

Abstract
Hypersensitivity pneumonitis is an interstitial lung disease that results from repeated pulmonary exposure to various organic antigens, including Saccharopolyspora rectivirgula (SR, the causative agent of farmer's lung disease). Although the contributions of pro-inflammatory mediators to the disease pathogenesis are relatively well documented, the mechanism(s) involved in initiation of pro-inflammatory responses against the causative microorganisms, and the contribution of signaling molecules involved in host immune defense have not been fully elucidated. In the present study, we found that SR induces activation of protein kinase D1 (PKD1) in lung cells in vitro and in vivo. Activation of PKD1 by SR was dependent on MyD88. Inhibition of PKD by pharmacological PKD inhibitor Gö6976, and silencing of PKD1 expression by siRNA, revealed that PKD1 is indispensable for SR-mediated activation of MAPKs and NF-κB and expression of various pro-inflammatory cytokines and chemokines. In addition, compared to controls, mice pretreated with Gö6976 showed significantly suppressed alveolitis and neutrophil influx in bronchial alveolar lavage fluid and interstitial lung tissue, and substantially decreased myeloperoxidase activity in the lung after pulmonary exposure to SR. These results demonstrate that PKD1 is essential for SR-mediated pro-inflammatory immune responses and neutrophil influx in the lung. Our findings also imply the possibility that PKD1 might be one of the critical factors that play a regulatory role in development of hypersensitivity pneumonitis caused by microbial antigens, and that inhibition of PKD1 activation could be an effective way to control microbial antigen-induced hypersensitivity pneumonitis.
Keywords: Protein kinase D1, MyD88, Cytokines/Chemokines, Inflammation, Saccharopolyspora rectivirgula, hypersensitivity pneumonitis
Introduction
Hypersensitivity pneumonitis, also known as extrinsic allergic alveolitis or organic dust pneumoconioses, is a granulomatous, immune-mediated inflammatory lung disease caused by intense or repeated inhalation of antigenic organic particles in a variety of environmental settings (1). The most common inciting antigens are thermophilic actinomycetes, such as Saccharopolyspora rectivirgula (SR), that cause farmer's lung disease. The pathogenesis of hypersensitivity pneumonitis is complex and the mechanisms involved remain incompletely understood. Following antigen sensitization, pro-inflammatory mediators lead to accumulation of inflammatory cells in the lung and complex mechanisms involving interactions between cells-antigens, cells-cells and cells-inflammatory mediators occur (2). Following inhalation, hypersensitivity pneumonitis-causing antigens bind to pattern recognition receptors (PRRs) on/in innate immune cells and trigger PRR-signaling pathways, and/or soluble antigens form immune complexes with circulating IgG that trigger the complement activation cascade or the antibody-specific Fc receptor-signaling pathway (1, 3-5). These pathways lead to expression of numerous inflammatory and chemotactic factors, such as IL-1β, IL-6, IL-8, IL-12, TNFα, IFNγ, RANTES, CCL18, KC, MCP-1 and MIP-2. These inflammatory and chemotactic factors contribute to the recruitment of inflammatory cells, such as neutrophils, macrophages and lymphocytes, into the lungs and to the generation of the inflammatory environment (1, 6-12). IL-12 promotes the differentiation of CD4+ Th0 lymphocytes into Th1 lymphocytes (10, 13). Recruited neutrophils and Th1 cells produce IFNγ (14, 15). IFNγ further stimulates alveolar macrophages to produce greater amounts of IL-1β and TNFα (16). Thus, continued antigen exposures result in a positive feedback loop, leading to increased production of IL-1β and TNFα by alveolar macrophages and increased production of IFNγ by activated Th1 cells (10, 17-19). Animal studies have shown that hypersensitivity pneumonitis is associated with the overproduction of IFNγ, and that IL-10 counteracts the pro-inflammatory effects of IFNγ and modulates the severity of hypersensitivity pneumonitis (6, 10, 20). Taken together, these findings suggest that the early inflammatory response to hypersensitivity pneumonitis-causing antigens results in production of chemokines and pro-inflammatory cytokines that likely play a role in the induction and progression of the disease. Although the role of inflammatory mediators and the contributions of immune cells in the development of hypersensitivity pneumonitis are intensively studied and relatively well documented, the mechanisms involved in the initiation of the host pro-inflammatory responses to the hypersensitivity pneumonitis-inciting agents, such as SR, are not completely understood. Identification of host innate immune receptors that are involved in initial detection of the inciting antigens of hypersensitivity pneumonitis, and careful investigation of signal transduction mediated by those innate immune receptors will provide new and important insight into the disease pathogenesis.
Cells in the innate immune system express a family of evolutionarily conserved PRRs, such as Toll-like receptors (TLR), NACHT-leucine-rich repeat-family proteins, helicase domain-containing antiviral proteins, and cytosolic DNA sensors (21-26). Recognition of the evolutionally conserved structures in microorganisms (pathogen-associated molecular pattern; PAMP), including lipopolysaccharide (LPS), peptidoglycan (PGN) and microbial nucleic acids, by PRRs on/in innate immune cells initiates the complex signaling cascades leading to secretion of pro-inflammatory cytokines and mediators. The most extensively studied PRRs are TLRs. TLRs are Type I transmembrane proteins containing N-terminal leucine-rich repeats that are responsible for binding to PAMPs, a transmembrane domain, and a C-terminal Toll/interleukin-1 receptor (TIR) domain that is responsible for signaling. There have been at least 13 TLRs identified to date in mice and 10 in humans (27). Each TLR forms either homodimers or heterodimers and has different binding specificity to PAMPs (28). After binding to a specific PAMP, TLRs recruit one or more TIR domain-containing adaptor molecules that orchestrate a complexity of down-stream signaling cascades and biologic outcomes. Among the five identified TIR domain-containing adaptor molecules, myeloid differentiation factor 88 (MyD88) is utilized by all TLRs except TLR3 (29, 30). In MyD88-dependent TLR signaling, recruitment of MyD88 to a TLR through interaction between TIR domains leads to the recruitment of IL-1 receptor-associated kinase (IRAK) family members IRAK4 and IRAK1 to the TLR/MyD88 signaling complex through interaction between the death domains of MyD88 and of IRAKs (22, 31, 32). IRAK1 becomes rapidly phosphorylated by IRAK4, resulting in recruitment of TNFα receptor-associated factor 6 (TRAF6) to the receptor complex (31, 33-35). Phosphorylated IRAK1 and TRAF6 are thought to dissociate from the receptor, which is followed by TRAF6 autoubiquitination with K63-linked polyubiquitin chains and subsequent polyubiquitination of IRAK1 by TRAF6 (36-39). Ubiquitinated TRAF6 binds to and activates a signaling complex composed of TGFβ-activated kinase 1 (TAK1) and TAK1-binding protein 2 (TAB2), which initiate signaling cascades that lead to activation of NF-κB and mitogen-activated protein kinases (MAPKs), and subsequent expression of pro-inflammatory cytokines and chemokines (32, 39-44). In addition to this well-known MyD88-dependent pathway, we recently found that a serine/threonine kinase protein kinase D1 (PKD1) is activated by all TLRs that transduce their signal through MyD88 (45, 46). PKD1 is recruited to and activated in the TLR/MyD88 receptor complex via interaction with IRAK4, IRAK1 and TRAF6. PKD1 is required for ubiquitination of TRAF6 and subsequent activation of TAK1. Activation of PKD1 by TLRs is essential for MyD88-dependent expression of pro-inflammatory cytokines and mediators in innate immune cells. Using an animal model of hypersensitivity pneumonitis, a recent study has demonstrated that initial expression of pro-inflammatory cytokines and chemokines in the lung and subsequent neutrophil infiltration into the lung after pulmonary exposure to SR are largely dependent on MyD88 (4). These findings suggest a possibility that SR may activate PKD1, and PKD1 may play a regulatory role in SR-induced pro-inflammatory responses. In the present study we investigated whether SR, a microbial antigen that causes farmer's lung disease, induces activation of PKD1 in various innate immune cells in the lung in vitro and in vivo in an MyD88-dependent manner, and whether PKD1 plays a biologic role in SR-mediated pro-inflammatory responses in vitro and in vivo.
Materials and Methods
Mice
C57BL/6 mice at 4-5 weeks of age were obtained from The Frederick Cancer Research and Development Center, National Cancer Institute (Frederick, MD) and were used within 3 weeks. MyD88 gene-deficient (MyD88-/-) mice were provided by Dr. S. Akira (Osaka University, Osaka, Japan). All animal care and housing requirements set forth by the National Institutes of Health Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources were followed, and animal protocols were reviewed and approved by the University of Tennessee Animal Care and Use Committee.
Cells and culture conditions
Bone marrow-derived macrophages (BMDM) were isolated as described (45, 46). Murine cell lines RAW264.7 (macrophages), AMJ2-C11 (alveolar macrophages), MPRO (premyelocytes), and MLE12 (bronchial epithelial cells) were purchased from American Type Culture Collection (ATCC, Manassas, VA). FLAG-tagged PKD-expressing RAW264.7 cells were generated as described (46). BMDM, RAW264.7 and FLAG-tagged PKD-expressing RAW264.7 cells were cultured in D-MEM supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS). AMJ2-C11 cells were cultured in D-MEM supplemented with 5% (v/v) heat-inactivated FCS. MLE cells were cultured in DMEM/F-12 supplemented with 2% (v/v) heat-inactivated FCS. MPRO cells were cultured in Iscoves modified Dulbecco medium supplemented with 5% (v/v) heat-inactivated FCS and 10 ng/ml of GM-CSF. Morphologic differentiation of MPRO promyelocytes to neutrophils (MPRO-Neutrophils) were induced by supplementing 10 μM of retinoic acid (R2625, Sigma, St. Louis, MO) for 3 days. To confirm morphologic changes, cells were stained with Diff-Quik (IMEB Inc., San Marcos, CA) according to the manufacturer's protocol. All cells were incubated in the indicated FCS-containing medium supplemented with 1.5 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified incubator with 5% CO2. All culture reagents were purchased from Invitrogen (Carlsbad, CA) and Sigma (St. Louis, MO).
SR-antigen, toll-like receptor (TLR) ligands, and reagents
Saccharopolyspora rectivirgula (SR; strain designation A1313; ATCC number 29034) was purchased from ATCC. SR was grown in trypticase soy broth at 55°C in a shaking incubator for 2 days, centrifuged, and washed three times with endotoxin-free water. The bacterial cells were lysed by sonication and then lypophilized. The lyophilized bacterial pellet (SR-antigen) was reconstituted in endotoxin-free saline at a concentration of 10 mg/ml and kept at -80°C until used. Reconstituted SR-antigen (SR-Ag) had no detectable endotoxin by Limulus assay (Sigma, St. Louis, MO). Nuclease-resistant phosphorothioate oligodeoxynucleotides 1826 (CpG DNA) were purchased from Coley Pharmaceutical Group (Kanata, Ontario, Canada). PGN and poly(I:C) (PIC) were purchased from Invivogen (San Diego, CA). PMA, Gö6976 and Gö6983 were purchased from Calbiochem (La Jolla, CA).
Saccharopolyspora rectivirgula-antigen (SR-Ag) exposure protocol, bronchoalveolar lavage (BAL), and lung cell isolation
Female mice (C57BL/6, MyD88-/-; 3 to 5 mice/group), were exposed intranasally to SR-Ag (200 μg) or endotoxin-free saline. In some experiments, C57BL/6 mice were administered DMSO (vehicle), Gö6976 (2.3 mg/kg body weight), or Gö6983 (2.3 mg/kg body weight) by intraperitoneal injection and intranasal inhalation at 4 h and 1 h prior to SR-Ag exposure, and then exposed intranasally to either endotoxin-free saline or SR-Ag (200 μg) plus DMSO, Gö6976 (1.15 mg/kg body weight), or Gö6983 (1.15 mg/kg body weight). Mice were sacrificed at designated time points (3, 6, or 24 h). Unless indicated, control and SR-Ag-exposed mice were analyzed individually. BAL was performed by intratracheal injection of 1 ml PBS into the lungs with immediate vacuum aspiration. The typical amount of fluid recovered was approximately 70 % of the input. The recovered BAL fluid (BALF) was centrifuged. The supernatants were kept at -80°C until used for detection of cytokines and chemokines. The cells recovered from BALF were used to determine the degree of alveolitis. To obtain interstitial lung tissue cells, lungs were perfused with PBS to remove blood and lung lobes were removed from the mouse. Lung tissues were digested with collagenase (20 U/ml) and DNase I (40 μg/ml) for 45 min at 37°C. Cells were freed by mechanical disruption using a Stomacher tissue processor (Seward, UK). Mononuclear cells were isolated at the 40/80% interface following discontinuous Fico/Lite-M (Atlanta Biologicals, Lawrenceville, GA) gradient centrifugation and used for flow cytometric analysis and total cell counts.
Small interfering RNA (siRNA) transfection
AMJ2-C11 cells or MLE12 cells were plated at 2 × 106 cells/10 ml in a 100-mm Petri dish and incubated overnight and then transfected with 100 nM of non-target siRNA (Dharmacon, Lafayette, CO) or PKD1-specific siRNAs (5′-CTCCTGATGTCTAAGGTGA-3′ and 5′-CCATTGATCTTATCAATAA-3′) using lipofectamine (Invitrogen) according to the manufacturer's protocol.
In vitro kinase assays
Each FLAG-tagged PKD protein in whole cell lysates was immunoprecipitated with anti-FLAG Ab. The resulting immune complexes were subjected to in vitro kinase assay using Syntide-2 (Sigma) as a PKD substrate, as previously described (47).
RT-PCR, enzyme-linked immunosorbent assay (ELISA), and Western blot assay
Levels of the selected cytokine and chemokine genes in cells or lung tissue, concentrations of the selected cytokines and chemokines in culture supernatants or BALF, and levels or phosphorylation status of specific proteins in whole cell extracts were analyzed by RT-PCR, ELISA, and Western blot assay, respectively, as described previously (46, 48). Actin or GAPDH was used as a loading control for all RT-PCR. Actin was used as a loading control for all Western blot assays. All primers for RT-PCR were purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and sequences of primers are listed in Table 1 or described previously (45, 46, 48). All recombinant murine cytokines and MIP-2 and antibodies (Abs) specific for murine cytokines or MIP-2 were purchased from BD Biosciences (San Diego, CA), R&D Systems (Minneapolis, MN), or eBioscience (San Diego, CA). Ab specific for TLR9 was purchased from IMGENEX (San Diego, CA). Abs specific for TLR4, MyD88, TRAF6, PKD, IκBα, IκBβ or actin were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All phospho-specific Abs were purchased from Cell Signaling (Beverly, MA).
Table 1. Sequences of primers.
| Gene | Forward | Reverse |
|---|---|---|
| IL-23p19 | AGTCCTGACTGGGCTCAGTG | CAAAGACCCGGGCAGCTATG |
| IL-1β | TCAGGCAGGCAGTATCACTC | TGCTTGGGATCCACACTCTC |
| IFNγ | CATCAACTATAAGCAGCTCCA | TTCAAGTGGAGAGCAGTTGAG |
| MIP-1α | CACCACTGCCCTTGCTGTTC | TCGCTGCCTCCAAGACTCTC |
| MIP-1β | CATGAAGCTCTGCGTGTCTG | CTCTCCTGAAGTGGCTCCTC |
| MIP-2 | CAGCCACACTTCAGCCTAGC | TCCTTTCCAGGTCAGTTAGC |
| Eotaxin | GCTCCACAGCGCTTCTATTC | GGCTCTGGGTTAGTGTCAAG |
| LIX | GCGGTTCCATCTCGCCATTC | GAACACTGGCCGTTCTTTCC |
| KC | CGCCTATCGCCAATGAGCTG | TGTCAGAAGCCAGCGTTCAC |
| ITAC | CGGCTGCTGAGATGAACAGG | GGGCTCACAGTCAGACGTTC |
| Mig | CTCAGCTCTGCCATGAAGTC | TTGAACGACGACGACTTTGG |
| TLR2 | TCATTAGAATTCTTAGACC | CACCCGGATCCCTGTACTGGAC |
| TLR4 | GGGTCACAGAATTCTCAGCG | CAAGAGTGCTGAGGGAATAC |
Determination of alveolitis
The degree of the alveolitis was determined by counting the number of interstitial lung tissue cells or live cells recovered in BALF using trypan blue exclusion. Cellular composition of the alveolitis was determined by differential staining of the BAL cells with Diff-Quik according to the manufacturer's protocol. Differential cell counts were made on a total of 300 cells/sample using standard morphological criteria to determine the percent of lymphocytes, eosinophils, neutrophils, and macrophages per total cell count.
Multiplex sandwich immunoassay
Unconcentrated BALF samples were incubated in triplicate with beads coupled with the specific Abs to the selected chemokines (KC and MCP-1). The beads were then incubated with biotinylated Abs followed by streptavidin-phycoerythrin. The fluorescence was measured with a BioPlex array reader (Bio-Rad Laboratories, Hercules, CA). Chemokine standards (Bio-plex Pro™ mouse cytokine standard group I 23-plex, Bio-Rad Laboratories) ranging from 5 pg/ml to 20,000 pg/ml were prepared to determine the concentration of chemokines in the samples. For data analysis, a curve fit was applied to the standards and the sample concentrations extrapolated from the standard curve using the Logistic 5PL model in the BioPlex manager 3.0 software (Bio-Rad Laboratories). Multiplex kits for chemokines were purchased from Bio-Rad Laboratories.
Measurement of myeloperoxidase (MPO) activity
One lung lobe from each mouse was homogenized at a concentration of 50 mg/ml in hexadecyl trimethylammonium bromide buffer. The resulting lung homogenates were centrifuged at 16,000 × g for 20 min. After centrifugation, the supernatants were mixed with 50 nmol phosphate buffer containing O-dianisidine hydrochloride and H2O2. The samples were read spectrophotometically at 460 nm for 1 – 20 min.
Lung histology
The left lung lobes removed from the mice were fixed in 10% neutral buffered formalin, dehydrated, and embedded in paraffin. The paraffin-embedded lungs were sectioned longitudinally at 5 μm and then stained with hematoxylin and eosin (H&E). To determine inflammatory cell influx into the interstitial lung tissue, digital images (40× objective) of whole H&E stained slides were captured using the Aperio ScanScope®XT Slide Scanner (Aperio Technologies, Inc. Vista CA) system.
Flow cytometric analysis
Isolated lung mononuclear cells were stained with fluorochrome-conjugated antibodies to identify neutrophils (CD45+/Ly6G+/7/4+) and macrophages (CD45+/Ly6G -/F4/80+). To exclude dead cells from the analysis, cells were also stained with the DNA binding dye DAPI. A minimum of 30,000 live events/sample were collected on a BD Biosciences LSR II flow cytometer and analyzed using FACS DIVA software (BD Biosciences). A gate was set to exclude dead cells (DAPI+) and an additional gate was set to include only CD45+ hematopoietic cells. The gated cells were analyzed to determine the percentage of each cell population per total CD45+ cells from lungs of individual mice in each group.
Statistical analysis
All in vitro experiments were repeated 3 times before analysis. Data were expressed as mean ± SD. The differences between the control and experimental groups were evaluated using Student's t-test. Statistical differences with p < 0.05 and p < 0.005 are indicated as * and **, respectively, and considered significant.
Results
Saccharopolyspora rectivirgula antigen (SR-Ag) induces activation of PKD1 in innate immune cells in the lung in vitro and in vivo
Recently, we found that all TLR ligands (with the exception of the TLR3 ligand PIC) and pro-inflammatory cytokines IL-1β and IL-18 induce activation of PKD1, and that PKD1 is essential for MyD88-dependent pro-inflammatory gene expression in innate immune cells (45, 46). However, it is currently not known whether PKD1 is activated in innate immune cells in response to any whole microorganism or complex components of microorganisms, or what role it may play in the pro-inflammatory response to these stimuli. Therefore, we investigated whether S. rectivirgula, an inciting antigen for farmer's lung disease, induces activation of PKD1 in innate immune cells. RAW264.7 cells stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with SR-Ag. As demonstrated in Figure 1A, SR-Ag induced kinase activity and phosphorylation of PKD1. However, SR-Ag did not induce kinase activity and phosphorylation of either PKD2 or PKD3. Of note, control PMA induced both kinase activity and phosphorylation of all three family members of PKD. We further investigated whether SR-Ag induces activation of PKD1 and/or other signaling modulators, such as MAPKs and NF-κB, in innate immune cells involved in lung inflammation in vitro and in vivo. First, we stimulated AMJ2-C11 (a murine alveolar macrophage cell line), MPRO-Neut (neutrophils derived from murine premyelocytes, MPRO), and MLE12 (a murine bronchial epithelial cell line) cells with various concentrations of SR-Ag. As shown in Figure 1B, SR-Ag induced activation of PKD1, as judged by phosphorylation at serine 744/748 and 916 residues, in all three types of cells. In addition, SR-Ag induced activation of MAPKs (JNK, ERK, and p38), as judged by phosphorylation, and transcription factor NF-κB, as evidenced by degradation of IκBα and IκBβ, in those cells. Second, to further investigate whether SR-Ag induces activation of PKD1 and/or those signaling modulators in the lung in vivo, C57BL/6 mice were exposed intranasally to SR-Ag and then whole lung lysates were prepared. As demonstrated in Figure 1C, SR-Ag inhalation resulted in activation of PKD1, JNK, ERK and p38, and phosphorylation of IκBα (indication of NF-κB activation) in the lung of C57BL/6 mice. Taken together, these results demonstrated that SR-Ag induces activation of PKD1 and other signaling modulators (MAPKs and NF-κB) critical for expression of pro-inflammatory genes in cells involved in inflammation in the lung in vitro and in vivo.
Figure 1. SR-Ag induces activation of PKD1 in innate immune cells in the lung in vitro and in vivo.

(A) RAW264.7 cells (murine macrophages) stably expressing empty vector, FLAG-tagged PKD1, FLAG-tagged PKD2, or FLAG-tagged PKD3 were stimulated with medium, SR-Ag (2 μg/ml), or PMA (10 ng/ml) for 45 min. Whole cell lysates were prepared and each PKD family protein was immunoprecipitated with anti-FLAG Ab. Kinase activity of PKDs was analyzed in vitro using syntide-2 as a PKD substrate (Top). Expression and phosphorylation status of PKDs were analyzed by immunoblotting with anti-FLAG or anti-phospho-PKD Abs (pPKDs744/748, pPKDs916) (Bottom). (B) AMJ2-C11 (alveolar macrophages), MPRO-Neut (neutrophils developed from MPRO), and MLE12 (bronchial epithelial cells) cells were stimulated with medium or the indicated concentration of SR-Ag. Phosphorylation status of PKD1, JNK, p38 and ERK and degradation of IκBα and IκBβ were analyzed by Western blot (Top). MPRO cells and MPRO-Neut cells were stained with Diff-quick (Bottom). MPRO-Neut cells showed standard morphologic features of neutrophils. (C) C57BL/6 mice were intranasally exposed to saline or SR-Ag (200 μg) for 3 h. Lung lysates were prepared and activation status of PKD1, JNK, p38 and ERK and phosphorylation of IκBα in lung lysates was detected by Western blot. Actin was used as a loading control. Each lane represents an individual mouse. All experiments were done two to four times with similar results.
SR-Ag induces activation of PKD1, MAPKs, and NF-κB through a pathway involving MyD88
As with other microorganisms, various components of S. rectivirgula might interact with various PRRs on/in the host innate immune cells. A previous study demonstrated that MyD88 plays a substantial role in SR-Ag-induced production of pro-inflammatory cytokine TNFα and chemokine MIP-2, and recruitment of neutrophils into the lungs (4). Therefore we investigated whether SR-Ag-mediated PKD1 activation is dependent on MyD88. BMDMs isolated from wild-type control or MyD88-/- mice were stimulated with SR-Ag, and activation of PKD1 and MAPKs and degradation of IκBα (as an indication of NF-κB activation) were assayed. As shown in Figure 2A, SR-Ag, as well as CpG DNA, induced phosphorylation of PKD1, JNK, ERK and p38, and degradation of IκBα in BMDMs isolated from wild-type mice. SR-Ag failed to induce activation of PKD1, JNK, ERK and p38, and degradation of IκBα in MyD88-/- BMDMs, indicating that SR-Ag induces activation of these signaling modulators via an MyD88-dependent pathway. Of note, PMA-induced activation of PKD and other signaling modulators was unaffected in MyD88-/- macrophages. We further investigated whether SR-Ag induces activation of PKD1 and/or those signaling modulators in a manner dependent on MyD88 in the lung in vivo. Wild-type control or MyD88-/- mice were intranasally exposed to saline or SR-Ag. Lung lysates were prepared and activation of PKD1 and MAPKs was detected. SR-Ag consistently failed to induce activation of PKD1, MAPKs (JNK, ERK and p38) and NF-κB (judged by phosphorylation of IκBα) in the lungs of MyD88-/- mice (Fig. 2B). Taken together, these results indicate that SR-Ag leads to the activation of PKD1, JNK, ERK, p38 and NF-κB by utilizing MyD88, the common signaling adaptor molecule for the TLR/IL-1R superfamily.
Figure 2. SR-Ag induces activation of PKD1 and MAPKs through a TLR2, TLR9, and MyD88-dependent manner.
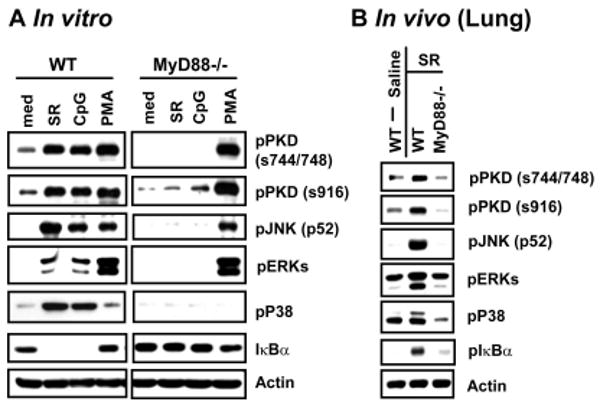
(A) BMDMs from C57BL/6 (WT) or MyD88-gene deficient (MyD88-/-) mice were stimulated with medium, SR-Ag (5 μg/ml), CpG DNA (12 μg/ml), or PMA (10 ng/ml) for 45 min. (B) C57BL/6 (WT) or MyD88-/- mice were intranasally exposed to saline or SR-Ag (200 μg) for 3 h. Phosphorylation status of PKD1, and MAPKs (JNK, ERK, p38) in lung tissue lysates was detected by Western blot analysis. Actin was used as a loading control. Experiments were done two to three times with similar results.
Effect of pharmacological PKD inhibitor Gö6976 on SR-Ag-mediated activation of MAPKs and NF-κB and expression of cytokines and chemokines in vitro
Although the pharmacological inhibitor that specifically inhibits only PKD1 has yet to be developed, careful use of several pharmacological inhibitors can provide insight to the biologic role of PKD1. Pharmacological PKD/PKC inhibitor Gö6976 selectively inhibits PKCα (IC50 = 2.3 nM), βI (IC50 = 6.2 nM), and PKD (IC50 = 20 nM) but does not inhibit PKCδ, ε, and ζ. Pharmacological PKC inhibitor Gö6983 selectively inhibits PKCα (IC50 = 7 nM), β (IC50 = 7 nM), γ (IC50 = 6 nM), δ (IC50 = 10 nM), and ζ (IC50 = 60 nM) but does not inhibit PKD, and is used together with Gö6976 to differentiate PKD from other PKC isozymes (49, 50). Recently, we found that Gö6976 selectively inhibits TLR ligand-mediated PKD1 activation in vitro and in vivo, while Gö6983 has no effect (45, 46). To investigate the biologic role of SR-Ag-activated PKD1, cells (AMJ2-C11, MPRO-Neut, and MLE12) were pretreated with vehicle (DMSO), PKD inhibitor Gö6976 or PKC inhibitor Gö6983, and then stimulated with SR-Ag. Activation of PKD1 by SR-Ag in these cells was inhibited by Gö6976, but not by Gö6983 (Fig. 3A). In contrast, activation of PKDs by PMA, which occurs in a PKC-dependent manner, was suppressed similarly by either Gö6976 or Gö6983 in all three types of cells. In addition, phosphorylation of pan PKCs (an indication of activation of conventional PKC isozymes) by PMA in AMJ2-C11 cells and MPRO-Neut cells was ablated by either Gö6976 or Gö6983. These results demonstrate that both Gö6976 and Gö6983 effectively inhibit conventional PKC isozymes, but only Gö6976 effectively and selectively inhibits SR-Ag-mediated PKD1 activation, which presumably occurs through the conventional PKC-independent pathway. We further investigated whether this inhibition of PKD1 activation affects SR-Ag-mediated activation of MAPKs and NF-κB. As shown in Figure 3A, SR-Ag failed to induce activation of JNK, ERK and p38, or degradation of IκBα and IκBβ in all three types of cells pretreated with Gö6976. In contrast, SR-Ag-mediated activation of JNK, ERK and p38, and degradation of IκBα and IκBβ in these cells was not affected by the presence of Gö6983. As expected, Gö6976 and Gö6983 inhibited PMA-induced activation of JNK and ERK in all three types of cells to a similar degree. Neither Gö6976 nor Gö6983 inhibited activation of JNK, ERK, p38, or STAT1, or degradation of IκBα and IκBβ induced in response to PIC, which does not activate PKD1, in AMJ2-C11 and MPRO-Neut cells, indicating that neither Gö6976 nor Gö6983 was toxic to cells at the concentration used in this experiment. Of note, MLE12 cells did not respond to PIC. Although we cannot completely rule out the possible involvement of other PKC isozymes on SR-Ag-mediated activation of MAPKs and NF-κB in these types of cells, our results suggest that Ca++-dependent conventional PKCs and PKCδ and ζ (which can be inhibited by Gö6983) may be not critical for SR-Ag-mediated activation of MAPKs and NF-κB. As mentioned above, PKD family members are kinases whose activation is differently affected by Gö6976 and Gö6983 (45, 46). Although Gö6976 inhibits activation of all three PKD family members, PKD1 may be the kinase whose activity was inhibited by Gö6976 in those cells stimulated with SR-Ag, because SR-Ag activates only PKD1, not PKD2 or PKD3. Therefore, these results imply that PKD1 plays an indispensable role in SR-Ag-mediated activation of MAPKs and NF-κB. Since MAPKs and NF-κB play a pivotal regulatory role in expression of various cytokines and chemokines that are critical for SR-Ag-mediated leukocyte recruitment and proinflammatory responses, we investigated whether PKD1 plays a role in SR-Ag-induced expression of selected pro-inflammatory cytokines and chemokines in AMJ12-C11, MPRO-Neut, and MLE12 cells. Figure 3B shows that SR-Ag, as well as TLR2 ligand PGN or TLR9 ligand CpG DNA, induced expression of cytokines TNFα, IL-1β, IL-6, IL-10, IL-12p40, and IL-23p19, and chemokines MIP-1α, MIP-1β, MCP-1, MIP-2, KC, IP-10, RANTES, ITAC and LIX in AMJ2-C11 cells and MPRO-Neut cells. In MLE12 cells, SR-Ag or PGN induced expression of cytokines IFNγ, IL-6, and IL-23p19 and chemokines MIP-1α, MCP-1, MIP-2, KC, IP-10, RANTES, ITAC, Eotaxin, LIX, and Mig. MLE12 cells did not respond to CpG DNA. Expression of these cytokines and chemokines in response to SR-Ag, as well as PGN or CpG DNA, was ablated in the presence of PKD inhibitor Gö6976 in all three types of cells. In contrast, PKC inhibitor Gö6983 did not affect expression of these cytokines and chemokines in response to SR-Ag, PGN or CpG DNA, in any of the cells. Similar to mRNA expression, production of selected cytokine (TNFα, IL-6, IL-10, IL-12, IFNγ) and chemokine (MIP-2) proteins in response to SR-Ag, as well as to PGN or CpG DNA, was almost completely, if not completely, inhibited by Gö6976, but not by Gö6983, in all three types of cells (Fig. 3C). These results suggest that PKD family members (presumably PKD1) play a critical role in SR-Ag-induced expression of cytokines and chemokines. Of note, control stimulus IFNγ, which does not activate PKD1 but is critical for the pathogenesis of hypersensitivity pneumonitis, induced expression of cytokines TNFα in AMJ12-C11 cells and MPRO-Neut cells and IFNγ in MLE12 cells (Fig. 3B). IFNγ also induced expression of chemokines IP-10, RANTES, ITAC, and Mig in AMJ2-C11 cells and IP-10, RANTES, ITAC, Eotaxin, and Mig in MPRO-Neut cells and MLE12 cells. Neither Gö6976 nor Gö6983 affected IFNγ-mediated expression of cytokines and chemokines in all three types of cells. Gö6976 or Gö6983 also failed to inhibit IFNγ-mediated production of TNFα protein in AMJ2-C11 cells and MPRO-Neut cells (Fig. 3C). These results indicate that neither Gö6976 nor Gö6983, at the concentration used for these experiments, is toxic to or affects the basic biology of cells. Collectively, these results suggest that PKD1 plays an indispensable role in SR-Ag-mediated activation of MAPKs and NF-κB, and subsequent expression and production of cytokines/chemokines.
Figure 3. Effect of pharmacological PKC/PKD inhibitor Gö6976 on SR-Ag-mediated activation of MAPKs and NF-κB and expression of cytokines and chemokines in vitro.

AMJ2-C11, MPRO-Neut, and MLE12 cells were pretreated with DMSO, Gö6976 (250 ng/ml), or Gö6983 (250 ng/ml) for 1 h and then stimulated with medium, SR-Ag (1 μg/ml for AMJ2-C11, 5 μg/ml for MPRO-Neut, 10 μg/ml for MLE12), PMA (10 ng/ml), PIC (100 μg/ml), CpG DNA (12 μg/ml for AMJ2-C11, 24 μg/ml for MPRO-Neut and MLE12), PGN (1 μg/ml for AMJ2-C11, 2 μg/ml for MPRO-Neut, 4 μg/ml for MLE12) or IFNγ (10 ng/ml) for 45 min (Panel A), 2 h (for IFNγ in Panel B), 4 h (Panel B), or 24 h (Panel C). Phosphorylation of pan-PKCs, PKD1, JNK, p38, ERK, and STAT1 and the presence of IκBα and IκBβ in cell lysates were detected by Western blot analysis (Panel A). Messenger RNA levels of the indicated cytokines and chemokines in cells were analyzed by RT-PCR (Panel B). Levels of the cytokines and chemokines in culture supernatants were analyzed by ELISA (Panel C). Data represent the mean concentration (pg/ml) ± SD of triplicates. Actin was used as a loading control. Experiments were done three times with similar results.
PKD1 is required for SR-Ag-induced activation of MAPKs and NF-κB, and expression of inflammatory cytokines and chemokines in vitro
To further confirm the findings with pharmacological PKD inhibitor Gö6976 that PKD1 is required for SR-Ag-mediated innate immune cell activation, we silenced PKD1 expression in AMJ2-C11 and MLE12 cells by transiently transfecting these cells with non-target siRNA (NT siRNA; control cells) or PKD1-specific siRNA (PKD1-siRNA; PKD1-knockdown cells). Expression of PKD1 mRNA and protein was almost completely inhibited in PKD1-knockdown cells (Fig. 4A and 4B). In contrast, mRNA and protein levels of other genes tested in PKD1-knockdown cells were comparable to those in control cells. In addition, SR-Ag failed to induce phosphorylation of PKDs in PKD1-knockdown cells (Fig. 4C), further supporting our finding that SR-Ag activates PKD1, but not PKD2 or PKD3. These results demonstrate that PKD1-siRNA specifically and effectively silenced PKD1 expression in these cells. Using these PKD1-knockdown cells, we further investigated whether PKD1 plays a role in SR-Ag-mediated activation of signaling modulators and subsequent cytokine/chemokine expression and production. Compared to control cells, activation of MAPKs (JNK, ERK and p38) and transcription factor NF-κB (judged by degradation of IκBα and IκBβ) in response to SR-Ag were ablated in PKD1-knockdown alveolar macrophages and bronchial epithelial cells (Fig. 4C). In addition, mRNA expression of selected cytokines (TNFα, IFNγ, IL-1β, IL-6, IL-10, IL-12p40, IL-23p19) and chemokines (MIP-1α, MIP-1β, MIP-2, MCP-1, MCP-2, KC, IP-10, RANTES, ITAC, LIX, Mig) in response to SR-Ag were ablated in these PKD1-knockdown cells (Fig. 4D). Furthermore, cytokine (TNFα, IFNγ, IL-6, IL-10, IL-12) and chemokine (MIP-2, MCP-1, KC) protein production in response to SR-Ag was almost completely, if not completely, inhibited in these PKD1-knockdown cells (Fig. 4E). Of note, activation of STAT1, gene expression, and protein production in response to IFNγ were not altered in these PKD1-knockdown cells, indicating that the absence of PKD1 specifically affects the SR-Ag-mediated signaling pathway but not the general biology of the cell. These results demonstrate that PKD1 plays an indispensable role in SR-Ag-mediated activation of MAPKs and NF-κB and subsequent expression and production of inflammatory cytokines/chemokines.
Figure 4. PKD1 is essential for SR-induced activation of MAPKs and NF-κB and subsequent expression and production of cytokines and chemokines in vitro.

AMJ2-C11 and MLE12 cells were transiently transfected with 100 nM of non-target siRNA (NT siRNA; control) or PKD1-specific siRNA (PKD1-siRNA; PKD1-knockdown) using lipofectamine. (A) Messenger RNA levels of the indicated genes were analyzed by RT-PCR. (B) PKD1 protein levels of the indicated genes were examined using Western blot analysis. (C-E) Control or PKD1-knockdown cells were stimulated with medium, SR-Ag, or IFNγ for 45 min (Panel C), 2 h (for IFNγ in Panel D), 4 h (Panel D), or 24 h (Panel E). Activation status of PKD1, MAPKs, STAT1, and NF-κB (assessed by degradation of IκBα and IκBβ) was detected by Western blot (Panel C). Messenger mRNA levels of the indicated cytokines and chemokines were analyzed by RT-PCR (Panel D). Levels of cytokines and chemokines in culture supernatants were analyzed by ELISA (TNF-α, IFNγ, IL-6, IL-12, MIP-2) or multiplex sandwich assay (KC, MCP-1) (Panel E). Data represent the mean concentration (pg/ml) ± SD of triplicates. Actin and GAPDH were used as loading controls. All experiments were done three times with similar results.
Inhibition of PKD1 activation by Gö6976 in mice prior to intranasal exposure to SR-Ag results in decreased expression of pro-inflammatory cytokines/chemokines in lungs
Initial pro-inflammatory responses in the lung after pulmonary exposure to S. rectivirgula play a critical role in initiation of farmer's lung disease, a prototype hypersensitivity pneumonitis (1). Because inhibition of PKD1 activation by Gö6976 or inhibition of PKD1 expression by siRNA resulted in inhibition of SR-Ag-mediated activation of MAPKs and NF-κB, and the subsequent expression and production of various cytokines and chemokines in types of cells critical for lung inflammatory responses, we further investigated whether PKD1 plays a role in SR-Ag-induced pro-inflammatory responses in the lung in vivo. To inhibit activation of PKD1, we pretreated C57BL/6 mice with Gö6976. C57BL/6 mice pretreated with Gö6976 were exposed intranasally to SR-Ag and then examined for pro-inflammatory responses and development of alveolitis in the lungs. As shown in Figure 5A, systemic administration of Gö6976 effectively inhibited SR-Ag-induced activation of PKD1 in lung cells. In contrast, control Gö6983 did not inhibit SR-Ag-induced activation of PKD1 in lung cells. Similar to what we observed in vitro, SR-Ag failed to induce activation of MAPKs (JNK, ERK and p38) and phosphorylation of IκBα in lung cells isolated from C57BL/6 mice pretreated with Gö6976. Activation of MAPKs and phosphorylation of IκBα in lung cells after exposure to SR-Ag were not suppressed by Gö6983. These results indicate that systemic administration of Gö6976 effectively inhibits SR-Ag-mediated PKD1 activation in the lung and that PKD1 plays a key role in SR-Ag-induced activation of MAPKs and NF-κB in the lung. Pro-inflammatory responses in the lungs of C57BL/6 mice exposed to SR-Ag were assessed by analyzing mRNA levels of the selected cytokines and chemokines in lungs and protein levels in bronchial lavage fluids. As demonstrated in figure 5B, SR-Ag induced expression of cytokines TNFα, IFNγ, IL-1β, IL-6, IL-10, IL-12p40, and IL-23p19 and chemokines IP-10, MCP-1, RANTES, MIP-1α, MIP-1β, MIP-2, KC, ITAC, Eotaxin and LIX in the lung. Although there were slight variations among individuals, compared to vehicle (DMSO)-pretreated control mice, mice pretreated with Gö6976 showed substantially suppressed expression of all cytokines and chemokines analyzed in response to SR-Ag in the lung. In contrast, mRNA expression levels of these cytokines and chemokines in response to SR-Ag in mice pretreated with Gö6983 were comparable to those in vehicle-pretreated control mice. Cytokine (TNFα and IL-6) and chemokine (KC, MIP-2, MCP-1) levels in BALF were substantially increased in mice exposed to SR-Ag (Fig. 5C). However, they were significantly decreased in mice pretreated with PKD inhibitor Gö6976, but not in mice pretreated with PKC inhibitor Gö6983. These results indicate that PKD1 is essential for the SR-Ag-induced pro-inflammatory responses in the lung.
Figure 5. Effects of PKD inhibitor Gö6976 on SR-Ag-mediated pro-inflammatory responses in the lung.

C57BL/6 mice were administered DMSO, Gö6976 or Gö6983 (2.3 mg/kg body weight) by intraperitoneal injection and intranasal inhalation at 4 h and 1 h prior to SR-Ag exposure. The mice were exposed intranasally to either saline or SR-Ag (200 μg) plus DMSO, Gö6976 or Gö6983 (1.15 mg/kg body weight) for 3 h (Panel A), 6 h (Panels B and C), or 24 h (for MCP-1 in Panel C). (A) Lung lysates were prepared from one lung lobe and phosphorylation status or expression levels of the indicated proteins were detected by Western blot assay. Each lane represents an individual mouse. (B) Total RNA was purified from one lung lobe and mRNA levels of the indicated gene were analyzed by RT-PCR. Each lane represents an individual mouse. Actin was used as a loading control. (C) Levels of the indicated cytokines and chemokines in BALF were analyzed by ELISA (TNFα, IL-6, MIP-2) or multiplex sandwich assay (KC, MCP-1). Data represent the mean concentration (pg/ml) ± SD of 3 mice/group. Gö6976/SR-Ag or Gö6983/SR-Ag groups were compared to the control DMSO/SR-Ag group. Statistically significant differences are indicated (*p < 0.05). Experiments were done twice with similar results.
Inhibition of PKD1 activation by Gö6976 in mice prior to intranasal exposure to SR-Ag results in decreased alveolitis and neutrophil recruitment in the lung
Since our results showed that SR-Ag fails to induce expression and production of chemokines that attract leukocytes into the lungs in mice pretreated with PKD inhibitor Gö6976, we further investigated whether inhibition of PKD1 also prevents leukocyte infiltration into the lung in response to SR-Ag exposure. As expected, at 24 h post-exposure to SR-Ag, mice exhibited dramatic increases in total BAL cell numbers and total interstitial lung cell numbers (alveolitis) compared to control mice exposed to saline (Table 2 and Fig. 6A). Histological section of lungs from SR-Ag-exposed mice also showed the presence of extensive mononuclear cell infiltration in the lungs compared to saline-exposed mice (Fig. 6B). Neutrophils were the predominant cell type recovered from airways and lung tissues isolated from mice exposed to SR-Ag (Table 2 and Fig. 6C). In addition, SR-Ag-exposed mice showed significant increases in MPO activity in the lung, reflecting increased neutrophil influx (Fig. 6D). In contrast, PKD inhibitor Gö6976 pretreatment ablated SR-Ag-mediated induction of alveolitis and neutrophil influx in airways and interstitial lung tissues (Table 2 and Fig. 6A-6C). Also, the Gö6976-pretreated mice showed significantly decreased MPO activity in the lung after SR-Ag exposure compared to the vehicle-pretreated mice, reflecting significantly decreased neutrophil influx into the lung (Fig. 6D). Of note, PKC inhibitor Gö6983 pretreatment did not have any significant effect on SR-Ag-induced alveolitis and neutrophil influx in the airways and interstitial lung tissues. These results indicate that PKD1 is essential for SR-Ag-induced alveolitis, and that pharmacological inhibitors that suppress PKD1 activity can be useful therapeutic agents for SR-induced hypersensitivity pneumonitis.
Table 2. Comparison of cells in BAL fluid from mice at 24 h post-exposure.
C57BL/6 mice were administered DMSO (vehicle), Gö6976 (2.3 mg/kg body weight), or Gö6983 (2.3 mg/kg body weight) by intraperitoneal injection and intranasal inhalation at 4 h and 1 h prior to SR-Ag exposure, and then exposed intranasally to either endotoxin-free saline or SR-Ag (200 μg) plus DMSO, Gö6976 (1.15 mg/kg body weight), or Gö6983 (1.15 mg/kg body weight). Mice were euthanized at 24 h after SR-Ag exposure. Bronchial alveolar lavage (BAL) was performed and the cells recovered. The degree of the alveolitis was determined by counting the number of live cells recovered in BAL fluid using trypan blue exclusion. Cellular composition of the alveolitis was determined by differential staining of the BAL cells with Diff-Quik. Differential cell counts were made on a total of 300 cells/sample using standard morphological criteria to determine the percent of lymphocytes, eosinophils, neutrophils, and macrophages per total cell count.
| Pretreatment | Stimulation | Total cells (104)a | % Macrophagesb | % Neutrophils | % Lymphocytes |
|---|---|---|---|---|---|
| DMSO | Saline | 1.2 ± 1.17 | 89.0 ± 3.20 | 5.1 ± 1.50 | 6.0 ± 4.70 |
| SR-Ag | 73.7 ± 25.85 | 15.4 ± 7.60 | 79.5 ± 6.00 | 5.1 ± 1.70 | |
| Gö6976 | Saline | 2.5 ± 2.03 | 74.1 ± 3.15 | 9.1 ± 7.93 | 18.6 ± 8.60 |
| SR-Ag | 17.0 ± 15.24 * | 61.7 ± 3.30** | 28.9 ± 1.00** | 9.4 ± 4.40 | |
| Gö6983 | Saline | 2.6 ± 1.97 | 64.8 ± 8.27 | 20.6 ± 11.95 | 14.5 ± 4.67 |
| SR-Ag | 73.2 ± 3.88 | 22.5 ± 11.70 | 70.6 ± 10.90 | 7.0 ± 0.80 |
Mean number of cells (104) ± S.D. derived from five individual mice per group.
Mean percentage of cells.
p < 0.05
p < 0.005. Gö6976/SR-Ag or Gö6983/SR-Ag groups were compared to control DMSO/SR-Ag group.
Figure 6. PKD inhibitor Gö6976 pretreatment suppressed SR-Ag-induced alveolitis and neutrophil recruitment.

C57BL/6 mice (n = 5/group) were administered DMSO, Gö6976 or Gö6983 (2.3 mg/kg body weight) by intraperitoneal injection and intranasal inhalation at 4 h and 1 h prior to SR-Ag exposure. The mice were exposed intranasally to either saline or SR-Ag (200 μg) plus DMSO, Gö6976 or Gö6983 (1.15 mg/kg body weight) for 24 h. (A) Interstitial lung tissue cells were isolated from whole lung from each mouse. Cells were counted to determine the degree of alveolitis (mean total cells ± S.D. from 5 mice/group) using trypan blue exclusion. Gö6976/SR-Ag or Gö6983/SR-Ag groups were compared to the control DMSO/SR-Ag group. Statistically significant differences are indicated (**p < 0.005). (B) Representative H&E stained lung sections from mice exposed to the indicated stimuli. The Aperio ScanScope®XT Slide Scanner system was used to capture whole-slide digital images with a 40 × objective. The area inside the boxes in the second column is 20 × magnified and then presented in the third column. (C) Interstitial lung tissue cells were isolated from whole lung and flow cytometry was performed. Expression of Ly6G, 7/4, and F4/80 was analyzed using a live gate set on CD45+ cells. (D) MPO activity in lung homogenates was measured spectrophotometrically at 460 nm for 1 – 20 min. Data represent the mean absorbance ± S.D. from 5 mice/group. Statistically significant differences, as compared to the DMSO/SR-Ag-treated control, are indicated (**p < 0.005).
Discussion
Using pharmacological PKD inhibitor Gö6976, several recent studies suggested that PKD family proteins might be involved in certain cytokine production mediated through TLR4- and TLR5-signaling (33, 50, 51). Using more defined approaches, we demonstrated that various synthetic or purified ligands for TLR/IL-1R superfamily members that utilize the TIR domain-containing adaptor molecule MyD88 induce activation of PKD1 in human and murine macrophages (45, 46). PKD1 activation by TLR ligands is dependent on MyD88, IRAK4, and IRAK1, but independent of TRAF6. Although the precise role of PKD1 in the TLR/IL-1R signaling pathway is currently unknown, PKD1 appears to be involved in MyD88-dependent ubiquitination of TRAF6 and subsequent activation of TAK1, which eventually leads to the activation of MAPKs and NF-κB. Activation of PKD1 by TLRs is essential for MyD88-dependent expression of pro-inflammatory cytokines and mediators in innate immune cells. Currently it is not known whether any microorganism induces activation of PKD1 or whether PKD1 plays a regulatory role in inflammatory responses induced by pathogenic microorganisms. We demonstrate here that the farmer's lung disease-inciting agent S. rectivirgula induces activation of PKD1, and that PKD1 accounts for SR-Ag-mediated activation of MAPKs and NF-κB and subsequent expression and production of inflammatory cytokines and chemokines that are critical for recruitment of leukocytes. Indeed, SR-Ag-mediated induction of alveolitis was ablated in the lungs of mice pretreated with a pharmacological inhibitor of PKD, pointing to the essential role of PKD1 in pulmonary inflammation in response to SR-Ag.
The precise mechanism through which innate and adaptive immune cells are activated and recruited into the lungs during the course of hypersensitivity pneumonitis, and the contribution of different types of innate immune cells to the initiation and progress of the disease have yet to be elucidated. Although the initial responding innate immune cells in the lungs after exposure to S. rectivirgula have yet to be identified, it is expected that lung residential dendritic cells/macrophages and bronchial epithelial cells might be the type of cells that provide the first line of defense in the lung. Airway epithelial cells play critical roles in homeostasis and host defense by acting as a physical barrier, secreting and modifying the airway surface liquid, and removing particulates via muco-ciliary transport (52). In addition, they detect and respond to PAMPs, and secrete various cytokines and chemokines that are critical for innate host defense. A previous study has demonstrated that epithelial cell line A549 expresses and releases IL-8 in response to S. rectivirgula (7). Our results also showed that SR-Ag can directly act on and induce activation of PKD1, MAPKs, and transcription factor NF-κB and expression and production of pro-inflammatory cytokines and chemokines in bronchial epithelial cells, as well as alveolar macrophages and neutrophils. Profiles of cytokines and chemokines expressed in bronchial epithelial cells in response to SR-Ag are different from those in alveolar macrophages or neutrophils. We were not able to detect expression of TNFα, IL-1β, IL-10, and IL-12 in MLE12 cells in response to SR-Ag under our experimental conditions. However, MLE12 cells express IFNγ, IL-6, IL-23p19, MIP-1α, MIP-2, MCP-1, KC, IP-10, RANTES, ITAC, Eotaxin, LIX, and Mig in response to SR-Ag. These cytokines and chemokines are known to be critical for Th17 cell development and recruitment of macrophages, neutrophils, and T cells. These results suggest that airway epithelial cells might play more than a negligible role in the development and progress of hypersensitivity pneumonitis caused by pulmonary exposure to S. rectivirgula.
Similar to various synthetic or purified TLR ligands, activation of PKD1 by SR-Ag in vivo and in vitro is also dependent on MyD88 signaling, indicating that recognition of SR-Ag by one or more of the MyD88-dependent TLR/IL-1R superfamily members account for PKD1 activation. Using TLR-overexpressing HeLa cells, a previous study showed that SR-Ag is recognized by TLR2, but not by TLR4, TLR5, or TLR7 (4). However, experiments using TLR2-/- mice revealed that TLR2 only partially accounts for MIP-2 production and is not required for production of TNFα and KC and infiltration of neutrophils into the lungs following SR-Ag exposure (4). In agreement with these previous findings, we also found that the contribution of TLR2 to SR-Ag-mediated activation of PKD1, as well as activation of MAPKs and NF-κB, in vitro and in vivo is only partial to minimal under our experimental conditions (data not shown), indicating involvement of additional PRRs in SR-Ag-mediated activation of those signaling modulators. It is possible that genomic DNA released from S. rectivirgula can be detected by TLR9 and leads to activation of PKD1 and other downstream signaling modulators. However, we found that SR-Ag-mediated activation of PKD1, MAPKs and NF-κB in TLR9-/- mice is comparable to that in wild-type mice (data not shown). These results indicate that the contribution of TLR9 to SR-Ag-mediated activation of those signaling modulators is minimal to none, although we cannot completely rule out the possibility that TLR2 compensates for the lack of TLR9 and vice versa. Assessment of PKD1 activation in response to SR-Ag in TLR2/TLR9 double knockout mice might provide insight. To the best of our knowledge, natural ligands for TLR10-13 are currently not identified. It is possible that one or more of those TLRs may contribute to detection of SR-Ag. Another possibility is that inflammasome-activating PRRs detect parts of SR-Ag and lead to the maturation and secretion of pre-existing precursors of IL-1, IL-18, and/or IL-33. The resulting mature IL-1 family cytokines may be responsible for MyD88-dependent activation of PKD1. Further investigations on these aspects are warranted.
Farmer's lung disease, a prototypical hypersensitivity pneumonitis, is caused by repeated pulmonary exposure to S. rectivirgula present in moldy hay (1). The initial stage (or acute form) of the disease is characterized by increased production of pro-inflammatory cytokines and chemokines and subsequent alveolitis, accompanied by an influx of cells predominantly composed of neutrophils. A recent study demonstrated that initial expression of pro-inflammatory cytokines and chemokines in the lung and subsequent neutrophil infiltration into the lung after pulmonary exposure to SR-Ag are dependent on MyD88 (4). Recently we found that PKD1 is indispensable for MyD88-dependent activation of MAPKs and NF-κB and expression and production of pro-inflammatory cytokines and chemokines, but dispensable for MyD88-dependent type I IFN expression (45, 46). These findings suggest the possibility that PKD1 might play a role in SR-Ag-induced pro-inflammatory response in the lung. Indeed, SR-Ag failed to induce activation of MAPKs and NF-κB and expression and production of pro-inflammatory cytokines and chemokines in PKD1-knockdown cells in vitro. These findings demonstrate that PKD1 activation in innate immune cells in response to SR-Ag is essential for induction of pro-inflammatory cytokines and chemokines that are necessary for recruitment of neutrophils and lymphocytes into the site of SR-Ag exposure. Pharmacological inhibitors that inhibit the function or activation of a specific signaling molecule can be useful tools to uncover the biological role of the signaling molecule, as well as effective therapeutic agents if the signaling molecule is involved in the pathogenesis of certain diseases. Although the pharmacological inhibitor that specifically inhibits only PKD1 has yet to be developed, Gö6976 has been shown to effectively inhibit PKD1 activation by ligands for TLR/IL-1R superfamily members (33, 45, 46, 50, 51). Similar to these previous findings, SR-Ag-mediated PKD1 activation was selectively inhibited by Gö6976, but not by Gö6983, in vitro and in vivo. This indicates that SR-Ag-mediated PKD1 activation is independent of conventional PKC and that Gö6976 can be a useful agent to suppress SR-Ag-mediated PKD1 activation and downstream biologic events. Indeed, in vivo activation of MAPKs and NF-κB and expression and production of cytokines/chemokines in the lung and subsequent neutrophil infiltration into the lung after SR-Ag exposure were significantly suppressed by systemic inhibition of PKD1 activity using Gö6976. Although long-term treatment with Gö6976 would require caution and careful monitoring of adverse effects that might develop, a single-course treatment of Gö6976 at the concentration we used in this experimental setting does not appear to be toxic to mice. Gö6976-pretreated mice showed normal TRIF-dependent responses to PIC or LPS (45). Collectively, our results indicate that activation of PKD1 by SR-Ag is a necessary step leading to activation of MAPKs and NF-κB, expression and production of various cytokines/chemokines, and inflammatory cell infiltration into the lungs. Our results also suggest that PKD1 might be an attractive candidate as a therapeutic target for hypersensitivity pneumonitis caused by microbial antigens. Considering the increasing roles of MyD88 in acute and chronic inflammatory diseases and the critical role of PKD1 in MyD88-dependent pro-inflammatory gene expression, development of an effective and specific pharmacological inhibitor for PKD1, or a pathway-specific inhibitor for PKD1, is desired.
In summary, we found that in addition to alveolar macrophages and neutrophils, bronchial epithelial cells also respond directly to SR-Ag and produce various cytokines and chemokines (IL-6, IL-23, IFNγ, KC, MIP-1α, MIP-2, MCP-1, KC, IP-10, RANTES, ITAC, Eotaxin, LIX and Mig) that are critical for recruitment of leukocytes and development of the Th17 response and hypersensitivity pneumonitis. In addition, we demonstrated that SR-Ag induces activation of PKD1, but not PKD2 or PKD3, in various types of innate cells in the lung through an MyD88-dependent mechanism. Activation of PKD1 is required for expression of pro-inflammatory cytokines and chemokines in bronchial epithelial cells, alveolar macrophages and neutrophils, and for neutrophil infiltration into the lung in response to SR-Ag exposure. Taken together, our findings indicate that PKD1 might be an attractive molecular target for therapy of microbial antigen-induced hypersensitivity pneumonitis.
Acknowledgments
We thank Dr. S. Akira (Osaka Univ., Osaka, Japan) for kindly providing TLR9-/- and MyD88-/- mice. We thank Dr. Anand Kulkarni (Director, Tissue Services Core, UTHSC, Memphis) for scanning histology slides using Scanscope®XT, and Dr. Karen Whittington (Memphis V.A. Medical Center) for technical assistance. The Flow Cytometry Facility in Research Service at the Memphis V.A. Medical Center is also gratefully acknowledged. We also thank Mrs. Andrea Patters for her excellent assistance with preparation of the manuscript.
Footnote: A.K.Y. was supported by the Children's Foundation Research Center at Le Bonheur Children's Medical Center, and grants from NIH (AI053137) and the Children's Foundation of Memphis. Y.I.K. and J.E.P. were supported by grants from Le Bonheur Children's Medical Center. D.D.B. was supported by a grant from the Department of Veterans Affairs and E.A.F was supported by a grant from NIH (HL084172). Animal experiments were supported in part by a transgenic mice program grant from the Children's Foundation of Memphis. Tissue Services Core at UTHSC was supported by a grant from NCRR 1S10RR025665-01. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, the Department of Veterans Affairs, the Children's Foundation of Memphis, or Le Bonheur Children's Medical Center.
Abbreviations
- PRRs
pattern recognition receptors
- IFN
interferon
- IL
interleukin
- TLR
Toll-like receptors
- PAMP
pathogen-associated molecular pattern
- MyD88
myeloid differentiation factor 88
- TIR
Toll/interleukin-1 receptor
- IRAK
IL-1 receptor-associated kinase
- TRAF6
TNFα receptor-associated factor 6
- TAK1
TGFβ-activated kinase 1
- TAB2
TAK1-binding protein 2
- MAPKs
mitogen-activated protein kinases
- LPS
lipopolysaccharide
- PGN
peptidoglycan
- SR-Ag
Saccharopolyspora rectivirgula-antigen
- BAL
bronchoalveolar lavage
- BALF
bronchoalveolar lavage fluid
- siRNA
small interfering RNA
- PKD
protein kinase D
- MPO
myeloperoxidase
- H&E
hematoxylin and eosin
- JNK
c-Jun-N-terminal kinase
- ERK
extracellular signal-regulated kinase
- NF-κB
nuclear factor-κB
- IκB
inhibitor of κB
- STAT1
signal transducer and activator of transcription 1
- BMDM
bone marrow-derived macrophages
- ELISA
enzyme-linked immunosorbent assay
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- DMSO
dimethyl sulphoxide
- IP-10
interferon-inducible protein-10
- MIP
macrophage inflammatory protein
- MCP
monocyte chemoattractant protein
- ITAC
IFN-inducible T cell alpha chemoattractant
- Mig
monokine induced by IFNγ
- KC
keratinocyte-derived chemokine
- LIX
lipopolysaccharide-induced CXC chemokine
- RANTES
regulated on activation normal T cell expressed and secreted
References
- 1.Girard M, Lacasse Y, Cormier Y. Hypersensitivity pneumonitis. Allergy. 2009;64:322–334. doi: 10.1111/j.1398-9995.2009.01949.x. [DOI] [PubMed] [Google Scholar]
- 2.Lacasse Y, Israel Assayag E, Laviolette M, Cormier Y. Clinical and immunopathological aspects of hypersensitivity pneumonitis. Rev Mal Respir. 2004;21:769–781. doi: 10.1016/s0761-8425(04)71418-7. [DOI] [PubMed] [Google Scholar]
- 3.Losa Garcia JE, Rodriguez FM, Martin de Cabo MR, Garcia Salgado MJ, Losada JP, Villaron LG, Lopez AJ, Arellano JL. Evaluation of inflammatory cytokine secretion by human alveolar macrophages. Mediators Inflamm. 1999;8:43–51. doi: 10.1080/09629359990711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nance SC, Yi AK, Re FC, Fitzpatrick EA. MyD88 is necessary for neutrophil recruitment in hypersensitivity pneumonitis. J Leukoc Biol. 2008;83:1207–1217. doi: 10.1189/jlb.0607391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shijubo N, Imai K, Shigehara K, Hirasawa M, Tsujisaki M, Hinoda Y, Abe S. Soluble intercellular adhesion molecule-1 (ICAM-1) in sera and bronchoalveolar lavage (BAL) fluids of extrinsic allergic alveolitis. Clin Exp Immunol. 1995;102:91–97. doi: 10.1111/j.1365-2249.1995.tb06641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudmundsson G, Bosch A, Davidson BL, Berg DJ, Hunninghake GW. Interleukin-10 modulates the severity of hypersensitivity pneumonitis in mice. Am J Respir Cell Mol Biol. 1998;19:812–818. doi: 10.1165/ajrcmb.19.5.3153. [DOI] [PubMed] [Google Scholar]
- 7.Gudmundsson G, Hunninghake GW. Respiratory epithelial cells release interleukin-8 in response to a thermophilic bacteria that causes hypersensitivity pneumonitis. Exp Lung Res. 1999;25:217–228. doi: 10.1080/019021499270277. [DOI] [PubMed] [Google Scholar]
- 8.Gudmundsson G, Monick MM, Hunninghake GW. IL-12 modulates expression of hypersensitivity pneumonitis. J Immunol. 1998;161:991–999. [PubMed] [Google Scholar]
- 9.Israel-Assayag E, Dakhama A, Lavigne S, Laviolette M, Cormier Y. Expression of costimulatory molecules on alveolar macrophages in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1999;159:1830–1834. doi: 10.1164/ajrccm.159.6.9810087. [DOI] [PubMed] [Google Scholar]
- 10.Schuyler M, Gott K, Cherne A. Mediators of hypersensitivity pneumonitis. J Lab Clin Med. 2000;136:29–38. doi: 10.1067/mlc.2000.107694. [DOI] [PubMed] [Google Scholar]
- 11.Schuyler M, Gott K, Cherne A. Experimental hypersensitivity pneumonitis: role of MCP-1. J Lab Clin Med. 2003;142:187–195. doi: 10.1016/S0022-2143(03)00107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ismail T, McSharry C, Boyd G. Extrinsic allergic alveolitis. Respirology. 2006;11:262–268. doi: 10.1111/j.1440-1843.2006.00839.x. [DOI] [PubMed] [Google Scholar]
- 13.Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol. 2001;108:661–670. doi: 10.1067/mai.2001.119570. [DOI] [PubMed] [Google Scholar]
- 14.Nance S, Cross R, Yi AK, Fitzpatrick EA. IFN-gamma production by innate immune cells is sufficient for development of hypersensitivity pneumonitis. Eur J Immunol. 2005;35:1928–1938. doi: 10.1002/eji.200425762. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki H, Ando M, Brazer W, Center DM, Cruikshank WW. Polarized type 1 cytokine profile in bronchoalveolar lavage T cells of patients with hypersensitivity pneumonitis. J Immunol. 1999;163:3516–3523. [PubMed] [Google Scholar]
- 16.Suga M, Yamasaki H, Nakagawa K, Kohrogi H, Ando M. Mechanisms accounting for granulomatous responses in hypersensitivity pneumonitis. Sarcoidosis Vasc Diffuse Lung Dis. 1997;14:131–138. [PubMed] [Google Scholar]
- 17.Denis M, Bisson D. Antigen-induced alveolitis: cytokine production in a mouse model. Inflammation. 1995;19:157–177. doi: 10.1007/BF01534459. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds SP, Jones KP, Edwards JH, Davies BH. Immunoregulatory proteins in bronchoalveolar lavage fluid. A comparative analysis of pigeon breeders' disease, sarcoidosis and idiopathic pulmonary fibrosis. Sarcoidosis. 1989;6:125–134. [PubMed] [Google Scholar]
- 19.Salvaggio JE. Recent advances in pathogenesis of allergic alveolitis. Clin Exp Allergy. 1990;20:137–144. doi: 10.1111/j.1365-2222.1990.tb02658.x. [DOI] [PubMed] [Google Scholar]
- 20.Gudmundsson G, Hunninghake GW. Interferon-gamma is necessary for the expression of hypersensitivity pneumonitis. J Clin Invest. 1997;99:2386–2390. doi: 10.1172/JCI119420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 22.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 23.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 25.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 28.Netea MG, Van der Meer JW, Kullberg BJ. Recognition of pathogenic microorganisms by Toll-like receptors. Drugs Today (Barc) 2006;42 A:99–105. [PubMed] [Google Scholar]
- 29.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 30.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 31.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci U S A. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 33.Jeohn GH, Cooper CL, Jang KJ, Liu B, Lee DS, Kim HC, Hong JS. Go6976 inhibits LPS-induced microglial TNFalpha release by suppressing p38 MAP kinase activation. Neuroscience. 2002;114:689–697. doi: 10.1016/s0306-4522(02)00356-1. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 35.Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J Biol Chem. 2004;279:40653–40658. doi: 10.1074/jbc.M402666200. [DOI] [PubMed] [Google Scholar]
- 36.Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol Cell Biol. 2008;28:3538–3547. doi: 10.1128/MCB.02098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem. 2007;282:4102–4112. doi: 10.1074/jbc.M609503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 40.Hacker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med. 2000;192:595–600. doi: 10.1084/jem.192.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 42.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Yi AK, Yoon JG, Krieg AM. Convergence of CpG DNA- and BCR-mediated signals at the c-Jun N-terminal kinase and NF-kappaB activation pathways: regulation by mitogen-activated protein kinases. Int Immunol. 2003;15:577–591. doi: 10.1093/intimm/dxg058. [DOI] [PubMed] [Google Scholar]
- 44.Yi AK, Yoon JG, Yeo SJ, Hong SC, English BK, Krieg AM. Role of mitogen-activated protein kinases in CpG DNA-mediated IL-10 and IL-12 production: central role of extracellular signal-regulated kinase in the negative feedback loop of the CpG DNA-mediated Th1 response. J Immunol. 2002;168:4711–4720. doi: 10.4049/jimmunol.168.9.4711. [DOI] [PubMed] [Google Scholar]
- 45.Park JE, Young In Kim, Ae Kyung Yi. Protein kinase D1 is essential for MyD88-dependent TLR signaling pathway. J Immunol. 2009;182:6316–6327. doi: 10.4049/jimmunol.0804239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park JE, Kim YI, Yi AK. Protein kinase D1: a new component in TLR9 signaling. J Immunol. 2008;181:2044–2055. doi: 10.4049/jimmunol.181.3.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 48.Yeo SJ, Yoon JG, Hong SC, Yi AK. CpG DNA induces self and cross-hyporesponsiveness of RAW264.7 cells in response to CpG DNA and lipopolysaccharide: alterations in IL-1 receptor-associated kinase expression. J Immunol. 2003;170:1052–1061. doi: 10.4049/jimmunol.170.2.1052. [DOI] [PubMed] [Google Scholar]
- 49.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 50.Ivison SM, Graham NR, Bernales CQ, Kifayet A, Ng N, Shobab LA, Steiner TS. Protein kinase D interaction with TLR5 is required for inflammatory signaling in response to bacterial flagellin. J Immunol. 2007;178:5735–5743. doi: 10.4049/jimmunol.178.9.5735. [DOI] [PubMed] [Google Scholar]
- 51.Song MJ, Wang YQ, Wu GC. Lipopolysaccharide-induced protein kinase D activation mediated by interleukin-1beta and protein kinase C. Brain Res. 2007;1145:19–27. doi: 10.1016/j.brainres.2007.01.128. [DOI] [PubMed] [Google Scholar]
- 52.Kato A, Schleimer RP. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol. 2007;19:711–720. doi: 10.1016/j.coi.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]