The data for BDC-2.5 and PD-12.4.4 are from 4 experiments (1-4 replicates per experiment), for BDC-10.1 from two experiments (1-2 replicates per experiment), and for BDC-5.10.3 from one experiment with a single sample. Error bars (SEM) are shown for the BDC-2.5 and PD-12.4.4 data.
A new peptide mimotope for three diabetogenic T cell clones
In a parallel set of studies we identified a new peptide mimotope for the three BDC clones in a library of peptides covalently bound to I-Ag7 and displayed on the surface of insect cells via baculovirus, as previously described18,19. Based on studies of crystal structures of peptides bound to I-Ag7 (refs. 20,21), the peptide amino acids in the baculovirus encoded library (~107 independent clones) were minimally varied at the four major peptide–I-Ag7-binding positions: p1 (Arg or Leu), p4 (Leu or Val), p6 (Leu or Val), and p9 (Gly or Glu). The amino acids at p–1 (p minus 1), p2, p3, p5, p7 and p8 were fully randomized to all 20 amino acids. Insect cells were infected with the library at a multiplicity of infection of <1 such that most infected cells expressed a single member of the library. The few infected cells expressing an I-Ag7-peptide combination capable of binding a fluorescent, soluble multimeric version of BDC-2.5 TCR (see Methods) were isolated using flow cytometry (Fig. 4a, panel 1). Positive cells were used to create a new enriched viral stock. This experimental cycle was performed twice more, producing a highly enriched population of viruses that expressed I-Ag7-peptide combinations, most of which bound the BDC-2.5 TCR (Fig. 4a, panel 2). Cloned viruses from this enriched population were retested for BDC-2.5 TCR binding. The viral DNA was sequenced for all TCR-binding clones and encoded a single peptide sequence, RLGLWVRME, which we refer to as pS3 (Fig. 4a, panel 3).
Figure 4.
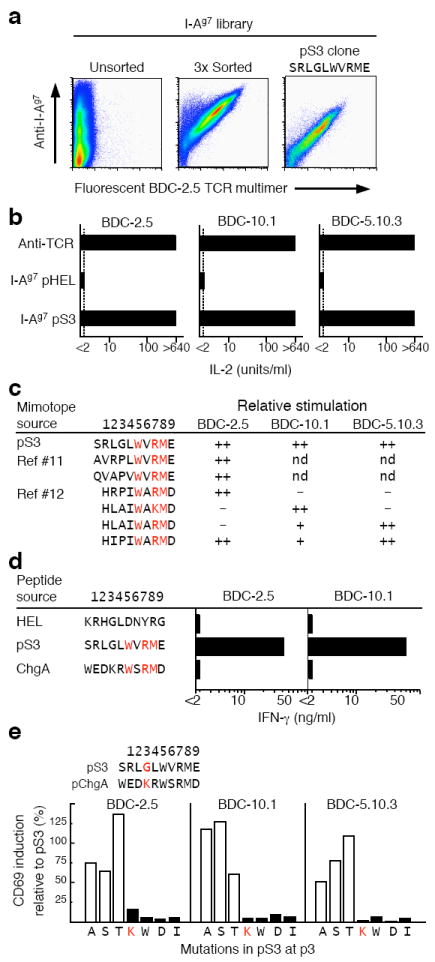
Mimotope peptide antigens for the BDC T cells suggest the region of ChgA encoding the epitope for the BDC T cells. (a) A fluorescent, oligomerized, soluble BDC-2.5 TCR was used to enrich from a baculovirus library a virus encoding an I-Ag7-mimotope (pS3) that forms a strong ligand for the BDC-2.5 TCR (see Methods for details). The data are from a single flow cytometry experiment performed after the sorting and cloning of the library were completed. SF9 cells were infected with either the unsorted library (left panel), 3 times sorted library (middle panel) or pS3 clonal virus (right panel) and analyzed for I-Ag7 expression vs. binding of the TCR reagent. (b) BDC T cell hybridomas were stimulated in culture either with immobilized H597 anti-TCR Cβ Mab or with CD80+ICAM-expressing SF9 cells infected with virus encoding I-Ag7 with a HEL peptide or I-Ag7 with pS3. IL-2 production was assayed after 24 h. Results are representative of three experiments, each assayed in duplicate. (c) The sequence and activity of the pS3 mimotope were compared to those previously identified using other library techniques11,12. The reported potency of the mimotopes in stimulating the 3 BDC T cell clones is represented qualitatively: ++, very strong stimulation; +, modest stimulation; −, little or no stimulation, and is based upon the results shown in panel b for pS3 and as previously reported11,12 for the other mimotopes. Motif positions p5, p7, p8 are highlighted in red. (d) Baculovirus encoding membrane-anchored I-Ag7 covalently bound to each of three peptides (pHEL, pS3, and the ChgA-derived peptide, WEDKRWSRMD) were prepared. CD80+ICAM-expressing SF9 cells were infected with the viruses and tested for stimulation of IFN-γ production by the BDC-2.5 and BDC-10.1 T cell clones. Results are from a single experiment performed in duplicate. (e) Mutation of pS3 p3 glycine to other amino acids. The effect of the mutations on early activation of the three BDC hybridomas was assessed by CD69 surface expression by flow cytometry. The results are shown as the percent of cells expressing CD69 relative to those activated with the unmutated pS3 peptide. The sequences of the pS3 and ChgA peptide are shown, highlighting the amino acids at the p3 position in red. The results are from a single experiment.
T cell hybridomas bearing TCR from either the BCD-2.5, BDC-10.1 or BDC-5.10.3 T cell clones were tested for their ability to recognize the covalent pS3–I-Ag7 complex using CD80+ICAM-expressing insect cells as artificial APCs18,19 (Fig. 4b). The three hybridomas were maximally activated by the pS3 mimotope, bolstering the idea that all three T cells were reactive to the same self-antigen and that the pS3 sequence might resemble that of the natural antigen.
Two prior studies had used the technique of positional scanning peptide libraries to identify antigen mimotopes for one or more of these three BDC T cell clones11,12 (Fig. 4c). A striking feature of these mimotopes and pS3 was a common WX(R/K)M(D/E) motif in amino acids p5-p9 of the peptides. Previous searches of databases with these other mimotope sequences had failed to turn up the natural source of the antigen11,12. However, an examination of the ChgA sequence identified this motif in the C-terminal portion of protein and suggested that the probable core peptide epitope (p–1 to p9) in ChgA was aa353–362 (WEDKRWSRMD). We incorporated a DNA sequence encoding this peptide into the baculovirus I-Ag7 construct and the resulting virus was used to infect CD80+ICAM-expressing insect cells. Cells infected with pHEL–I-Ag7 and pS3–I-Ag7 served as negative and positive controls, respectively. These cells were tested for activation of the BDC-2.5 and BDC-10.1 T cell clones by measuring production of IFN-γ (Fig. 4d). As expected, the pHEL–I-Ag7 expressing cells did not activate either clone and the pS3–I-Ag7 cells strongly activated both clones. Unexpectedly, the pChgA–I-Ag7 cells failed to stimulate either T cell clone.
Since this was the only sequence in ChgA with any homology to the antigen mimotopes, we examined the mimotope sequences more closely (Fig. 4c). Although the N-terminal sequences of the mimotopes (p–1 or p1 to p4) vary considerably, they all have a small non-charged amino acid at p3 (Gly, Ala, or Pro). On the other hand, the ChgA peptide had a large, positively charged amino acid (Lys) at this position. We postulated that this Lys could be sterically blocking epitope recognition by T cells. This idea was strengthened by a mutational study of this position in the pS3 mimotope in which we tested variants of pS3 with the Gly at p3 mutated to many other amino acids (Fig. 4e). Single substitutions with amino acids with small side chains (Ala, Ser, or Thr) preserved the ability of the mimotope to stimulate all three BDC T cell hybridomas. However, changing this amino acid to Lys, the p3 amino acid of the ChgA peptide, or to several other amino acids with large side chains, eliminated the activation of all three BDC T cell clones.
Identification of a natural antigenic epitope from ChgA
Our results suggested that the N-terminal part of the ChgA peptide interferes with T cell recognition, leading us to consider a naturally processed ChgA-derived peptide, WE14 (WSRMDQLAKELTAE)13. This peptide lacks the first 5 amino acids (p–1 to p4) of the previously tested ChgA peptide (Fig. 4d), but retains the common mimotope motif, with additional amino acids present at the C-terminus (Fig. 5a). This peptide was predicted to bind poorly to I-Ag7 because placement of the WSRMD pentapeptide in the p5 to p9 position would only partially fill the peptide binding groove, eliminating many of the usually conserved interactions between MHC class II molecules and peptide involving p–1 to p4.
Figure 5.
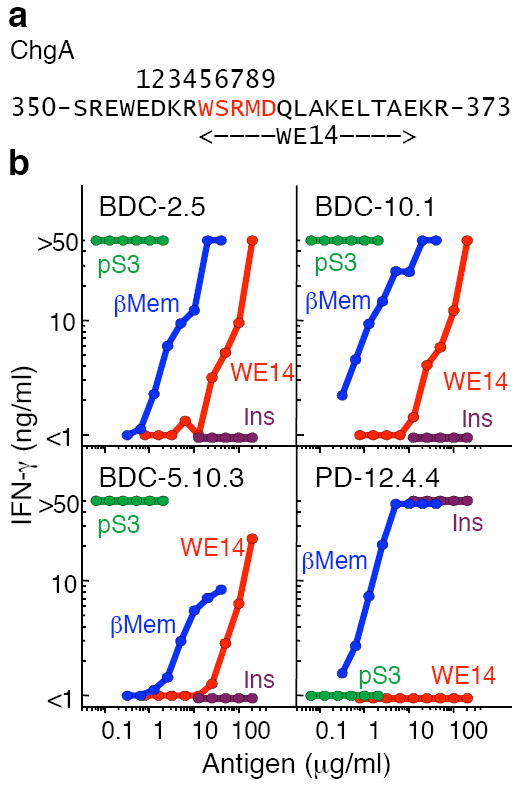
The ChgA derived peptide, WE14, activates all three BDC T cells. (a) A portion of the chromogranin A (ChgA) amino acid sequence with the WE14 peptide indicated. Putative positions in the I-Ag7 peptide-binding groove (positions 1–9) are shown and the motif common to the antigen peptide mimotopes is highlighted in red. (b) IFN-γ response (ng/ml) of the BDC-2.5, BDC-10.1, BDC-5.10.3 and PD-12.4.4 T cell clones stimulated by various peptide concentrations of pS3 (green), WE14 (red), INS2 B9-23 (SHLVEALYLVCGERG) (purple) and beta cell tumor membrane preparation (β-Mem) (blue). Peptide antigen was titrated in each assay and the data is representative of at least two separate experiments with single measurements at each peptide concentration.
We tested a soluble synthetic version of WE14 for its ability to activate the three BDC T cell clones, comparing it to the pS3 mimotope and the control beta cell tumor antigen preparation (Fig. 5b). The pS3 mimotope potently stimulated all three BDC clones maximally at all concentrations tested. All three clones also responded to the beta cell antigen preparation. WE14 peptide also stimulated all three BDC clones, confirming that elimination of the ChgA amino acids predicted to occupy p1 to p4 in the I-Ag7 binding groove was necessary for T cell recognition. As a negative control, we tested the insulin-reactive T cell clone, PD-12.4.4 and its insulin-derived peptide epitope, B:9-23 (ref. 17). As expected, PD-12.4.4 responded to the insulin peptide and beta cell antigen preparation, but not to pS3 or either ChgA peptide. The synthetic WE14 peptide was also considerably less potent than the beta cell antigen preparation, suggesting that the natural version of this peptide may be subject in vivo to some alternate form of processing or to post-translational modification.
To confirm the unique binding register of the WE14 peptide, we performed experiments with a series of peptides that were N-terminal extensions and/or C-terminal deletions of WE14 (Fig. 6). Peptides were evaluated for their ability to stimulate the BDC-2.5 T cell clone (Fig. 6a) or to bind to I-Ag7 as judged by the inhibition of the binding of a biotinylated control peptide, pHEL (Fig. 6b). The pS3 mimotope was used as the positive control peptide and an irrelevant peptide from moth cytochrome c, pMCC, was used as a negative control. The data from these experiments (Fig. 6a, 6b) were used to calculate the stimulatory or binding capacity of the test peptides relative to WE14 (Fig. 6c).
Figure 6.
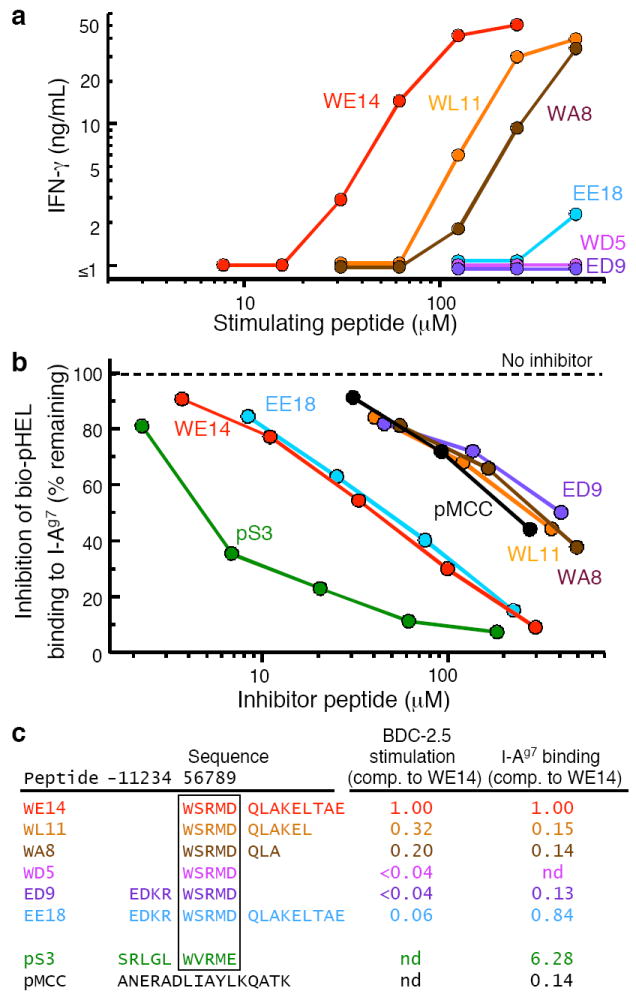
Precise processing of the WE14 peptide required for optimal presentation by I-Ag7. (a) IFN-γ response of the BCD-2.5 T cell clone to varying concentrations (5–500 μM) of ChgA-derived peptides. Data are representative of two separate experiments containing single measurements at each peptide concentration. (b) ChgA derivative peptides were tested for their ability to compete with a biotinylated HEL peptide (bio-pHel) for binding to soluble I-Ag7. The pS3 mimotope was used as a positive control peptide and an I-Ek binding peptide from moth cytochrome c (pMCC) was used as negative control peptide. The data (the average of two experiments with very similar results) are presented as the amount of biotinylated HEL peptide bound to I-Ag7 (expressed as the percent of that bound in the absence of an inhibitor peptide) vs. the concentration of inhibitor peptide. (c) A multiple regression program (MKASSAY, available on request) was used to analyze the stimulation and inhibition curves in (a) and (b), treating them as a series of parallel polynomial curves. The results are presented as the stimulatory or inhibitory activity of the peptides relative to WE14.
Sequential truncation of WE14 (WL11, WA8, WD5) from the C-terminus resulted in decreasing stimulatory activity (Fig. 6a,c). The shortest peptide (WD5), that contained only the WX(R/K)M(E/D) motif, lacked any stimulatory activity. The detrimental effect of these truncations was even more pronounced in the I-Ag7 binding assay (Fig. 6b,c). WE14 bound to I-Ag7, although not nearly as well as the pS3 mimotope. However, the truncation of as few as 3 C-terminal amino acids from WE14 (WL11) reduced binding to a level indistinguishable from the negative control peptide. These data indicate that the C-terminal 9 amino acids of WE14 were required for optimal binding and stimulation by the peptide.
Extension of the WE14 or WD5 peptide at the N-terminus was also informative. Extending WE14 by 4 amino acids (EE18) had no effect on I-Ag7 binding (Fig. 6b,c), but eliminated the BDC-2.5 response (Fig. 6a,c), consistent with the idea that additional N-terminal amino acids are incompatible with T cell recognition. Likewise, extending the WD5 peptide by the same 4 amino acids (ED9) also produced a peptide that failed to stimulate BDC-2.5 (Fig. 6a,c). Similar results were seen with extensions of WD5 by 1 (RD6), 2 (KD7) or 3 (DD8) amino acids (data not shown). Surprisingly however, the ED9 peptide, despite its length, did not bind to I-Ag7 (Fig. 6b,c). This result, and because EE18 binds to I-Ag7 no better than WE14 itself, suggests that these 4 amino acids were unable to fill the p1-p4 portion of I-Ag7 binding groove properly, most likely because of a problem with the p4 Arg present at that anchor position. The simplest interpretation of our results is that WE14 employs an unusual means of binding to I-Ag7 via interaction of its C-terminal 9 amino acids with a site outside of the normal I-Ag7 binding groove.
DISCUSSION
The BDC CD4+ T cell clones are highly active in the acceleration or induction of diabetes in vivo. However, their usefulness has been somewhat limited because their antigenic target was not known. We show here that the source of the antigen for a major cohort of these clones is ChgA, a protein found in the secretory granules of pancreatic beta cells and other neuroendocrine tissues22,23. We also show that WE14, a natural 14 amino acid cleavage product of ChgA13, when presented by I-Ag7 APCs, activated the ChgA-specific T cell clones in vitro.
In previous studies11,12 and in our work reported here, various types of peptide libraries were screened to identify peptide mimotopes for one or more of the BDC T cell clones. These libraries were all constructed to contain peptides that would bind well to I-Ag7 by placing suitable anchor residues at the usual p1, p4, p6 and p9 positions of the peptide. Amino acids at other peptide positions were randomized. All of these studies identified mimotopes with similar sequences from p5 to p9 - WX(R/K)M(E/D), but the sequences varied greatly from p1 to p4.
The identification of WE14 as the active peptide in ChgA was therefore surprising, since placement of its common motif (WSRMD) in the same positions in the I-Ag7-binding groove as predicted by the mimotopes would leave unoccupied any position prior to p5. In such a case, the peptide would be predicted to bind very poorly, if at all, to I-Ag7, not only because of the lack of the major p1 and p4 anchor amino acids, but also because a number of normally highly conserved H-bonds to the peptide backbone would be missing. Nevertheless, our data show that the WE14 peptide bound I-Ag7and potently stimulated all three of the BDC clones. The key to its activity lies in the C-terminal 9 amino acids of the peptide, which were found to be critical for binding to I-Ag7. Truncation of even 3 C-terminal amino acids severely reduced I-Ag7 binding and diminished the T cell response. These data strongly suggest that the C-terminus of WE14 interacts with I-Ag7 at a site outside of the normal peptide binding groove, compensating for the lack of the p1-p4 portion of the peptide. While there are other examples of peptide amino acids flanking the binding groove contributing to MHC class II binding and T cell recognition24-26, the requirement for such a long stretch of flanking amino acids is unprecedented. Furthermore, adding amino acids from the ChgA sequence to the N- terminus of the WE14 peptide inhibited, rather than enhanced, peptide presentation and was not able to restore I-Ag7 binding or T cell activation to the WD5 peptide, which contained the core WE14 WSRMD recognition motif.
We interpret these results to suggest that generation of a functional antigen from ChgA requires removal or alteration of these amino acids to avoid interference with TCR binding to the peptide–I-Ag7 complex. This idea can explain why so many different sequences unrelated to ChgA have been found for the N-terminal part of the antigen mimotopes and why some sequences are recognized by some of the T cell clones, but not others. Depending on the particular TCR, various peptide sequences, but not that of ChgA itself, can fulfill the function of avoiding conflict with the TCR in this region. In addition, since these peptides were all constructed or selected for optimal p1 and p4 amino acids to promote strong I-Ag7 binding, they do not require a C-terminal extension like that of WE14, thus providing an explanation for why the various library strategies did not produce mimotopes that suggested WE14 as the source of the antigen.
T cell peptide epitopes that do not fill the MHC class II groove are not unprecedented in autoimmunity. In the mouse model of EAE, the N-terminal peptide of myelin basic protein is a major T cell epitope, but structural studies have concluded that the natural form of this peptide that is recognized by T cells does not fill the beginning of the I-Au binding groove27,28. In these studies, an active variant of the peptide that filled the rest of the groove with small amino acids was used. In the structure of a TCR bound to this complex, relatively little contact was made to these extra amino acids.
The pS3 peptide mimotope is at least 1000 fold more potent than the synthetic WE14 peptide in activating the BDC clones. WE14 also is considerably less potent than the antigen preparation from the beta cell adenoma tumor, despite the fact that ChgA is only one protein species among many. This result may indicate that the naturally processed antigen may differ from the synthetic WE14 peptide in some way, for example due to some form of post-translational modification that improves either peptide binding to I-Ag7 or TCR binding to the complex. Our mass spectrometric data leave these possibilities open. For example, whereas previous studies have shown the presence of WE14 in the pancreas29, we did not identify this peptide directly in the purified antigenic fractions from beta tumor cells. Rather we found evidence for the presence of the C-terminal portion of ChgA that would contain this peptide, suggesting further processing or modification in antigen-presenting cells may be required to generate the active epitope.
The idea of post-translational modification of antigens has received considerable attention in T cell mediated inflammatory disease studies. For example, in rheumatoid arthritis, citrullination of arginines by peptidylarginine deiminases has been discussed as a possible mechanism for improving binding of self-peptides to DR4 by creating an improved p4 anchor residue30. Also, in celiac disease, tissue transglutaminase conversion of glutamine to glutamic acid in particular gluten peptides is important in creating new T cell epitopes, again by improving peptide binding to the relevant HLA-DQ alleles through changing anchor residues31,32. Both of these enzymes are induced locally by inflammation33,34, suggesting a mechanism by which enhanced antigen presentation can be induced locally in target tissues, but not in the thymus, allowing potentially pathogenic T cells to escape thymic deletion. The WE14 peptide has potential amino acids for both of these post-translational modifications, as well as others, such as lysine hydroxylation.
ChgA is widely expressed in neuroendocrine tissue and the WE14 cleavage product has been described not only in pancreatic islet beta cells, but also in other gastro-entero-pancreatic tissues such as the adrenal gland22. It is unclear why autoimmune attack of these other tissues is not seen in NOD mice. One possibility is that the potency of ChgA as an antigen in the pancreas might be dependent on a pancreas-specific post-translational modification of the relevant peptide, as described above. Alternatively, the selective destruction of beta cells in pancreatic islets has been attributed to their high sensitivity to inflammatory damage compared to other islet cells35. Perhaps other neuroendocrine cells are also more resistant to or protected from this type of immune damage.
The question arises as to why T cells specific for ChgA exist, given that the widespread expression of this protein might be expected to result in efficient deletion of ChgA-specific T cells during thymic development. In addition to tissue- or inflammation-specific processing or post-translational modifications, another possibility may be poor thymic expression. In a study of regulation of mRNA expression by the autoimmune regulator AIRE in thymus medullary epithelium, Chga mRNA was not detected with or without AIRE36, so there may not be sufficient ChgA in the thymus to mediate deletion.
While caution is always advisable in extending findings in mouse models of autoimmunity to human disease, the available data suggest that the target autoantigens are sometimes the same. For example, considerable overlap between mouse and human autoantigens is thought to be involved in several autoimmune diseases, including multiple sclerosis (myelin basic protein), rheumatoid arthritis (collagen) and lupus erythematosis (DNA and chromatin), as well as T1D (insulin). ChgA is highly expressed in human pancreatic cells and the human form of WE14 peptide has a sequence that is nearly identical to that of mouse37. Furthermore, the similarity in binding and presentation of peptides between I-Ag7 and the human DQ alleles associated with T1D38 suggests that WE14 may be presented by MHC class II molecules in T1D susceptible humans. This idea is worth pursuing.
METHODS
Mice
NOD and NOD RIPTag mice were bred and maintained in the Biological Resource Center at National Jewish Health, Denver CO. Chga−/− mice (Chga+/– background strain 129/SvJ backcrossed to C57BL/6J) were generated in the animal facilities at the University of California, San Diego15. All experimental procedures were in accordance with Institutional Animal Care and Use Committee guidelines and approved by the NJH Animal Care and Use Committee.
Antigen purification and mass spectrometric analysis
Enrichment of membrane proteins from beta cells isolated from NOD RIPTAg adenomas has been previously described4. Membrane protein preparations were solublized for 1 h at 4 °C in detergent-containing buffer (20 mM Tris pH 8.0, 1% Octyl-β-Glucoside) followed by centrifugation at 18,400 × g, (10 min, 4°C) to remove insoluble debris. Protein content was determined using a Micro BCA kit (Pierce). Size Exclusion (SE) chromatography was carried out on a Superdex™ 200 16/60 column (Amersham Biosciences) at 21°C (flow rate 1 ml/min, fraction size 1.25 ml, injection volume 2.0 ml) using SE buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.4 mM Tween 20). Peak antigenic fractions were dialyzed overnight (16 h, 20 mM Tris pH 6.5, 4°C) using Tube-O-DIALYZER™ (1K, GBiosciences) and then separated on a HiTrap™ Q HP column (GE Healthcare) at 21°C (flow rate 1 ml/min, fraction size 1.0 ml, injection volume 2.0 ml) applying a 20 min linear NaCl gradient after 10 min (Buffer A: 20 mM Tris pH 6.5, Buffer B: 20 mM Tris pH 6.5, 1 M NaCl). Fractions were concentrated and desalted on CBED spin columns (Norgen Biotek Corporation) using the protocol for acidic proteins described by the manufacturer. Tricine Tris gel electrophoresis was carried out on a 16.5% precast criterion gel (Bio-RAD) applying an initial 65 mA current for 10 min followed by a 35 mA current for 6 h. The gel was stained using SilverSNAP® stain (Thermo Scientific).
A standard protein identification strategy was performed using mass spectrometry39. Briefly, proteins were digested with trypsin and extracted peptides were chromatographically resolved on-line using a C18 column and 1200 series high performance liquid chromatography (HPLC, Agilent Technologies) and analyzed using a 6340 LCMS ion trap mass spectrometer in the National Jewish Health Facility. Raw data was extracted and searched against the SwissProt or NCBI databases using the Spectrum Mill search engine (Rev A.03.03.038 SR1, Agilent Technologies). Data was evaluated and protein identifications were considered significant if the following confidence thresholds were met: minimum of 2 peptides per protein, protein score > 20, individual peptide scores of at least 10, and Scored Percent Intensity (SPI) of at least 70%. A reverse (random) database search was simultaneously performed and manual inspection of spectra was used to validate the match of the spectrum to the predicted peptide fragmentation pattern.
Assays for antigen
Antigenicity of islet cells, cellular and biochemical fractions, peptides, or insect cells expressing I-Ag7-peptide constructs, was assessed through responses of T cell clones or hybridomas made by fusing T cell clones to the TCR- version40 of T cell lymphoma, BW5147. T cell clone cultures typically contained 2 × 104 responder T cells, 2.5 × 104 NOD peritoneal exudate cells (PEC) as APCs, and antigen (SEC/IEX fractions, peptides, islet cells); all assays were performed with β-Mem as a positive control. IFN-γ was determined by specific ELISA (BD Biosciences). For cultures with T cell hybridomas, antigen–MHC activation was assessed by IL-2 production measured by a bioassay using the HT-2 T cell line41. Synthetic peptides were either produced in the Molecular Resource Center at National Jewish Health or obtained from CHI Scientific.
Expression of I-Ag7 and the BDC-2.5 TCR in baculovirus
Membrane-anchored and soluble I-Ag7 stabilized with an acid-base leucine zipper were expressed with a covalently attached peptide in baculovirus infected insect cells as previously described19,42-44. The soluble I-Ag7 was purified from culture supernatants by immunoaffinity and size exclusion chromatography. A soluble version of the BDC-2.5 was also produced with baculovirus as previously described19,42-44 and was isolated from culture supernatants by immunoaffinity (using anti-Cβ) and size exclusion chromatography.
Production and screening of a baculovirus encoded I-Ag7-peptide library
Details for creating baculovirus encoded peptide-MHC class II libraries and screening these libraries have been previously described18,19. In the case of I-Ag7 the peptide library was randomized at positions at p1, p2, p3, p5, p7 and p9 using the codons NN[G/C]. Variations allowed at the four anchor positions were: p1:Arg/Ile (A[G/T]A), p4 and p6:Leu/Val ([T/G]TG), p9:Gly/Glu (G[G/A]A). The PCR DNA fragment encoding the library was cloned directly into baculovirus DNA already encoding the I-Ag7 genes, attached via a linker to the N-terminus of the β chain. The ligated DNA was transfected into insect cells to produce a high titer baculovirus stock (~107 independent clones). Insect cells infected with the library at a multiplicity of infection of <1 were analyzed by flow cytometry for cells that expressed I-Ag7 (OX-6 Mab, BD-Pharmingen) and also bound a multivalent TCR reagent consisting of the soluble BDC-2.5 TCR captured by a biotinylated monoclonal anti-Cα, ADO-304, bound to Alexafluor-647 labeled streptavidin (Molecular Probes). Cells binding both reagents were sorted and incubated with more SF9 insect cells to expand the enriched virus. The infection, analysis and sorting enrichment were performed twice more. The virus was then cloned and insect cells infected with individual virus clones were tested as before for I-Ag7 expression and BDC-2.5 TCR binding. The peptide sequence encoded in the positive clones was determined.
Assay of peptide binding to I-Ag7
Soluble I-Ag7 with covalently attached pHEL was treated with thrombin to cleave the linker attaching the peptide to the I-Ag7 β chain 42. Samples (0.5 μg) were incubated with a soluble biotinylated version of pHEL, Biotin-GGGMKRHGLDNYRGYSL, (11 μM), either alone or in the presence of various concentrations of potential competitors peptides in 15 μl of pH 5.6 buffer overnight at 21°C. The sample was diluted to 100 μl of PBS in a well of a 96-well ELISA plate coated with an I-Ag7 monoclonal antibody, OX6 (BD Pharmaceuticals). The captured I-Ag7 was washed several times with PBS and the bound bio-pHEL detected with akaline phosphatase coupled Extravadin (Sigma) and o-nitrophenol phosphate.
Acknowledgments
This work was supported by grants from the National Institutes of Health: RO1 DK50561 (KH), T32 AI007405 (TD), BioResources Core of DERC P30 DK057516 (KH), 5 U19-AI050864 (BS, JWK), RO1 AI17134 (BS, PM, JWK), RO1 AI18785 (BS, PM, JWK); from the National Center for Research Resources: S10RR023703 (NR); and from the Juvenile Diabetes Research Foundation: 1-2008-132 (KH, NR). We also thank P. Pratt for technical assistance.
Footnotes
AUTHOR CONTRIBUTIONS BDS, TD, NR, RR, JWK and KH designed the experiments. BDS, TD, RLP, MA performed most of the experiments assisted by GB, BB, and FC. JDP initially suggested ChgA as a candidate autoantigen. SKM provided the Chga−/− and Chga+/+ mice. BDS, TD, NR, RR, PM, JWK and KH analyzed and interpreted the data. BDS, TD, JWK and KH wrote the manuscript and prepared the figures. NR, RR and PM helped edit the manuscript.
References
- 1.Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T-cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA. 1989;86:8000–8004. doi: 10.1073/pnas.86.20.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haskins K. Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol. 2005;87:123–162. doi: 10.1016/S0065-2776(05)87004-X. [DOI] [PubMed] [Google Scholar]
- 3.Bergman B, Haskins K. Islet-specific T-cell clones from the NOD mouse respond to β-granule antigen. Diabetes. 1994;43:197–203. doi: 10.2337/diab.43.2.197. [DOI] [PubMed] [Google Scholar]
- 4.Bergman B, McManaman JL, Haskins K. Biochemical characterization of a beta cell membrane fraction antigenic for autoreactive T cell clones. J Autoimmun. 2000;14:343–351. doi: 10.1006/jaut.2000.0377. [DOI] [PubMed] [Google Scholar]
- 5.Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A β chain is unique. Proc Natl Acad Sci USA. 1987;84:2435–2439. doi: 10.1073/pnas.84.8.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prochazka M, Leiter EH, Serreze DV, Coleman DL. Three recessive loci required for insulin-dependent diabetes in nonobese diabetic mice. Science. 1987;237:286–289. doi: 10.1126/science.2885918. [DOI] [PubMed] [Google Scholar]
- 7.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 8.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 9.Pauza ME, et al. T-cell receptor transgenic response to an endogenous polymorphic autoantigen determines susceptibility to diabetes. Diabetes. 2004;53:978–988. doi: 10.2337/diabetes.53.4.978. [DOI] [PubMed] [Google Scholar]
- 10.Burton AR, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes. 2008;57:1321–1330. doi: 10.2337/db07-1129. [DOI] [PubMed] [Google Scholar]
- 11.Judkowski V, et al. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida K, et al. Evidence for shared recognition of a peptide ligand by a diverse panel of non-obese diabetic mice-derived, islet-specific, diabetogenic T cell clones. Int Immunol. 2002;14:1439–1447. doi: 10.1093/intimm/dxf106. [DOI] [PubMed] [Google Scholar]
- 13.Curry WJ, et al. WE-14, a chromogranin a-derived neuropeptide. Ann N Y Acad Sci. 2002;971:311–316. doi: 10.1111/j.1749-6632.2002.tb04485.x. [DOI] [PubMed] [Google Scholar]
- 14.Hamaguchi K, Gaskins HR, Leiter EH. NIT-1, a pancreatic beta-cell line established from a transgenic NOD/Lt mouse. Diabetes. 1991;40:842–849. doi: 10.2337/diab.40.7.842. [DOI] [PubMed] [Google Scholar]
- 15.Mahapatra NR, et al. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest. 2005;115:1942–1952. doi: 10.1172/JCI24354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portela-Gomes GM, Gayen JR, Grimelius L, Stridsberg M, Mahata SK. The importance of chromogranin A in the development and function of endocrine pancreas. Regul Pept. 2008;151:19–25. doi: 10.1016/j.regpep.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25:1056–1062. doi: 10.1002/eji.1830250430. [DOI] [PubMed] [Google Scholar]
- 18.Crawford F, Huseby E, White J, Marrack P, Kappler JW. Mimotopes for alloreactive and conventional T cells in a peptide-MHC display library. PLoS Biol. 2004;2:523–533. doi: 10.1371/journal.pbio.0020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford F, et al. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol Rev. 2006;210:156–170. doi: 10.1111/j.0105-2896.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 20.Corper AL, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288:505–511. doi: 10.1126/science.288.5465.505. [DOI] [PubMed] [Google Scholar]
- 21.Latek RR, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 2000;12:699–710. doi: 10.1016/s1074-7613(00)80220-4. [DOI] [PubMed] [Google Scholar]
- 22.Gleeson CM, Curry WJ, Johnston CF, Buchanan KD. Occurrence of WE-14 and chromogranin A-derived peptides in tissues of the human and bovine gastro-entero-pancreatic system and in human neuroendocrine neoplasia. J Endocrinol. 1996;151:409–420. doi: 10.1677/joe.0.1510409. [DOI] [PubMed] [Google Scholar]
- 23.Curry WJ, et al. Chromogranin A and its derived peptides in the rat and porcine gastro-entero-pancreatic system. Expression, localization, and characterization. Adv Exp Med Biol. 2000;482:205–213. [PubMed] [Google Scholar]
- 24.Carson RT, Vignali KM, Woodland DL, Vignali DAA. T cell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity. 1997;7:387–399. doi: 10.1016/s1074-7613(00)80360-x. [DOI] [PubMed] [Google Scholar]
- 25.Arnold PY, et al. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J Immunol. 2002;169:739–749. doi: 10.4049/jimmunol.169.2.739. [DOI] [PubMed] [Google Scholar]
- 26.Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol. 2007;178:6051–6057. doi: 10.4049/jimmunol.178.10.6051. [DOI] [PubMed] [Google Scholar]
- 27.Maynard J, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 28.He XL, et al. Structural snapshot of aberrant antigen presentation linked to autoimmunity: the immunodominant epitope of MBP complexed with I-Au. Immunity. 2002;17:83–94. doi: 10.1016/s1074-7613(02)00340-0. [DOI] [PubMed] [Google Scholar]
- 29.Curry WJ, Johnston CF, Shaw C, Buchanan KD. Colocalization of WE-14 immunostaining with the classical islet hormones in the porcine pancreas. Adv Exp Med Biol. 1997;426:139–144. doi: 10.1007/978-1-4899-1819-2_18. [DOI] [PubMed] [Google Scholar]
- 30.Hill JA, et al. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. 2003;171:538–541. doi: 10.4049/jimmunol.171.2.538. [DOI] [PubMed] [Google Scholar]
- 31.Tollefsen S, et al. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest. 2006;116:2226–2236. doi: 10.1172/JCI27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hovhannisyan Z, et al. The role of HLA-DQ8 β 57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature. 2008;456:534–538. doi: 10.1038/nature07524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foulquier C, et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 2007;56:3541–3553. doi: 10.1002/art.22983. [DOI] [PubMed] [Google Scholar]
- 34.Ientile R, Caccamo D, Griffin M. Tissue transglutaminase and the stress response. Amino Acids. 2007;33:385–394. doi: 10.1007/s00726-007-0517-0. [DOI] [PubMed] [Google Scholar]
- 35.Mathews CE, et al. Mechanisms underlying resistance of pancreatic islets from ALR/Lt mice to cytokine-induced destruction. J Immunol. 2005;175:1248–1256. doi: 10.4049/jimmunol.175.2.1248. [DOI] [PubMed] [Google Scholar]
- 36.Anderson MS, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 37.Curry WJ, Shaw C, Johnston CF, Thim L, Buchanan KD. Isolation and primary structure of a novel chromogranin A-derived peptide, WE-14, from a human midgut carcinoid tumour. FEBS Lett. 1992;301:319–321. doi: 10.1016/0014-5793(92)80266-j. [DOI] [PubMed] [Google Scholar]
- 38.Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-Ag7 molecules show common sequence specificity. J Clin Invest. 2005;115:2268–2276. doi: 10.1172/JCI25350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 40.White J, et al. Two better cell lines for making hybridomas expressing specific T cell receptors. J Immunol. 1989;143:1822–1825. [PubMed] [Google Scholar]
- 41.Walker E, Warner NL, Chesnut R, Kappler J, Marrack P. Antigen-specific, I region-restricted interactions in vitro between tumor cell lines and T cell hybridomas. J Immunol. 1982;128:2164–2169. [PubMed] [Google Scholar]
- 42.Kozono H, White J, Clements J, Marrack P, Kappler J. Production of soluble MHC class II proteins with covalently bound single peptides. Nature. 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 43.Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8:675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- 44.Kappler J, White J, Kozono H, Clements J, Marrack P. Binding of a soluble αβ T-cell receptor to superantigen/major histocompatibility complex ligands. Proc Natl Acad Sci USA. 1994;91:8462–8466. doi: 10.1073/pnas.91.18.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]