

Abstract
Purpose
Most neuroblastoma patients over 18 months of age at diagnosis present disseminated disease. The presence of neuroblastoma cells in bone marrow can be used to evaluate the response to treatment. It is possible that alterations in certain tumour cells might confer a selective advantage over tumour dissemination process, and probably be helpful in the clonal selection of tumour-specific cells that could originate metastasis.
Methods
We performed real-time quantitative PCR to identify the presence of disseminated tumour cells in bone marrow samples, and we used MSP to analyse the methylation profile of 20 genes putatively implied in dissemination.
Results
We described epigenetic alterations in the methylated status of certain genes in disseminated tumour cells from bone marrow. Those cases with high rate of hypermethylation showed an increased probability of relapse during or after treatment. We found significantly poor prognosis in event-free survival in cases with hypermethylation of TMS1, MGMT and RARβ2 genes.
Conclusion
We could not confirm the presence of a specific methylation profile in disseminated neuroblastoma tumour cells, but a high accumulation of epigenetic events in those cells is associated with a high risk of relapse, independently of MYCN amplification.
Keywords: Neuroblastoma, Methylation, Prognosis factor, Disseminated disease, Metastasis, TMS1
Introduction
Childhood neuroblastoma (NB) exhibits contrasting patterns of clinical behaviour, ranging from spontaneous regression to rapid progression and death despite intensive treatment (Schwab et al. 2003).
Classically, neuroblastoma prognosis is based on two clinical parameters: patient age at diagnosis and tumour stage (Brodeur et al. 1988). Several established molecular prognostic parameters, such as DNA content (ploidy) (Kaneko et al. 1987), MYCN oncogene amplification (Seeger et al. 1985), allelic loss in the short arm of chromosome 1 (1p) (Christiansen and Lampert 1988), 11q (Attiyeh et al. 2005) and gain of genetic material in chromosome 17 (Caron 1995) have been progressively introduced as further prognostic indicators.
NB is characterised by extensive genetic, morphologic and clinical heterogeneity and is presented from benign to highly aggressive forms (Maris et al. 2007). Most children over 1 year of age present disseminated disease identified in bone marrow (BM), distant lymph nodes, bone, liver and/or other organs by imaging and/or histological examination. Despite the use of dose-intensive chemotherapy regimens for more than half of those children with stage 4 disease, prognosis remains poor.
The presence of NB cells detected in BM by cytology and molecular biology is one of the most powerful markers of poor prognosis in NB (Burchill 2004). Metastasis to the BM is a hallmark of high risk in NB and predicts poor prognosis for most children. However, children with stage 4S disease may present with BM metastases but still have a good outcome.
Epigenetic modifications, particularly the methylation of cytosines 5′ of guanine residues (CpGs) in gene promoter regions are essential regulatory mechanism for normal cell development. DNA methylation can inactivate tumour suppressor genes by altering gene transcription and by favouring C > T transitions in somatic and germline cells (Zhu and Yao 2007).
Here, we hypothesised that there are different genetic and epigenetic alterations between tumour cells from primary tumours and those tumour cells from BM metastasis. These alterations in certain tumour cells might confer a selective advantage over tumour dissemination process, and may help in the clonal selection of specific tumour cells that could originate the metastasis. We evaluated the presence of specific hypermethylation in disseminated NB disease and its putative role on several genes involved in different cellular pathways.
Patients and methods
Clinical samples
Tumour samples were collected from 11 NB stage 4 patients at diagnosis before treatment. NB staging was established according to the International Neuroblastoma Staging System (INSS) (Brodeur et al. 1988 ). The 22 BM samples were collected from these NB patients at diagnosis. All of these 33 samples were available for real-time quantitative PCR.
Preparation of total RNA
Ficoll gradient centrifugation was used to isolate mononuclear cells from BM specimens, as recommended by the manufacturer (Lymphoprep AXIS-SHIELD PoC AS). Once purified, mononuclear cells were lysed in RLT (RNasy kit; Qiagen) and immediately stored at −80°C. Tumour samples were also lysed in RLT after mechanical disaggregation and stored at −80°C. Total RNA was isolated with a RNasy kit (Qiagen), following the manufacturer’s recommendations.
RT-PCR
Total RNA was reverse transcribed in a final volume of 40 μL, following the manufacturer’s guidelines (Geneamp Gold RNA PCR Core Kit, Applied Biosystems). The reaction mixture was incubated at 25°C for 15 min and at 42°C for 30 min.
Real-time quantitative PCR
Relative quantification of DCX, TH and GAPD mRNA was achieved by means of the ABI Prism 7000 Sequence Detection System (Applied Biosystems). TaqMan MGB probes and primers were used to measure the DCX, TH and GAPD expression. All expression assays have FAM reporter dye at the 5′ end and a non-fluorescent quencher at the 3′ end of the probe. GAPD was used as an endogenous reference to control the difference in RNA extraction and cDNA synthesis. The assays’ references were: Hs00167057_m1, Hs00165941_m1 and Hs99999905_m1, respectively, for DCX, TH and GAPD genes. Samples were always run in duplicate PCR experiments. The initial PCR started using the ABI Prism 7000 Sequence Detector with a 50°C, 2-min step to optimise UNG activity, followed by a 95°C, 10-min step to activate AmpliTaq Gold DNA polymerase and UNG deactivation. Then, 50, two-step cycles were performed, one at 95°C for 15 s and other at 60°C for 1 min. The entire PCR took 2 h and 11 min to complete, with no post-PCR handling.
Relative quantification
The amount of both DCX and TH was measured in reference to the housekeeping GAPD gene, also studied in duplicate by obtaining the ΔC T values in the following way: the mean C T for the GAPD was subtracted from the mean C T of marker gene (Oltra et al. 2005).
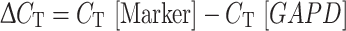 |
We established all results lower than ΔC T = 10 for at least one molecular marker as high level of BM infiltration.
Methylation-specific PCR (MSP)
Sodium bisulphite modification was carried out using the EpiTect system (Qiagen) according to the manufacturer’s instructions.
MSP analysis was performed to discriminate the methylation status of the 20 genes studied and classified according to their implication in cellular pathways: (1) angiogenesis: PTEN; (2) DNA repairing: MGMT, RIZ1 and hMLH1; (3) matrix adhesion: CD44 and THBS1; (4) drug resistance: GSTP1 and CFTR; (5) signal transduction: DR4, BLU, NORE1A and RARβ2; (6) apoptosis: CASP8, TMS1 and APAF1; (7) cell cycle: RB, EMP3, CCND2, RASSF1A and SYK.
Genomic DNA purified from peripheral blood of a healthy voluntary donor was used as a positive control for the unmethylated alleles, and this DNA treated in vitro with SssI methyltransferase (New England Biolabs) was used as a positive control for methylated alleles. A blank control containing all the PCR components, except template DNA, was also included in all the experiments. Reaction products were separated by electrophoresis on a 12% polyacrylamide gel, stained with silver nitrate.
A preliminary study was performed in order to check/verify the detection of hypermethylated DNA for each gene from disseminated tumour cells among non-methylated DNA of healthy cells. For this purpose, we detected hypermethylation in a 1/1,000 dilution of DNA previously treated with SssI among DNA from healthy donors.
Statistical analysis
Frequency analyses were performed by applying either the Fisher’s exact test or the Chi-square test. Survival time was measured from the event-free survival (EFS) date of diagnosis to first event (relapse or death by any cause). Survival analysis was performed to compare overall survival (OS) and EFS between groups using the Log-rank method. Stepwise Cox proportional hazards model (successive exclusion of the covariate with highest P value, if it is greater than 0.05) was applied to test the independent significance of different genetic and clinical parameters on patient outcome. These statistical analyses were performed using SPSS, version 12.0.
Results
For the present study, we selected tumours from 11 NB patients collected at diagnosis and before treatment, and their paired BM samples. For the methylation studies, we selected BM samples with ΔC T < 10 for TH and DCX gene markers.
In these matched samples from primary tumours and highly infiltrated BM, we studied the methylation pattern of 20 genes involved in different cellular pathways. We found no evidence of aberrant hypermethylation for the SYK gene in any BM and tumoral samples. In general, we found more evidence of hypermethylation in the primary tumour than in BM, but in three genes (GSTP1, MGMT and PTEN) BM samples showed epigenetic alterations in the disseminated tumour cells that where not seen in primary tumour cells (see Table 1).
Table 1.
Methylation percentage found in tumoral and BM samples for each gene classified according to their function
| Function | Gene | Methylation (%) | |
|---|---|---|---|
| Tumour | Bone marrow | ||
| Cell cycle | RASSF1A | 100 | 73 |
| EMP3 | 91 | 91 | |
| CCND2 | 91 | 91 | |
| RB | 9 | 0 | |
| SYK | 0 | 0 | |
| Apoptosis | CASP8 | 91 | 55 |
| APAF1 | 55 | 46 | |
| TMS1 | 27 | 36 | |
| Signal transduction | NORE1A | 64 | 82 |
| BLU | 73 | 91 | |
| RARβ2 | 55 | 55 | |
| DR4 | 55 | 36 | |
| Drug resistance | CFTR | 73 | 82 |
| GSTP1 | 0 | 9 | |
| Matrix adhesion | CD44 | 82 | 64 |
| THBS1 | 27 | 27 | |
| DNA repairing | RIZ1 | 45 | 64 |
| hMLH1 | 27 | 45 | |
| MGMT | 0 | 27 | |
| Angiogenesis | PTEN | 0 | 9 |
| Total | n = 11 | n = 11 | |
Almost all paired BM samples showed epigenetic alterations in the disseminated tumour cells. In some cases, the presence of these alterations was not found simultaneously in both kind of samples, but for the study we have considered the presence of hypermethylation in disseminated tumoral cells when at least one of the paired BM samples showed this aberrant methylation.
We observed aberrant hypermethylation in disseminated tumour cells and not in primary tumour cells in genes that are involved in DNA repair, cell cycle regulation, drug resistance, signal transduction, angiogenesis and apoptosis. Conversely, in those genes involved in matrix adhesion (THBS1 and CD44) we found no evidence of specific epigenetic alterations in disseminated tumour cells.
In order to analyse the putative influence of a high-hypermethylation pattern in our series, we studied the distribution of the hypermethylation profile of tumour and BM samples. Previously, we determined the existence of two different sample groups regarding the number of hypermethylated genes in a series of 82 primary tumours (work submitted). Applying the methylation rate obtained for our case series, it was not possible to identify two groups due to the small number of samples. For these reasons, we decided to use the same standard as in primary tumours considering the hypermethylation of more than 9 genes out of the 20 studied genes as threshold to define a high rate of hypermethylation in disseminated tumour cell samples. According to this classification, 54.5% (6/11) of BM samples were highly hypermethylated.
The analysis of hypermethylation rate in disseminated tumour cells opposite to age at diagnosis and MYCN amplification showed no significant results. In contrast, the high rate of hypermethylation in disseminated tumour cells was associated with relapse risk (P value = 0.0042) and seems to have influence in bad behaviour of overall survival of stage 4 patients in spite of the nonsignificant result (P value = 0.062) (see Fig. 1).
Fig. 1.

Event-free survival (left) and overall survival (right) in highly hypermethylated disseminated tumour cells from NB patients
We observed that the methylation degree in disseminated tumour cells was associated with disease progression; therefore we decided to evaluate the putative independence of this methylation degree as prognostic factor of the disease. We performed a survival analysis using Cox regression method, considering the methylation rate in disseminated tumour cells, as well as other established prognostic factors such as age at diagnosis and MYCN amplification. The results pointed out the high methylation rate in disseminated tumour cells as a variable with a high influence in relapse risk (P value = 0.02) (see Table 2).
Table 2.
Cox regression results on event-free survival analysis considering disseminated tumour cell hypermethylation, age at diagnosis and MYCN amplification
| Variables | Sig. | Exp(B) | 95.0% CI for Exp(B) | |
|---|---|---|---|---|
| Lower | Upper | |||
| Step 1 | ||||
| MYCN | 0.264 | 0.332 | 0.048 | 2.298 |
| Age at diagnosis | 0.895 | 1.185 | 0.096 | 14.622 |
| High hypermethylation DTC | 0.028 | 22.077 | 1.396 | 439.219 |
| Step 2 | ||||
| MYCN | 0.246 | 0.349 | 0.059 | 2.070 |
| High hypermethylation DTC | 0.014 | 20.237 | 1.854 | 220.920 |
| Step 3 | ||||
| High hypermethylation DTC | 0.020 | 13.118 | 1.496 | 115.050 |
DTC disseminated tumoral cells
On the other hand, we analysed the influence of the hypermethylated status for each gene on survival. We obtained significant association between hypermethylation in disseminated tumour cells and a high relapse risk in TMS1 (P value = 0.0046), MGMT (P value = 0.0317) and RARβ2 gene (P value = 0.0414) (see Fig. 2).
Fig. 2.

Event-free survival curves of TMS1 hypermethylation (left), MGMT hypermethylation (middle) and RARβ2 hypermethylation (right) in disseminated tumour cells of NB stage 4 patients
Discussion
We have studied the methylation profile of 20 genes involved in different cellular pathways from pair samples of primary tumour and BM. We tried to evaluate the existence of a hypermethylated phenotype specific for metastatic disease in NB, as well as the repercussion in disease progression of the hypermethylated status of each gene.
We found epigenetic alterations in the methylation pattern of all genes studied, except for SYK gene. Moreover, most of the epigenetic alterations observed in disseminated disease were also present in primary tumours.
The tumour series showed the higher methylation rate with respect to BM. This could be due to the fact that this kind of sample is composed almost entirely of tumour cells, which could acquire lots of molecular aberrations including the hypermethylation of regulatory regions in the genome. In disseminated tumour cells, the decreased methylation rate compared with primary tumour cells could be explained by the different methylation profile found in disseminated tumour cells.
The epigenetic alterations found in BM but not in the primary tumour mainly belong to genes involved in DNA repair, drug resistance, cell cycle regulation, apoptosis and signal transduction, which are mechanisms involved in tumour cell spreading. Nevertheless, the presence of these specific alterations in disseminated tumour cells points out that there is no specific mechanism epigenetically altered.
The high hypermethylation in disseminated tumour cells is significantly correlated with poor prognosis in EFS. In this way, we could suppose that disseminated tumour cells from BM of patients who have undergone a relapse showed a higher rate of epigenetic events that may confer to these cells, some advantages to metastasize and lead to disease progression. Given that all BM samples had been collected at diagnosis, this high rate of hypermethylation would indicate that the epigenetic alterations are a primary event in the tumour progression. Maybe the high amount of hypermethylation makes no possible the disseminated tumour cells to become a differentiated cell, remaining as undifferentiated and facilitating the metastasis process.
In the Cox regression, analysis we observed a high influence of methylation rate in disseminated tumour cells on EFS with respect to the other two established prognosis factors, age at diagnosis and MYCN amplification. The fact that the methylation variable has obtained the most significant values could be due to majority of our cases being classified as high risk, therefore values for the established prognosis factors would be biased. In spite of it, high hypermethylation in disseminated tumour cells would be considered independent of MYCN amplification and it could probably be useful to predict relapse behaviour.
On the other hand, despite the low number of cases available, we performed a survival analysis of each separate gene in the disseminated tumour cell samples and we found that the highest impact in EFS was from the hypermethylation of TMS1 gene. MGMT and RARβ2 were associated with poor prognosis in EFS too. In contrast, none of these genes had an impact in OS in the Kaplan–Meier analysis.
The TMS1 gene encodes an adaptor protein that promotes caspase-dependent apoptosis. The absence of TMS1 expression in some tumours is because its methylation contributes to carcinogenesis and cancer development (Liu et al. 2006; Martinez et al. 2007; Zhang et al. 2007). Some authors have described that the TMS1 proapoptotic gene was methylated in stage 4 NB tumours but not in stage 4S tumours (Alaminos et al. 2004). Our data therefore suggest the possibility that methylation-mediated silencing of TMS1 may confer a survival advantage to neuroblastoma cells by allowing disseminated tumour cells to evade apoptosis. In this way, hypermethylation of TMS1 gene has a wide impact on NB relapses because its silencing may lead to the disseminated tumour cells apoptosis favouring the proliferation in other tissues.
An interesting case is MGMT methylation, only detected in disseminated tumour cells in our series. In human cancer, the MGMT gene is not commonly mutated or deleted; thus, loss of MGMT function is most frequently due to epigenetic changes, specifically promoter region methylation (Qian and Brent 1997). Several tumours showed lack of expression of the MGMT gene associated with promoter hypermethylation (Alonso et al. 2003; Komine et al. 2003; Esteller et al. 1999). In NB, previous studies (Bello et al. 2004; Gonzalez-Gomez et al. 2003) reported MGMT methylation in 28% of analysed tumours, but in a recent study the promoter hypermethylation of this gene was 8% of primary tumours (Lázcoz et al. 2007).
In concordance with the results above, we have not only detected the absence of MGMT hypermethylation in primary tumour, but we have also found evidence of epigenetic alterations in highly infiltrated BM samples. Exclusive hypermethylation in disseminated tumour cells points out that this epigenetic alteration would be more usual in this kind of cells.
The RARβ2 gene encodes the receptor beta of retinoic acid, a member of the thyroid-steroid hormone receptor superfamily of nuclear transcriptional receptors. The RARβ2 binds to retinoic acid, the biologically active form of vitamin A, which mediates cellular signalling in embryonic morphogenesis, cell growth and differentiation. It is thought that this gene limits growth of many cell types by regulating gene expression and this expression is reduced in various human cancers as squamous cell carcinomas of the uterine cervix or breast cancer. It has been shown that its epigenetic inactivation can be an early event in these kinds of tumours (Arapshian et al. 2000; Ivanova et al. 2002).
Our results suggest that this gene could play an essential role inhibiting the spontaneous spreading of stage 4 neuroblastoma, probably by inducing neuroblast differentiation. The RARβ2 hypermethylation can decrease the amount of the receptor beta of retinoic acid, so disseminated tumoral cells undergo undifferentiation in response to retinoic acid stimuli. Moreover, high-risk neuroblastoma patients receive differentiation therapies with 13-cis retinoic acid after the multimodal therapy, inducing neuroblastoma cell growth arrest and differentiation and improving EFS. In this way, we could postulate that differentiation therapy outcome would be affected by the methylated status of RARβ2 gene, so this would increase the relapse risk of these patients.
In conclusion, we have observed the existence of epigenetic alterations in the methylation status of certain genes in disseminated tumour cells drawn from BM of stage 4 NB patients. Patients with a high rate of hypermethylation in disseminated NB had a major probability to relapse during or after treatment. We have not confirmed the existence of a specific hypermethylation profile in metastatic NB disease, eventhough we observed that the accumulation of epigenetic events in disseminated tumour cells is related to EFS. Moreover, among the studied genes we found that TMS1, RARβ2 and MGMT hypermethylation in disseminated tumour cells is related to risk of relapse, confirming the role of apoptosis and cell differentiation in disease progression. Hence, further studies on larger series should be performed to demonstrate the clinical, prognostic or therapeutical utility of methylation status of these genes and a high hypermethylation profile in disseminated tumour cells in high-risk neuroblastoma.
Acknowledgments
We are grateful to all the professionals who referred samples for these studies. We would also like to thank Desiree Ramal for her valuable help in the data manager task. English text was revised by the translation services of Fundación para la Investigación Hospital La Fe.
Conflict of interest statement
No conflict of interest has been declared.
References
- Alaminos M, Davalos V, Cheung NK, Gerald WL, Esteller M (2004) Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. J Natl Cancer Inst 96:1208–1219 [DOI] [PubMed] [Google Scholar]
- Alonso ME, Bello MJ, Gonzalez-Gomez P, Arjona D, Lomas J, de Campos JM, Isla A, Sarasa JL, Rey JA (2003) Aberrant promoter methylation of multiple genes in oligodendrogliomas and ependymomas. Cancer Genet Cytogenet 144:134–142 [DOI] [PubMed] [Google Scholar]
- Arapshian A, Kuppumbatti YS, Mira-y-Lopez R (2000) Methylation of conserved CpG cites neighboring the beta retinoic acid response element may mediate retinoic acid receptor beta gene silencing in MCF-7 breast cancer cells. Oncogene 19:4066–4070 [DOI] [PubMed] [Google Scholar]
- Attiyeh EF, London WB, Mossé YP, Wang Q, Winter C, Khazi D, McGrady PW, Seeger RC, Look AT, Shimada H, Brodeur GM, Cohn SL, Matthay KK, Maris JM, Children’s Oncology Group (2005) Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 353:2243–2253 [DOI] [PubMed] [Google Scholar]
- Bello MJ, Alonso ME, Aminoso C, Anselmo NP, Arjona D, Gonzalez-Gomez P, Lopez-Marin I, de Campos JM, Gutierrez M, Isla A, Kusak ME, Lassaletta L, Sarasa JL, Vaquero J, Casartelli C, Rey JA (2004) Hypermethylation of the DNA repair gene MGMT: association with TP53 G:C to A:T transitions in a series of 469 nervous system tumors. Mutat Res 554:23–32 [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D’Angio G, De Bernardi B, Evans AE, Favrot M, Freeman AI, Haase G, Hartmann O, Hayes FA, Helson L, Kemshead J, Lampert F, Ninane J, Ohkawa H, Philip T, Pinkerton CR, Pritchard J, Sawada T, Siegel S, Smith EI, Tsuchida Y, Voute PA (1988) International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6:1874–1881 [DOI] [PubMed] [Google Scholar]
- Burchill SA (2004) Micrometastases in neuroblastoma: are they clinically important? J Clin Pathol 57:14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron H (1995) Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med Pediatr Oncol 24:215–221 [DOI] [PubMed] [Google Scholar]
- Christiansen H, Lampert F (1988) Tumour karyotype discriminates between good and bad prognostic outcome in neuroblastoma. Br J Cancer 57:121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59:793–797 [PubMed] [Google Scholar]
- Gonzalez-Gomez P, Bello MJ, Lomas J, Arjona D, Alonso ME, Aminoso C, Lopez-Marin I, Anselmo NP, Sarasa JL, Gutierrez M, Casartelli C, Rey JA (2003) Aberrant methylation of multiple genes in neuroblastic tumours. relationship with MYCN amplification and allelic status at 1p. Eur J Cancer 39:1478–1485 [DOI] [PubMed] [Google Scholar]
- Ivanova T, Petrenko A, Gritsko T, Vinokourova S, Eshilev E, Kobzeva V, Kisseljov F, Kisseljova N (2002) Methylation and silencing of the retinoic acid receptor-beta 2 gene in cervical cancer. BMC Cancer 21:2–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Kanda N, Maseki N, Sakurai M, Tsuchida Y, Takeda T, Okabe I, Sakurai M (1987) Different karyotypic patterns in early and advanced stage neuroblastomas. Cancer Res 47:311–318 [PubMed] [Google Scholar]
- Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T (2003) Promoter hypermethylation of the DNA repair gene O6-methylguanine-DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low-grade diffuse astrocytomas. Brain Pathol 13:176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázcoz P, Muñoz J, Nistal M, Pestaña A, Encío IJ, Castresana JS (2007) Loss of heterozygosity and microsatellite instability on chromosome arm 10q in neuroblastoma. Cancer Genet Cytogenet 174:1–8 [DOI] [PubMed] [Google Scholar]
- Liu XF, Zhu SG, Zhang H, Xu Z, Su HL, Li SJ, Zhou XT (2006) The methylation status of the TMS1/ASC gene in cholangiocarcinoma and its clinical significance. Hepatobiliary Pancreat Dis Int 5:449–453 [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R, Cohn SL (2007) Neuroblastoma. Lancet 369:2106–2120 [DOI] [PubMed] [Google Scholar]
- Martinez R, Schackert G, Esteller M (2007) Hypermethylation of the proapoptotic gene TMS1/ASC: prognostic importance in glioblastoma multiforme. J Neurooncol 82:133–139 [DOI] [PubMed] [Google Scholar]
- Oltra S, Martinez F, Orellana C, Grau E, Fernandez JM, Cañete A, Castel V (2005) The doublecortin gene, a new molecular marker to detect minimal residual disease in neuroblastoma. Diagn Mol Pathol 14:53–57 [DOI] [PubMed] [Google Scholar]
- Qian XC, Brent TP (1997) Methylation hot spots in the 5′ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res 57:3672–3677 [PubMed] [Google Scholar]
- Schwab M, Westermann F, Hero B, Berthold F (2003) Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol 4:472–480 [DOI] [PubMed] [Google Scholar]
- Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D (1985) Association of multiple copies of the Nmyc oncogene with rapid progression of neuroblastomas. N Engl J Med 313:1111–1116 [DOI] [PubMed] [Google Scholar]
- Zhang C, Li H, Zhou G, Zhang Q, Zhang T, Li J, Zhang J, Hou J, Liew CT, Yin D (2007) Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J Pathol 212:134–142 [DOI] [PubMed] [Google Scholar]
- Zhu J, Yao X (2007) Use of DNA methylation for cancer detection and molecular classification. J Biochem Mol Biol 40:135–141 [DOI] [PubMed] [Google Scholar]