

Abstract
Parallels exist between drug-induced QT/QTc prolongation and shortening. However, these parallels are largely superficial and the experience with drug-induced QTc prolongation and its potential proarrhythmic link cannot be directly applied to drug-related QTc shortening. The congenital short QT syndrome (SQTS) is clearly much less prevalent than congenital, long QT syndrome, possibly some 1000 times. If the same discrepancy exists between arrhythmic susceptibility to drug-induced QTc prolongation and shortening, it is questionable whether regulatory burden should be imposed on drugs that might cause serious arrhythmia, once in many millions of exposures. Further, majority of torsadegenic drugs block the IKr current which is susceptible to the drug blockade because of the corresponding channel geometry. There is no parallel known for drug-induced QTc shortening. Also, all drugs that prolong QTc interval massively cause torsade de pointes tachycardia in more than exceptional isolated instances. On the contrary, digitalis that causes substantial QTc shortening is not known to trigger frequently ventricular arrhythmias. Moreover, most available population QTc data were obtained with Bazett's correction which produces erroneous QTc shortening at slow heart rates. Safety limits derived from such data are inappropriate. Because practically all new drugs undergo the so-called thorough QT study, drug-induced QTc shortening will not go unnoticed for any new pharmaceutical. Describing drug-related QTc shortening in the label seems sufficient to avoid treatment of the rare SQTS subjects. Intensive investigations of QTc-shortening drugs (similar to those of drugs with positive thorough QT studies) do not seem to be warranted.
This article is a commentary on Shah, pp. 58–69 of this issue and is part of a themed section on QT safety. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2010
Introduction
In his review on the potential implications of drug-induced QT interval shortening, Shah (2010) proposed a number of fairly plausible hypotheses and reasonably convincing theories. Unfortunately, as reviewed in this paper, the data supporting these theories and suggestions are either missing or potentially disputable. With equal persuasion, arguments can easily be proposed for the exact opposite. Because of this, a broader discussion of the concept of drug-induced QT interval shortening and of its implications for drug approval, therefore, seems to be warranted. Indeed, several aspects in Shah's review deserve deeper thought and consideration. As I will discuss below, the major problem, at present, with the concept of drug-induced QT shortening is the lack of systematic data and established facts. The area is therefore largely speculative. Thus, to facilitate the discussion, this paper sets out to argue the very opposite view.
Relationship to drug-induced QT prolongation
It has been well established that the potential for causing Torsade de Pointes (TdP) tachycardia should be fully assessed for drugs, which are found to prolong the heart rate corrected QT (QTc) interval. At the same time, it is also well known that QTc interval prolongation per se is not the core of the problem. Rather, drug-induced QTc prolongation is used in the regulatory review as a reasonable (and at present, the only practical plausible) clinical surrogate of drug-induced increase in repolarization heterogeneity, which is likely to be the true culprit and the true object of our concern. The surrogate is not perfect. While it is true that all the drugs that have so far been identified as causing TdP-type pro-arrhythmia in a meaningful number of cases do prolong the QTc interval substantially at high doses (e.g. when overdosed), there are also other compounds that cause some marginal QTc prolongation without inducing meaningfully frequent TdP pro-arrhythmia.
The frequency of the drug-related pro-arrhythmic episodes is actually of substantial importance. With some exaggeration, one can claim that every chemical more complex than sodium chloride or glucose will eventually lead to TdP or some other pro-arrhythmia in isolated cases of particularly sensitive individuals. In other words, the distinction between pro-arrhythmic and ‘safe’ drugs is not black and white. Rather, the incidence of pro-arrhythmic episodes creates a grey spectrum between drugs that cause pro-arrhythmia fairly frequently (e.g. quinidine; Selzer and Wray, 1964; Roden et al., 1986) and drugs that are very ‘safe’ and cause TdP only in very isolated instances (e.g. fexofenadine; Pinto et al., 1999; Pratt et al., 1999). Therefore, in addition to any risk–benefit considerations specific to individual drugs, a general threshold of regulatory ‘awareness’ must exist to decide on the highest frequency of pro-arrhythmia incidence that can be reasonably ignored during the approval of any drug. Otherwise, even drugs that cause pro-arrhythmia in extremely rare cases (i.e. a number of very safe drugs) would need to be correspondingly labelled with obvious counterproductive consequences. Hence, the regulatory discussion needs to distinguish between the ‘blame’ on the side of the drug and the unfortunate circumstance on the side of the very rare individual susceptibility. The awareness threshold must of course be fairly low to allow the pro-arrhythmic potential below the threshold to be ignored during drug approval. Only drugs that cause pro-arrhythmia more frequently than the threshold need to have their approval based on risk–benefit considerations (i.e. as far as their torsadegenic potential is concerned).
Thus, arguments about the number of drugs that have been reported to cause QTc prolongation have little if any meaning unless the distinction is made between those that cause pro-arrhythmia more frequently and those that cause pro-arrhythmia in isolated instances. Because of the present advances in the precision of electrocardiographic measurements, and because cardiac repolarization can be influenced by autonomic changes and many other mechanisms, some drug-induced QTc changes are likely to be found with many completely harmless compounds.
There are presently no studies and/or established knowledge that would offer the same link between QTc shortening, arrhythmia occurrence and a relevant threshold of regulatory awareness. While it is plausible to speculate that similar to QT interval prolongation, QT interval shortening might also increase the repolarization heterogeneity; this is far from established and could be entirely false. It is equally plausible to propose that drugs that shorten the QTc interval actually make the repolarization of different regions and strata of the ventricular myocardium more synchronized and less heterogeneous. Abolishing the prolonged action potential duration in the mid-myocardial regions that has been proposed to account for most of the repolarization heterogeneity should indeed make the repolarization process more uniform and less prone to after-depolarization or other arrhythmia-triggering phenomena.
Congenital syndromes
Shah is correct to point out that only a minority of subjects who experience drug-induced TdP pro-arrhythmia have been found to have known mutations of the congenital long QT syndrome (LQTS) type. It is of course also possible that so far undiscovered and unique mutations exist, making the patients more susceptible to TdP episodes. It is also true that most patients who experience drug-triggered TdP and who do not have an established LQTS mutation have other predisposing factors, making them more susceptible to increased repolarization heterogeneity, such as heart failure or chronic renal insufficiency. As the patients with heart failure and the patients with chronic renal insufficiency tend to have longer QT intervals (Voiculescu et al., 2006; Breidthardt et al., 2007; Madias, 2007; Watanabe et al., 2007; Kolo et al., 2008), it is far from obvious whether drug-induced QTc shortening would have the same epidemiological dimensions. It is just as likely that in patients with diminished repolarization reserve, shortening the action potential durations would make myocardial repolarization more stable. Hence, drugs that shorten ventricular repolarization might easily be beneficial to patients who are prone to arrhythmic episodes without congenital abnormality of myocardial ionic channels. For instance, in randomized controlled studies in patients with advanced chronic heart failure, atorvastatin was observed to shorten QTc interval (Vrtovec et al., 2005; Xie et al., 2009) while having clear beneficial effects.
Shah is also very right that the incidence of congenital short QT syndrome is presently practically unknown. Nevertheless, it seems obvious that the syndrome is substantially less prevalent than the congenital LQTS, easily by several magnitudes; last year, only a few tens of patients were reported to be diagnosed with congenital short QT syndrome worldwide (Zareba and Cygankiewicz, 2008). Consistent with population reports (Reinig and Engel, 2007), the incidence of clinically manifested congenital short QT syndrome is likely to be below one case in millions (i.e. some thousand times rarer than the congenital LQTS) (Zareba and Cygankiewicz, 2008). Indeed, in a study of more than 6.5 million ECG recordings from more than 1.7 million individuals, Iribarren et al. (2009) found long QTc intervals 511 times more frequently than short QTc intervals. Moreover, the majority of findings of the short QTc intervals were sporadic (i.e. other tracings in the same individual did not show the short QT). Only in one person (out of >1.7 million!) did they observe multiple short QTc readings.
Assuming that Shah is right and that there is a relationship between the congenital abnormalities and the incidence of drug-induced pro-arrhythmia, we need then to consider the question of an appropriate regulatory awareness threshold. Presently, the threshold of regulatory awareness or QTc prolongation set in the E14 guidance (ICH, 2005) is fairly low. Indeed, it needs to be so as, when considering biomarkers of drug safety, our tolerance for false positives is (and should be) much higher than our tolerance for false negatives. Thus, the E14 threshold translates approximately into the requirement to scrutinize compounds intensively if they might cause TdP pro-arrhythmia in more than about 1 in 105 to 106 exposures (Malik, 2003). This level of the threshold is based on the reported TdP incidences with drugs that, under therapeutic doses, lead to QTc prolongation just above the E14 threshold. Naturally, differences between reported incidences, true clinically manifested incidences and total incidences, including asymptomatic episodes need to be considered and can only be approximated. Nevertheless, the same would most likely apply to pro-arrhythmia caused by both QT lengthening and shortening.
Hence, considering the proportions between the incidence of congenital long QT and congenital short QT syndromes, it is highly questionable whether there should be any regulatory discussion about compounds that might lead to one fatal case of arrhythmia in many millions or tens of millions of exposures. Clearly, no patients should be subjected to undue risk if it can be avoided, but is seems more appropriate and much more practical (economically and otherwise) to identify the rarely occurring families with congenital short QT syndrome, and to treat them with all due safety rather than to impose yet another regulatory hurdle on all the broad spectrum of new chemical entities in pharmaceutical development. As discussed further, simple statement of QT shortening in the label seems to be quite enough, while extensive post-marketing investigations might only waste valuable resources that could be better utilized elsewhere.
Theoretically, it is also possible that there is no relationship between the incidence of congenital abnormalities and the number of drug-induced pro-arrhythmia cases, and that the number of arrhythmic episodes due to drug-induced QT shortening is substantially larger than the incidence of short QT interval in the general population. Nevertheless, it seems reasonable to expect that if this were the case, there would already be some clinical experience with such instances. This is clearly not the case.
Mechanisms of drug effects
The vast majority of compounds that have so far been implicated in TdP pro-arrhythmia have been found to block the delayed potassium rectifier current, IKr. This is not surprising because the IKr channel is known to have a peculiar geometry with a large vestibule that can easily be entered and consequently blocked by a number of different molecules. It is of course known that not all the drugs that block the IKr current have pro-arrhythmic properties. Nevertheless, in drugs that affect the delayed rectifier, the IKr blocking mechanism appears to be responsible for the dose relationship of the pro-arrhythmic effects. With IKr blocks, the clinical effects are more obvious with higher doses, irrespective of whether these are caused by supratherapeutic administration or by metabolic inhibition. This dose relationship is of substantial importance as many drugs with TdP liability are pro-arrhythmic mainly or solely at increased doses.
There is no such parallel known for drug-induced QTc shortening. The geometric properties of the IKr channel and of its large vestibule make the mechanisms of the drug-induced QTc interval prolongation and drug-induced QTc shortening clearly non-reciprocal. Similar considerations can easily be applied to other channels with blocks and augmentations not mirroring each other. Hence, our experience with the investigation of drug-induced QTc interval prolongation cannot be so easily translated into drug-induced QTc shortening by just assuming a simple reflection.
Mechanisms of ventricular fibrillation
Shah proposed that clinical cases of patients dying due to drug-induced QTc shortening are missed (i.e. not recognized as such) because they are hidden in fatal ventricular fibrillation. This theory is of course plausible, but not necessarily true as there are no data to support it.
There are many other mechanisms of possible drug actions that might be portrayed in similarly drastic scenarios. For instance, it is well known that reduced heart rate variability (HRV) is associated with increased risk of arrhythmic and other adverse events in cardiac patients. This does not necessarily mean that in addition to the thorough QTc studies, we should also conduct thorough HRV studies investigating each drug for its effects on cardiac autonomic regulation. The list of drugs that affect HRV is as long as that of drugs that change QTc interval duration. At the same time, the association between drug-related HRV reduction and increased risk does not appear to exist. As an example, in spite of its repeatedly established benefits (Perna et al., 1998; Tepel et al., 2008), amlodipine has been reported to reduce HRV (Zaliūnas et al., 2005).
Similarly, we might easily speculate that drugs that increase R wave voltage on the electrocardiogram (Pelliccia et al., 1991) or those that increase the fractionation of the ventricular electrogram (Saumarez and Grace, 2000) would be prone to pro-arrhythmic mechanisms similar to those in hypertrophic and dilated cardiomyopathies. In this proposal, there are substantial similarities to QTc shortening. The incidence of familial dilated cardiomyopathy (Codd et al., 1989; Manolio et al., 1992) is larger than that of congenital short QT syndrome, clinically healthy relatives of patients show subclinical abnormalities (Baig et al., 1998) and the disease is associated with a clear risk of fatal arrhythmias (Jordaens et al., 1996). Further, preclinical models identified R wave increases caused by drugs and chemicals of known or assumed pro-arrhythmic properties (Sandusky and Meyers, 1985; Omran and Abdel-Nabi, 1997; Watanabe et al., 1999). Nevertheless, while the hypotheses about adverse drug actions associated with these electrocardiographic changes might be as plausible as or even more plausible than the hypothesis of QTc shortening causing abrupt ventricular fibrillation, there is no evidence suggesting that we should take them seriously. Consequently, developing a concept of ‘thorough QRS complex studies’ in drug development, and suggesting, for instance, that a reasonable limit of R wave voltage increase is 200 µV with an upper confidence interval of 500 µV, and so on, would clearly be much more practically harmful than potentially helpful.
Existing experience with drug-induced QTc shortening
Although, as already stated, drug-induced QT interval prolongation is not necessarily equal to clinical pro-arrhythmia, every drug that has been found to prolong the QT interval substantially (i.e. much more than the present regulatory threshold specified in the E14 guidance) has also been found to cause TdP arrhythmia in an appreciable number of cases. (Jenzer and Hagemeijer, 1976; Davidenko et al., 1989; DeWitt and Waksman, 2004). There are no exceptions; even amiodarone, which is frequently misquoted as a counter-example, causes TdP frequently (Hohnloser et al., 1994; Tran et al., 1997; Yamada et al., 2001; Lim et al., 2006), although perhaps less frequently than other anti-arrhythmic drugs. Hence, without any exception, all drugs that prolong QT interval more than trivially do cause TdP arrhythmia with a frequency that is important both from a regulatory and clinical point of view.
On the contrary, the drug that comes first to mind when considering obvious and clear drug-induced QTc shortening is digitalis (Ahnve, 1985; Saner et al., 1988). It shortens the QTc substantially and in almost every patient. In spite of this, while digitalis may cause other arrhythmic complications, it is not known to trigger ventricular arrhythmias frequently (Gheorghiade et al., 2004). Only very sporadic and isolated reports of digitalis-related ventricular fibrillation are available (Garberoglio et al., 2007). As these cases appear much less frequently than, say, TdP induction by amiodarone, their very low incidence convincingly suggests that their principal mechanism must be different to the general, and thus much more frequent, QTc shortening.
Reports of QTc shortening
It has been repeatedly acknowledged that Bazett's correction leads to artificial and erroneous QTc shortening in the presence of bradycardia (Figure 1). Indeed, when using Bazett's correction, one finds not only that beta blockers shorten QTc interval (Malik, 2002), but the same applies to other drugs and conditions that lead to heart rate deceleration. Thus, many observations of QTc shortening deserve deeper scrutiny. For instance, in the study of 53 neonates with congenital hypothyroidism and 15 age-matched normal neonates for controls, Asami et al. (2001) found not only a mean 25 ms difference in QTc intervals, but also a mean 13 bpm difference in heart rates, with statistical significances much stronger for the heart rate than for the QTc comparison. They also applied Bazett's formula to the fairly high heart rates in neonates (means of 148 and 134 bpm in the comparisons) where Bazett's formula, together with many other correction formulae derived from adult populations, is clearly inappropriate. Besides, while heart rate showed statistically significant correlation with the serum levels of serum thyrotropin and free thyroxine, this was not the case with QT/QTc intervals. Thus, suggesting that there is evidence of QTc shortening due to congenital hypothyroidism is not substantiated.
Figure 1.
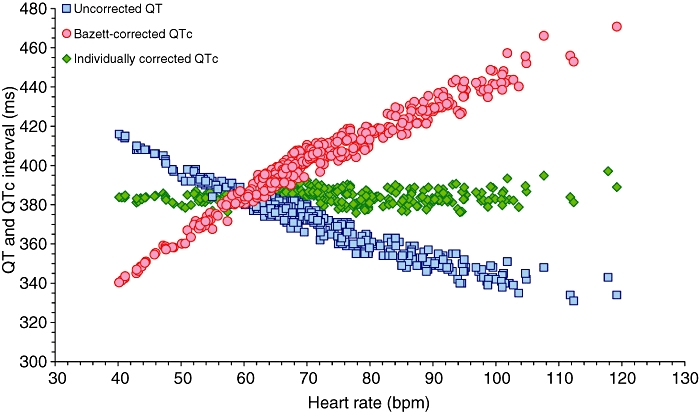
Example of a QT/heart rate and QTc/heart rate distribution in a drug-free recording of a healthy subject. The blue squares show the uncorrected QT interval. The red circles show the Bazett-corrected QTc interval. Note that the range of the QTc (B) values is larger than the range of QT values. While the accurate individualized heart rate correction (green diamonds) leads to a practically horizontal line of QTc ∼384 ms irrespective of heart rate, Bazett's correction leads to artificial QTc shortening at slow and QTc prolongation at fast heart rates. In this data set, the standard deviation of uncorrected QT intervals, Bazett-corrected QTc intervals and individually corrected QTc intervals is 17.2, 24.2 and 3.5 ms respectively.
Moreover, the differences in QTc reported by Shah (2010) as the support for his hypothesis of possible pro-arrhythmic signs are in many instances difficult to project into practise. For instance, the study of the electrocardiographic changes in patients with idiopathic ventricular fibrillation found a mean QTc shortening of 14 ms in male patients compared to controls (and a mean QTc prolongation of 9 ms in female patients), again all with Bazett's formula (Viskin et al., 2004). Similarly, the study showing association of short QTc interval with epilepsy reported a mean QTc of 420 ms in controls, 410 ms in patients with symptomatic epilepsy and 392 ms in patients with cryptogenic epilepsy (Teh et al., 2007). These differences are not only much smaller than the magnitude of QTc prolongation by some 80 ms (Honig et al., 1993) found in cases of drug-induced TdP (as well as smaller than Shah's proposed thresholds for concern), but also not very dissimilar from the QTc shortening seen frequently in thorough QTc studies on placebo, most likely because of autonomic conditioning effects (Figure 2).
Figure 2.
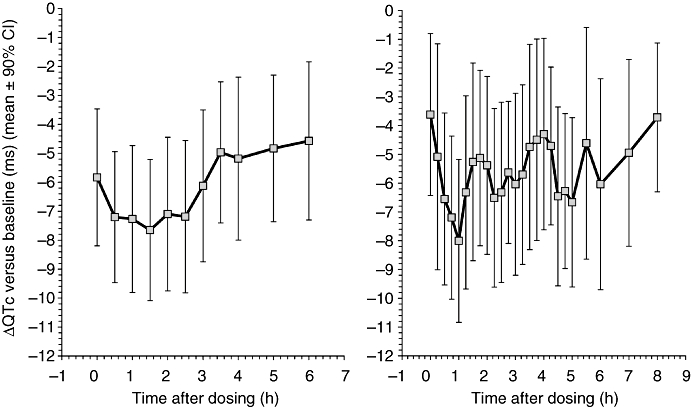
Examples of QTc shortening on placebo observed in two different unpublished thorough QTc studies. Each graph shows a section of changes in individually corrected QTc intervals versus baseline. These placebo-induced changes are likely to be caused by autonomic conditioning due to the stress of the closed and restricted environment of clinical units.
Evaluation of thorough QTc studies
While the thresholds of 5 and 10 ms for the QTc prolongation presently incorporated in the E14 guidance (ICH, 2005) have been derived from serious studies of existing data of drugs that do cause TdP pro-arrhythmia, the suggestions of the thresholds of −15 and −30 ms do not have the same credibility for QTc shortening. Shah derived them from simple comparisons of an assumed point estimate of a ‘normal’ QTc value of 430 ms and the thresholds for a significant QTc prolongation (>500 ms) and shortening (<320 ms). Unfortunately, the normal point estimate of 430 ms again comes from studies that used Bazett's correction. Because the majority of subjects, especially hospitalized patients (Reinig and Engel, 2007) have heart rates somewhat higher than 60 bpm, Bazett-derived QTc is overestimated. When using more accurate individual corrections, much lower normal QTc values are found. Indeed, in an electrocardiographic study of more than 11 000 subjects, Rautaharju and Zhang (2002) found the uncorrected QT intervals of around 410 ms at heart rate bins close to 60 bpm. Thus, the true normal point estimate of correctly calculated QTc is approximately just in the middle of the abnormality limits of 320 and 500 ms. Consequently, one could find the exact inversion of E14 limits appropriate. Nevertheless, this is, at present, clearly a step too far. While there are a number of TdP-pro-arrhythmic drugs that lead to small QTc increases, especially when administered at low doses (Malik and Camm, 2001), there is no such evidence whatsoever with drugs that lead to minor QTc shortening.
Moreover, the categorical analyses of the incidence of substantially prolonged (and likewise, substantially shortened) QTc interval are becoming presently rather outdated especially when new tight procedures for electrocardiographic processing are used in the evaluation of thorough QT/QTc studies. Pro-arrhythmic drugs are frequently found to prolong QT interval above the mean E14 threshold while not leading to a prolongation above 60 ms in any single electrocardiogram (Malik et al., 2009). Accurate electrocardiographic processing also allows conducting the thorough QTc studies with a small number of subjects, hence decreasing the probability that the study population would include an individual with particular susceptibility to drug effects. Thus, the QTc outlier analysis is presently of diminished value, especially if trying to detect drugs with less than monumental effects on cardiac repolarization.
Thorough QTc studies and QTc shortening
Practically, all new pharmaceuticals are now investigated in thorough QT/QTc studies, and this requirement is likely to remain in place for the foreseeable future. Unfortunately, expectations of replacing the QTc interval evaluation with other ECG biomarkers, such as T wave morphology changes, are failing to materialize (Malik, 2009). The observation of drug-induced QTc shortening will thus be made from time to time, and no drug that shortens ventricular repolarization will go unnoticed.
There are good regulatory reasons to request detailed ECG evaluations of drugs that marginally or moderately prolong QT interval (i.e. above the E14 threshold) during phase III clinical studies. However, it is not presently obvious whether anything useful would be gained from similar ECG evaluations of drugs that cause marginal or moderate QTc shortening. Potentially susceptible patients seem to occur so infrequently that they could be easily missed, even in the largest of clinical programmes. Risk–benefit evaluation is helped by evaluation of categorical QTc prolongations from phase III studies, but because of the likely low incidence, this would not be the case with QTc shortening. Hence, simple statement of the fact in the drug label would be enough so that when prescribing the drug, physicians are reminded about the very rarely occurring cases of congenital short QT syndrome. Apart from this basic statement of the fact in the label, there is presently no obvious reason to treat drugs that marginally or moderately shorten QTc any differently to drugs that have no effects on ventricular repolarization.
The recently approved label of rufinamide is an example of such a pragmatic approach. The rufinamide label rightly states that the degree of QT shortening induced by the drug is without any known clinical risk (Eisai, 2008).
Conclusion
All these comments do not mean to say that a massive effect on cardiac repolarization by a pharmaceutical compound should be taken lightly, irrespective of the direction of the QTc change. If a drug causes a truly large QTc shortening (e.g. by an average of 50 or 60 ms), its safety must be seriously considered because cardiac electrophysiology is clearly dramatically affected. With much lesser changes, the situation is very different. Mechanisms that can lead to small QTc decreases are manifold, and their safety implications are either none or poorly understood. It would of course be wrong to state that drug-induced QTc shortening is not an issue. Similar to the opposite view, there is no evidence for such a statement. Nevertheless, before we embark on regulatory implications and before we build yet another hurdle to drug approval (with potentially practical and economic implications), much more evidence is needed, so that we are not just dealing with a purely hypothetical situation, a mirage. It would be most unfortunate if drug-induced QTc shortening becomes important for the pure and simple reason that it is perceived to be important.
References
- Ahnve S. QT interval prolongation in acute myocardial infarction. Eur Heart J. 1985;6:85–95. doi: 10.1093/eurheartj/6.suppl_d.85. [DOI] [PubMed] [Google Scholar]
- Asami T, Suzuki H, Yazaki S, Sato S, Uchiyama M. Effects of thyroid hormone deficiency on electrocardiogram findings of congenitally hypothyroid neonates. Thyroid. 2001;11:765–768. doi: 10.1089/10507250152484600. [DOI] [PubMed] [Google Scholar]
- Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. doi: 10.1016/s0735-1097(97)00433-6. [DOI] [PubMed] [Google Scholar]
- Breidthardt T, Christ M, Matti M, Schrafl D, Laule K, Noveanu M, et al. QRS and QTc interval prolongation in the prediction of long-term mortality of patients with acute destabilised heart failure. Heart. 2007;93:1093–1097. doi: 10.1136/hrt.2006.102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd MB, Sugrue DD, Gersh BJ, Melton LJ. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- Davidenko JM, Cohen L, Goodrow R, Antzelevitch C. Quinidine-induced action potential prolongation, early after depolarizations, and triggered activity in canine Purkinje fibers: effects of stimulation rate, potassium, and magnesium. Circulation. 1989;79:674–686. doi: 10.1161/01.cir.79.3.674. [DOI] [PubMed] [Google Scholar]
- DeWitt CR, Waksman JC. Pharmacology, pathophysiology and management of calcium channel blocker and beta-blocker toxicity. Toxicol Rev. 2004;23:223–238. doi: 10.2165/00139709-200423040-00003. [DOI] [PubMed] [Google Scholar]
- Eisai. Banzel™ (rufinamide) 2008. Product information. http://www.banzel.com/Docs/Pdf/BanzelPI.pdf[accessed on 13 September 2009]
- Garberoglio L, Giustetto C, Wolpert C, Gaita F. Is acquired short QT due to digitalis intoxication responsible for malignant venticular arrhythmias? J Electrocardiol. 2007;40:43–46. doi: 10.1016/j.jelectrocard.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Adams KFJ, Colucci WS. Digoxin in the management of cardiovascular disorders. Circulation. 2004;109:2959–2964. doi: 10.1161/01.CIR.0000132482.95686.87. [DOI] [PubMed] [Google Scholar]
- Hohnloser SH, Klingenheben T, Singh BN. Amiodarone-associated proarrhythmic effects: a review with special reference to Torsade de Pointes tachycardia. Ann Intern Med. 1994;121:529–535. doi: 10.7326/0003-4819-121-7-199410010-00009. [DOI] [PubMed] [Google Scholar]
- Honig PK, Wortham DC, Zamani K, Conner DP, Mullin JC, Cantilena LR. Terfenadine–ketoconazole interaction. Pharmacokinetic and electrocardiographic consequences. JAMA. 1993;269:1513–1518. [PubMed] [Google Scholar]
- ICH. International Conference on Harmonization ICH Note for Guidance on the Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-antiarrhythmic Drugs (ICH E14) 2005. http://www.fda.gov/RegulatoryInformation/Guidances/ucm129335.htm[accessed on 11 July 2009. [PubMed]
- Iribarren C, Round AD, Lyon LL, Terdiman JF, Stang PD. Descriptive epidemiology of short QT interval in a cohort of 1.7 million persons. Eur Heart J. 2009;30 Abst suppl 697. [Google Scholar]
- Jenzer HR, Hagemeijer F. Quinidine syncope: Torsade de Pointes with low quinidine plasma concentrations. Eur J Cardiol. 1976;4:447–451. [PubMed] [Google Scholar]
- Jordaens L, de Pauw M, Caes F. Familial dilated cardiomyopathy and spontaneous ventricular arrhythmias. Am J Cardiol. 1996;78:102–104. doi: 10.1016/s0002-9149(96)00510-3. [DOI] [PubMed] [Google Scholar]
- Kolo PM, Opadijo OG, Omotoso AB, Balogun MO, Araoye MA, Katibi IA. Prevalence of QTc prolongation in adult Nigerians with chronic heart failure. West Afr J Med. 2008;27:69–73. [PubMed] [Google Scholar]
- Lim HE, Pak HN, Ahn JC, Song WH, Kim YH. Torsade de Pointes induced by short-term oral amiodarone therapy. Europace. 2006;8:1051–1053. doi: 10.1093/europace/eul118. [DOI] [PubMed] [Google Scholar]
- Madias JE. The resting electrocardiogram in the management of patients with congestive heart failure: established applications and new insights. Pacing Clin Electrophysiol. 2007;30:123–128. doi: 10.1111/j.1540-8159.2007.00586.x. [DOI] [PubMed] [Google Scholar]
- Malik M. The imprecision in heart rate correction may lead to artificial observations of drug induced QT interval changes. Pacing Clin Electrophysiol. 2002;25:209–216. doi: 10.1046/j.1460-9592.2002.00209.x. [DOI] [PubMed] [Google Scholar]
- Malik M. Drug-induced QT interval prolongation – why is it of concern? 2003. http://www.fda.gov/ohrms/dockets/ac/03/slides/4000S1_05_Malik_files/frame.htm[accessed on 13 September 2009]
- Malik M. Drug-induced changes in the T-wave morphology. Drug Saf. 2009;32:613–617. doi: 10.2165/00002018-200932070-00007. [DOI] [PubMed] [Google Scholar]
- Malik M, Camm AJ. Evaluation of drug-induced QT interval prolongation: implications for drug approval and labelling. Drug Saf. 2001;24:323–351. doi: 10.2165/00002018-200124050-00001. [DOI] [PubMed] [Google Scholar]
- Malik M, Hnatkova H, Schmidt A, Smetana P. Electrocardiographic QTc changes due to moxifloxacin infusion. J Clin Pharmacol. 2009;49:674–683. doi: 10.1177/0091270008330984. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, et al. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop) Am J Cardiol. 1992;69:1458–1466. doi: 10.1016/0002-9149(92)90901-a. [DOI] [PubMed] [Google Scholar]
- Omran MA, Abdel-Nabi IM. Changes in the arterial blood pressure, heart rate and normal ECG parameters of rat after envenomation with Egyptian cobra (Naja haje) venom. Hum Exp Toxicol. 1997;16:327–333. doi: 10.1177/096032719701600606. [DOI] [PubMed] [Google Scholar]
- Pelliccia F, Critelli G, Cianfrocca C, Nigri A, Reale A. Electrocardiographic correlates with left ventricular morphology in idiopathic dilated cardiomyopathy. Am J Cardiol. 1991;68:642–647. doi: 10.1016/0002-9149(91)90358-r. [DOI] [PubMed] [Google Scholar]
- Perna GP, Valle G, Cianfrone N, Luca GD, Amico C, Coli C. Amlodipine in ischaemic left ventricular dysfunction with mild to moderate heart failure. Clin Drug Investig. 1998;16:289–296. doi: 10.2165/00044011-199816040-00003. [DOI] [PubMed] [Google Scholar]
- Pinto YM, van Gelder IC, Heeringa M, Crijns HJ. QT lengthening and life-threatening arrhythmias associated with fexofenadine. Lancet. 1999;353:980. doi: 10.1016/s0140-6736(99)01009-0. [DOI] [PubMed] [Google Scholar]
- Pratt C, Brown AM, Rampe D, Mason J, Russell T, Reynolds R, et al. Cardiovascular safety of fexofenadine HCl. Clin Exp Allergy. 1999;29(Suppl 3):212–216. doi: 10.1046/j.1365-2222.1999.0290s3212.x. [DOI] [PubMed] [Google Scholar]
- Rautaharju PM, Zhang ZM. Linearly scaled, rate-invariant normal limits for QT interval: eight decades of incorrect application of power functions. J Cardiovasc Electrophysiol. 2002;13:1211–1218. doi: 10.1046/j.1540-8167.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- Reinig MG, Engel TR. The shortage of short QT intervals. Chest. 2007;132:246–249. doi: 10.1378/chest.06-2133. [DOI] [PubMed] [Google Scholar]
- Roden DM, Woosley RL, Primm RK. Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am Heart J. 1986;111:1088–1093. doi: 10.1016/0002-8703(86)90010-4. [DOI] [PubMed] [Google Scholar]
- Sandusky GE, Jr, Meyers DB. Toxicology of indecainide hydrochloride in mice, rats, and dogs. Fundam Appl Toxicol. 1985;5:175–181. doi: 10.1016/0272-0590(85)90062-4. [DOI] [PubMed] [Google Scholar]
- Saner HE, Pierach CA, Aeppli DM. Relation between serum digoxin concentration and the electrocardiogram. Clin Cardiol. 1988;11:752–756. doi: 10.1002/clc.4960111106. [DOI] [PubMed] [Google Scholar]
- Saumarez RC, Grace AA. Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res. 2000;47:11–22. doi: 10.1016/s0008-6363(00)00096-1. [DOI] [PubMed] [Google Scholar]
- Selzer A, Wray HW. Quinidine syncope. Paroxysmal ventricular fibrillation occurring during treatment of chronic atrial arrhythmias. Circulation. 1964;30:17–26. doi: 10.1161/01.cir.30.1.17. [DOI] [PubMed] [Google Scholar]
- Shah RR. Drug-induced QT interval shortening: potential harbinger of proarrhythmia and regulatory perspectives. Br J Pharmacol. 2010;159:58–69. doi: 10.1111/j.1476-5381.2009.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh HS, Tan HJ, Loo CY, Raymond AA. Short QTc in epilepsy patients without cardiac symptoms. Med J Malaysia. 2007;62:104–108. [PubMed] [Google Scholar]
- Tepel M, Hopfenmueller W, Scholze A, Maier A, Zidek W. Effect of amlodipine on cardiovascular events in hypertensive haemodialysis patients. Nephrol Dial Transplant. 2008;23:3605–3612. doi: 10.1093/ndt/gfn304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Chow MS, Kluger J. Amiodarone induced Torsades de Pointes with excessive QT dispersion following quinidine induced polymorphic ventricular tachycardia. Pacing Clin Electrophysiol. 1997;20:2275–2278. doi: 10.1111/j.1540-8159.1997.tb04249.x. [DOI] [PubMed] [Google Scholar]
- Viskin S, Zeltser D, Ish-Shalom M, Katz A, Glikson M, Justo D, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm. 2004;1:587–591. doi: 10.1016/j.hrthm.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Voiculescu M, Ionescu C, Ismail G. Frequency and prognostic significance of QT prolongation in chronic renal failure patients. Rom J Intern Med. 2006;44:407–417. [PubMed] [Google Scholar]
- Vrtovec B, Okrajsek R, Golicnik A, Ferjan M, Starc V, Radovancevic B. Atorvastatin therapy increases heart rate variability, decreases QT variability, and shortens QTc interval duration in patients with advanced chronic heart failure. J Card Fail. 2005;11:684–690. doi: 10.1016/j.cardfail.2005.06.439. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kubota I, Yamaki M, Tachibana H, Tomoike H. Direct effects of class I antiarrhythmic drugs on epicardial electrograms in dogs. Jpn Heart J. 1999;40:621–628. doi: 10.1536/jhj.40.621. [DOI] [PubMed] [Google Scholar]
- Watanabe E, Arakawa T, Uchiyama T, Tong M, Yasui K, Takeuchi H, et al. Prognostic significance of circadian variability of RR and QT intervals and QT dynamicity in patients with chronic heart failure. Heart Rhythm. 2007;4:999–1005. doi: 10.1016/j.hrthm.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Xie RQ, Cui W, Liu F, Yang C, Pei WN, Lu JC. Statin therapy shortens QTc, QTcd, and improves cardiac function in patients with chronic heart failure. Int J Cardiol. 2009 doi: 10.1016/j.ijcard.2008.11.030. Epub ahead of print] doi: 10.1016/j.ijcard.2008.11.030. [DOI] [PubMed] [Google Scholar]
- Yamada S, Kuga K, Yamaguchi I. Torsade de Pointes induced by intravenous and long-term oral amiodarone therapy in a patient with dilated cardiomyopathy. Jpn Circ J. 2001;65:236–238. doi: 10.1253/jcj.65.236. [DOI] [PubMed] [Google Scholar]
- Zaliūnas R, Brazdzionyte J, Zabiela V, Jurkevicius R. Effects of amlodipine and lacidipine on heart rate variability in hypertensive patients with stable angina pectoris and isolated left ventricular diastolic dysfunction. Int J Cardiol. 2005;101:347–353. doi: 10.1016/j.ijcard.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Zareba W, Cygankiewicz I. Long QT syndrome and short QT syndrome. Prog Cardiovasc Dis. 2008;51:264–278. doi: 10.1016/j.pcad.2008.10.006. [DOI] [PubMed] [Google Scholar]