

Abstract
Herpes simplex virus type 1 (HSV-1) immediate-early gene product ICP0 activates lytic infection and relieves cell-mediated repression of viral gene expression. This repression is conferred by preexisting cellular proteins and is commonly referred to as intrinsic antiviral resistance or intrinsic defense. PML and Sp100, two core components of nuclear substructures known as ND10 or PML nuclear bodies, contribute to intrinsic resistance, but it is clear that other proteins must also be involved. We have tested the hypothesis that additional ND10 factors, particularly those that are involved in chromatin remodeling, may have roles in intrinsic resistance against HSV-1 infection. The two ND10 component proteins investigated in this report are ATRX and hDaxx, which are known to interact with each other and comprise components of a repressive chromatin-remodeling complex. We generated stable cell lines in which endogenous ATRX or hDaxx expression is severely suppressed by RNA interference. We found increases in both gene expression and plaque formation induced by ICP0-null mutant HSV-1 in both ATRX- and hDaxx-depleted cells. Reconstitution of wild-type hDaxx expression reversed the effects of hDaxx depletion, but reconstitution with a mutant form of hDaxx unable to interact with ATRX did not. Our results suggest that ATRX and hDaxx act as a complex that contributes to intrinsic antiviral resistance to HSV-1 infection, which is counteracted by ICP0.
Herpes simplex virus type 1 (HSV-1) is an important human pathogen prevalent in a majority of the human population. Upon primary lytic infection of skin mucosa, the virus travels to neurons of the dorsal root ganglia, where it establishes lifelong latency (reviewed in reference 14). Subsequently, the virus reactivates periodically in the form of herpes labialis (commonly known as cold sores) in immunocompetent individuals, but it causes more-severe complications in people with suppressed immunity. HSV-1 pathogenesis is highly dependent on the switch between lytic and latent infection. The mechanisms underlying HSV-1 latency establishment, maintenance, and reactivation have been subjected to extensive research, but the cellular mechanisms involved remain incompletely understood. HSV-1 immediate-early (IE) protein ICP0 is a strong activator of viral gene expression that is required for both efficient lytic infection (reviewed in references 18 and 37) and productive reactivation from latency (18, 38, 39, 84, 97). HSV-1 mutants that fail to express ICP0 are highly sensitive to cell-mediated repression, especially during low-multiplicity infections of human diploid fibroblasts. Consequently, the mutant virus has a higher probability of establishing a quiescent infection that in some respects resembles latency (reviewed in reference 17). The cell-mediated repression of ICP0-null mutant HSV-1 by constitutively expressed cellular proteins has been dubbed intrinsic antiviral resistance or intrinsic defense, and analogous mechanisms are thought to operate during the early stages of human cytomegalovirus (HCMV) infection (87, 95). A major role of ICP0 during HSV-1 infection is to counteract cellular intrinsic antiviral resistance, but as yet, the cellular proteins that are involved in this process are incompletely understood and in some cases controversial. Therefore, further studies of the proteins that are involved in intrinsic cellular defenses against herpesvirus infection are required.
One aspect of the cellular mechanisms involved in intrinsic defense against HSV-1 gene expression is conferred by nuclear compartments known as ND10 or promyelocytic leukemia (PML) nuclear bodies. ND10 integrity is disrupted at early time points after wild-type (wt) HSV-1 infection (22, 31, 66) due to the degradation of the PML protein and the small ubiquitin modifier (SUMO)-modified forms of Sp100, which are both major ND10 components. These and a number of other cellular proteins are degraded, directly or indirectly, by the RING finger-mediated E3 ubiquitin ligase activity of ICP0 (5, 6, 12, 21, 27). We have reported previously that both PML and Sp100 contribute to cell-mediated repression of ICP0-null mutant HSV-1 gene expression (28, 30). In the absence of ICP0, these proteins are not degraded but instead are rapidly recruited to sites that are closely associated with parental viral genomes and early replication compartments. During wt HSV-1 infection, this redistribution is transient and difficult to detect because of the activities of ICP0. In the absence of ICP0, however, the ND10 proteins are stably retained at these virus-induced foci, suggesting the potential role of ND10 proteins in the initial repression of ICP0-null mutant viral genomes (25). Consistent with this hypothesis, the recruitment process is extremely rapid (26) and there is a close correlation between the abilities of mutant forms of ICP0 to inhibit this process and to stimulate lytic infection (29).
In this study, we have focused on two ND10 proteins that are components of a complex known to have chromatin modification and repressive activities, namely, hDaxx and ATRX (49, 94, 99). Daxx (hDaxx for human Daxx) was originally identified as a cytoplasmic Fas-interacting proapoptotic protein (100); however, antiapoptotic roles have also been proposed (1, 70). In later studies, hDaxx was found to be predominantly a nuclear protein that shuttles between heterochromatin and ND10 depending on the stage of the cell cycle (47). It has been shown to be involved in transcriptional repression of a number of cellular genes and in activation of apoptotic signaling (11, 43, 52, 60-62, 70, 71, 85). At its C terminus, hDaxx contains a SUMO interaction motif that is required for interaction with PML and is therefore essential for its localization to ND10 (60). Interaction with its SUMO-modified interaction partners is important for the roles of hDaxx in transcriptional regulation (44, 47, 55, 63, 69, 103). It has been reported that hDaxx itself can be modified by SUMO (51), although the precise roles of this modification in modulation of its transcriptional activities have yet to be established.
One of the many interaction partners of hDaxx is ATRX (α-thalassemia mental retardation X-linked), a chromatin-remodeling enzyme of the SWI/SNF family of ATP-dependent helicases (32, 99). ATRX is readily detected at ND10 and throughout the nucleoplasm and has been shown to localize to heterochromatin, where it interacts with hDaxx (49, 76, 94). In addition, ATRX associates with pericentric heterochromatin and methyl-CpG-binding protein (MeCP2) and is proposed to interact with HP1 (3, 15, 58, 59, 72). This evidence suggests a role for ATRX in chromatin remodeling, consistent with a report demonstrating its involvement in transcription repression (94). Despite the fact that no specific ATRX target genes have been identified, its structural characteristics suggest roles in direct chromatin interaction (2).
In the context of herpesvirus infections, the role of hDaxx in transcriptional repression has been subjected to more-thorough research than that of ATRX. During HCMV infection, the viral tegument protein pp71 interacts with hDaxx and promotes its degradation, a process that is important for the initiation of efficient HCMV gene expression (9, 10, 41, 45, 48, 80, 82). ATRX, on the other hand, is displaced from ND10 by pp71, which is one of the earliest consequences of HCMV infection detected by immunofluorescence. This displacement is functionally significant for the ability of pp71 to stimulate gene expression since replication of pp71-null mutant HCMV is enhanced substantially in ATRX-depleted cells (65). Therefore, ATRX and hDaxx have been confirmed as components of a cell-mediated intrinsic defense against HCMV infection, which is counteracted by pp71.
Because of the known relationships between ND10 proteins and herpesvirus genomes (25, 46), we hypothesized that ATRX and hDaxx may play similar roles in the repression of HSV-1 and HCMV gene expression. Since intrinsic cellular defenses are efficiently overcome by ICP0, we have used ICP0-null mutant HSV-1 in order to determine whether ATRX and hDaxx contribute to the cellular repression mechanism that occurs in the absence of ICP0. We used RNA interference (RNAi) to generate ATRX- and hDaxx-depleted cell lines, and we found a requirement for both ATRX and hDaxx for the fully efficient repression of ICP0-null mutant HSV-1 gene expression. The results indicate that the two proteins act as a complex in their role in intrinsic resistance to HSV-1 infection.
MATERIALS AND METHODS
Viruses and cells.
HSV-1 strain 17+ was the wt strain from which all mutant and variant viruses were derived. ICP0-null mutant dl1403 has been described previously (91). Viruses in1863 and dl1403/CMVlacZ were derived from the above-mentioned strains and contain the lacZ gene insertion into tk gene under the control of the HCMV promoter/enhancer (kindly provided by Chris Preston). HSV-1 mutant virus in1374 contains the tsK temperature-sensitive lesion in ICP4, a deletion of the ICP0 gene, and a mutation within VP16 that inactivates its ability to stimulate IE gene expression (80). All viruses were propagated in BHK (baby hamster kidney) cells and titrated in U2OS cells, in which ICP0 is not required for efficient replication of HSV-1 (101). Virus in1374 was propagated at the permissive temperature of 31°C in the presence of 2.5 mM hexamethylbisacetamide (80). Human foreskin diploid fibroblasts (HFs) and U2OS, HK-293T, HEp-2, and HeLa cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. BHK cells were grown in Glasgow modified Eagle's medium supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. HepaRG hepatocyte cells (33) were grown in William's medium E (WME) supplemented with 10% fetal bovine serum (FBS) Gold (PAA Laboratories, Ltd.), 2 mM glutamine, 5 μg/ml insulin, and 0.5 μM hydrocortisone. All cell growth media were supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin. Lentivirus-transduced cells were maintained with continuous antibiotic selection as specified.
Antibodies and enzymes.
All restriction enzymes and buffers were purchased from either Roche or New England Biolabs. T4 DNA ligase and λ-phosphatase were from New England Biolabs. Polyclonal rabbit (H300, sc-15408) and goat (D19, sc-10078) anti-ATRX antibodies were from Santa Cruz Biotechnology, and anti-ATRX mouse monoclonal 39F was kindly provided by Richard Gibbons (Oxford). Rabbit anti-hDaxx antibodies were from Sigma-Aldrich (D7810) or Upstate (07-471). Anti-hDaxx rabbit serum r1866 was previously described (77). Mouse anti-PML clone 5E10 (92) was a gift from Roel van Driel (Amsterdam, Netherlands), and anti-PML rabbit serum r8 was previously described (4). Rabbit anti-USP7 antibody was from Bethyl Laboratories (BL851), while anti-USP7 rabbit serum 201 has been described previously (23, 24). Anti-green fluorescent protein (anti-GFP) rabbit antibody was purchased from Abcam (ab290). Mouse anti-actin (AL-40) antibody was from Sigma-Aldrich. Mouse monoclonal antibodies for detection of ICP4 (58S) and UL42 (Z1F11) were used as described previously (8). Anti-ICP0 antibody was rabbit serum 190 (27). The secondary antibodies used for immunofluorescence were fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse immunoglobulin G (IgG; Sigma-Aldrich), goat Alexa 488-conjugated anti-mouse IgG (Invitrogen), goat Cy3-conjugated anti-mouse and Cy5-conjugated anti-rabbit IgGs (GE Healthcare), or donkey Alexa 647-conjugated anti-rabbit and Alexa 555-conjugated anti-mouse, anti-rabbit, and anti-goat IgGs (Invitrogen). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse antibodies for use in Western blotting were from Sigma-Aldrich.
Construction of an hDaxx-expression vector and hDaxx mutagenesis.
Plasmid pcDNA3-hDaxx expressing wt hDaxx (GenBank accession code NM_001350) was kindly provided by John Sinclair. Three DNA fragments encompassing the hDaxx cDNA were amplified in separate PCRs using three sets of primers 1 to 3 (Table 1) containing restriction sites which were unique for each fragment. Primer pairs 1 and 2 introduced a set of silent mutations into the hDaxx cDNA that render it resistant to the anti-hDaxx short hairpin RNA (shRNA) utilized for the depletion of endogenous hDaxx expressed in hDaxx-depleted cell line HALD2. Reaction products were purified, then digested with the indicated enzymes (Table 1), and ligated into vector plasmid pEYFP-C1. The enhanced yellow fluorescent protein (EYFP)-hDaxx fusion cDNA was excised with AgeI and EcoRI and ligated downstream of the HSV-1 gD (US6) gene promoter in a lentivirus vector plasmid expressing neomycin resistance. The resulting plasmid (pLNGY-hDaxx) was authenticated by sequencing the entire hDaxx cDNA region.
TABLE 1.
Primers used in construction of EYFP-hDaxx- and EYFP-hDaxx.ΔPAH1-expressing vectors
| Primer pair | Primer name | Sequencea (5′-3′) | Restriction site |
|---|---|---|---|
| 1 | DaxxN For | GTACTAACTCGAGCCATGGCCACCGCTAACAGC | XhoI |
| Daxx si-ve Rev | CTGCAGGAAAAGGAGTTAGATCTGAGCGAA | BglII | |
| 2 | Daxx si-ve For | GAGTTAGATCTGAGCGAATTGGATGACCCA | BglII |
| Daxx Rev | GCCATTCCACTAGGGCCCTCACCAGAATCC | ApaI | |
| 3 | DaxxC For | GGATTCTGGTGAGGGCCCTAGTGGAATGGC | ApaI |
| DaxxC Rev | GACCTGGAATTCCTAATCAGAGTCTGAGAG | EcoRI | |
| 4 | EYFP For | AGCTTAACCGGTCGCCACCATGGTGAGC | AgeI |
| Daxx ΔPAH Rev | GGCCCCATGAGGCTCAGAGGAGCTAGG | ||
| 5 | Daxx ΔPAH For | TCCTCTGAGCCTCATGGGGCCGCCAAGCTCTATGTCTAC | |
| DaxxC Rev | GACCTGGAATTCCTAATCAGAGTCTGAGAG | EcoRI |
Underlining indicates restriction sites.
For deletion of the ATRX interaction domain PAH1 from hDaxx, mutagenesis by the gene splicing approach (40) was used. In brief, a first round of PCR, using primer pairs 4 and 5 (Table 1), generated overlapping hDaxx cDNA fragments in a manner that removed hDaxx cDNA codons 47 to 121 from the products. A second round of PCR, using the products from the first round and the outside primers of pairs 4 and 5, was used for generating the final hDaxx.ΔPAH1 cDNA fragment. The final PCR fragments, encoding EYFP-hDaxx.ΔPAH1, were cleaved with AgeI and EcoRI to allow insertion into the lentiviral vector, as described above. Vectors pLNGY-hDaxx and pLNGY-hDaxx.ΔPAH1 were used for lentivirus production.
Lentiviral transduction.
Depletion of the endogenous ATRX and hDaxx proteins in HepaRG cells was achieved using lentivirus vectors pLKO-shATRX90, expressing an shRNA against ATRX (65), and pLKO-shDaxx2, expressing an shRNA against hDaxx (70, 82), respectively. Lentiviral supernatants were generated in HK-293T cells by cotransfection of these plasmids with helper and envelope plasmids pCMV.DR8.91 (kindly supplied by Didier Trono) and pVSV-G (BD Biosciences). Lentiviral transduction of HepaRG cells and production of control cells expressing an antiluciferase shRNA were as described previously (26). The cell lines were isolated after outgrowth of selection drug-resistant cells and hence are mixed cell populations rather than individual clonal isolates. The ATRX-depleted (HAA), hDaxx-depleted (HALD2), and control cells (HALL) (Table 2) were maintained in medium containing 0.5 μg/ml puromycin after initial selection using 1 μg/ml puromycin. For wt hDaxx and mutant hDaxx reconstituted cells, antibiotic selection of 0.5 μg/ml puromycin plus 0.5 mg/ml G418 after initial selection with 0.5 μg/ml puromycin plus 1 mg/ml G418 was used. Table 2 presents the nomenclature and details of the depleted and reconstituted cell lines obtained via lentiviral transduction methods.
TABLE 2.
Nomenclature for HepaRG-derived cell lines generated by lentiviral transduction
| Name | Phenotype | Drug(s) to which cells are resistant | Additional description |
|---|---|---|---|
| HALL | HepaRG shLuci | Puromycin | Depletion control |
| HAA | HepaRG shATRX90 | Puromycin | ATRX depletion line |
| HALD2 | HepaRG shDaxx2 | Puromycin | hDaxx depletion line |
| HL-E | HALL EYFP | Puromycin/neomycin | Depletion and EYFP expression control |
| HL-ED | HALL EYFP-hDaxx | Puromycin/neomycin | EYFP-hDaxx expression line |
| HD-E | HALD2 EYFP | Puromycin/neomycin | hDaxx depletion and EYFP expression control |
| HD-ED | HALD2 EYFP-hDaxx | Puromycin/neomycin | Cell line with endogenous hDaxx depletion and EYFP-hDaxx reintroduction |
| HD.ΔPAH | HALD2 EYFP-hDaxxΔPAH1 | Puromycin/neomycin | Cell line with endogenous hDaxx depletion and mutant EYFP-hDaxxΔPAH1 reintroduction |
FACS.
Enrichment of cells expressing EYFP fusion proteins after transduction with lentiviruses derived from plasmids of the pLNGY series was achieved by fluorescence-activated cell sorting (FACS). HepaRG (negative control) and lentivirus-transduced cells were harvested, washed twice, and resuspended in complete medium containing 200 units/ml penicillin, 200 μg/ml streptomycin, and 1% FBS Gold. The FACS service was kindly provided by Tom Gilbey (Beatson Institute for Cancer Research, Glasgow, Scotland) using a Becton Dickinson SORP FACSaria. EYFP-positive sorted cells were transferred to complete WME containing 10% FBS Gold. Puromycin and G418 selection was applied the following day.
Virus infections.
For all HSV-1 infection experiments, cells were seeded into 24-well plates at 1 × 105 cells per well and infected the following day. Virus was allowed to adsorb for 1 h in a low volume of medium with regular mixing and then overlaid with medium. For plaque assays, cells were infected with sequential virus dilutions of either in1863 or dl1403/CMVlacZ, overlaid with medium containing 1% human serum, and stained for β-galactosidase expression the following day, as described previously (50). For analysis of viral protein expression, cells were infected with wt HSV-1 strain 17+ or ICP0-null mutant dl1403 at a multiplicity of infection (MOI) of 2 PFU per cell and harvested at 0, 4, 6, and 8 h postinfection for Western blot analysis. For examination of cells by immunofluorescence, cells on coverslips were infected with HSV-1 strain 17+ at an MOI of 0.001 or with dl1403 at MOIs varying from 0.2 to 1.0. For assays of the establishment of quiescence, cells were infected with in1374 at an MOI of 5, virus was allowed to adsorb at 37°C, and then the cells were incubated overnight at a nonpermissive temperature (38.5°C). For determining the maximum potential of reactivation, samples of cells were also coinfected with HSV-1 ICP4 mutant virus tsK at an MOI of 1. The following day, cells were stained for β-galactosidase expression as described above.
Immunoprecipitation.
Uninfected or infected cell monolayers (equivalent to 4.5 × 106 cells) were harvested in 3 ml of immunoprecipitation (IP) lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 0.1% NP-40, 5% glycerol, 1 mM EDTA, 1 mM EGTA, 2 mM dithiothreitol [DTT]) supplemented with protease inhibitors (Roche). Cells were incubated on ice for 15 min prior to lysing by bath sonication. Cell debris were removed by centrifugation (25,000 rpm for 30 min at 4°C using a Beckman TL-100 Ultracentrifuge, rotor TLA100.2), and then the lysates were precleared by incubating them with protein G beads supplemented with salmon sperm DNA (Millipore) for 30 min at 4°C. After centrifugation to remove the beads and nonspecifically bound proteins, immunoprecipitation was carried out by incubating the lysates for 2 h at 4°C with 1 μg of rabbit antibodies per 750 μl lysate (anti-ATRX H300, anti-hDaxx 07-471, anti-USP7 serum 201 [irrelevant control], anti-ICP0 serum 190, or no antibody), utilizing continuous mixing. Immune complexes were captured by incubation with protein G beads for 90 min at 4°C, with continuous mixing. The beads were washed 3 times with 1 ml IP lysis buffer and once with 200 μl IP buffer before being resuspended in 30 μl sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. Samples were boiled for 5 min to dissociate complexes from the beads and then resolved on 6% polyacrylamide gels.
Phosphatase treatment.
Cells seeded into 24-well plates at a density of 1 × 105 cells per well and infected with appropriate amounts of wt or ICP0-null mutant virus, as described above, were harvested at the indicated times postinfection in 70 μl of IP lysis buffer without EDTA. For each 20 μl of cell lysate, 400 units of λ-phosphatase (New England Biolabs) were used. Reaction mixtures were supplemented with 1× phosphatase buffer and 2 mM MnCl2, incubated for 1 h at 30°C, and processed for Western blot analysis as described below.
Western blot analysis.
Whole-cell extracts were prepared by washing cells twice with phosphate-buffered saline (PBS) and then adding SDS-PAGE gel loading buffer, as described previously (8). Proteins were resolved on 7.5% or 6.0% polyacrylamide gels. ATRX and hDaxx were detected using mouse 39F and rabbit anti-hDaxx D7810 antibodies, respectively.
Immunofluorescence.
Cells seeded onto 13-mm coverslips were prepared for immunofluorescence analysis as described previously (20). The samples were analyzed using a Zeiss LSM 510 confocal microscope with 488-nm, 543-nm, and 647-nm laser lines, with each channel scanned separately under image capture conditions that eliminated channel overlap. The images were exported as TIFF files and then processed using Adobe Photoshop.
RESULTS
Expression of ND10 component proteins is cell type dependent.
Several studies have demonstrated that hDaxx contributes to cellular intrinsic resistance to HCMV infection by repressing viral IE gene expression, an effect that is counteracted by the viral tegument protein pp71 (9, 82, 86, 98). In a recent study, we found that the hDaxx interaction partner ATRX (94) plays a role in this process. The evidence for this conclusion is that ATRX is displaced from ND10 by pp71 before any effect on hDaxx can be discerned, and depletion of ATRX improves IE gene expression and replication of pp71 mutant HCMV (65). Although studies of this aspect of HCMV infection are quite well developed, the roles of these proteins in HSV-1 infection have not been investigated in any detail. In preliminary work, we assessed the expression levels of hDaxx, ATRX, and major ND10 proteins PML and Sp100 in several common laboratory cell types, including U2OS cells (in which the ICP0-null mutant defect is not exhibited) (101). These data are presented in Fig. 1.
FIG. 1.

Analysis of endogenous expression of ND10 proteins in cultured cell lines. Samples of the indicated cells were harvested, and whole-cell extracts derived from approximately 3 × 104 cells were loaded onto 7.5% or 6% polyacrylamide gels. The antibodies used for Western blot detection were anti-PML 5E10, anti-ATRX 39F, anti-Daxx D7810, anti-Sp100 SpGH, anti-actin AL-40, and anti-USP7 BL851. USP7 and actin were used as loading controls for 6% and 7.5% gels, respectively. The differences in USP7 expression were reproducible.
Both Sp100 and PML are expressed in all the cell lines tested; however, there are significant differences in the abundances of their various modified isoforms. Both proteins are apparently expressed at higher levels in HepaRG and HEp-2 cells, while PML is least well expressed in HeLa and U2OS cells. The reduced level of expression of PML in these cells appears to correlate with reduced SUMO modification of Sp100. These observations open the possibility that cell-type-specific variations in ND10 protein expression may contribute to differences in permissiveness to ICP0-null mutant HSV-1 infection. As previously demonstrated (85, 90), hDaxx is a phosphoprotein that appears on Western blots as a double band corresponding to its phosphorylated forms. While total expression levels of hDaxx were fairly uniform among this set of cells, the balances of phosphorylated forms differed. There were also differences in the levels of USP7 expression, which was initially used as a loading control in low-percentage polyacrylamide gels. A striking finding was that ATRX is not expressed in U2OS cells. This is unlikely to explain the efficient replication of ICP0-null mutant virus replication in these cells, because ATRX is expressed normally in HEp-2 and 293T cells, in which the ICP0-null mutant defect is less than 10-fold. However, based on the data presented on Fig. 1, it is possible that cell-type-specific differences in levels of ND10 protein expression and modification may contribute to various levels of permissiveness to wt and ICP0-null mutant HSV-1 infection.
ATRX and hDaxx are recruited to the sites of parental HSV-1 genomes and early replication compartments.
During HSV-1 infection, several ND10 components relocate to sites that are closely associated with viral genomes soon after they enter the nucleus. This is best visualized in cells at the edge of a developing plaque because they are infected at high multiplicity and in an asymmetric manner (25). ICP4 is used as a marker for HSV-1 genomes because of its strong interaction with viral DNA (25). In a cell infected with wt HSV-1, this redistribution is difficult to detect and short lived because it is rapidly inhibited by the activities of ICP0 (22, 67). In the absence of ICP0, however, the ND10 components are not dispersed or degraded and they become recruited to sites associated with viral genomes in a far more stable and long-lived manner. We confirmed that, as in human fibroblasts (65), ATRX and hDaxx colocalize with PML at ND10 in uninfected HepaRG cells (Fig. 2A and B). During wt HSV-1 infection, both ATRX and hDaxx become rapidly dispersed in the great majority of infected cells in which ICP4 is expressed at readily detectable levels (Fig. 2C and D). However, in rare cells at the edges of developing wt HSV-1 plaques, it was possible to detect redistribution of ATRX and hDaxx to asymmetric foci in the apparent absence of ICP4 expression (Fig. 2E and F). These cells are likely to be in the very earliest stages of infection, before ICP4 has accumulated to detectable levels, emphasizing the rapidity of the cellular response that leads to the accumulation of these proteins at sites associated with viral genomes. In the absence of ICP0, both ATRX and hDaxx relocalize in a more stable manner to novel sites associated with viral genomes and early replication compartments (Fig. 2G and H).
FIG. 2.

ATRX and hDaxx are recruited to the sites of incoming HSV-1 genomes in HepaRG cells. Immunofluorescence images of uninfected HepaRG cells (A and B) and cells infected with wt HSV-1 (MOI, 0.001) (C to F) and ICP0-null mutant HSV-1 (MOI, 0.5) (G to H) at the edge of a developing plaque. Cells were processed for immunofluorescence analysis at 24 h postinfection and stained for the indicated proteins. Uninfected cells (A and B) were costained for PML (5E10) and ATRX (H300) or hDaxx (07-471). Infected cells were costained for ICP4 (58S) and ATRX or hDaxx. For panel H, Daxx was detected with anti-rabbit r1866 serum. The secondary antibodies were FITC-conjugated anti-mouse (green) (A to H), Alexa 555-conjugated anti-rabbit (red) (A to F), or Cy3-conjugated anti-rabbit (red) (G to H) IgGs.
ATRX recruitment to sites associated with HSV-1 genomes requires hDaxx.
Previous studies have shown that hDaxx is required for ATRX localization to ND10 (49, 65, 94). To investigate whether hDaxx is also required for ATRX recruitment to the sites associated with ICP0-null mutant HSV-1 genomes, hDaxx-depleted HepaRG cells, named HALD2 (Table 2 gives a complete listing of the cell nomenclature used), were generated by lentivirus-mediated shRNA expression. Analysis by Western blotting and immunofluorescence confirmed substantial reductions in hDaxx expression in HALD2 cells compared to the levels for parental HepaRG (Fig. 2B) and control HALL cells (Fig. 3). Depletion of hDaxx did not affect the levels of ATRX expression, as analyzed by Western blotting (data not shown). Consistent with previous reports, ATRX localization to ND10 was abolished in hDaxx-depleted cells, whereas PML remained in apparently normal punctate foci (Fig. 3B). Examination of HALD2 cells at the peripheries of plaques formed by ICP0-null mutant HSV-1 revealed that ATRX was no longer recruited to ICP4 foci but that PML was recruited normally (Fig. 3C and data not shown). In contrast to hDaxx, PML was not required for recruitment of ATRX to the virus-induced foci (data not shown). If the recruitment of ATRX to sites associated with viral genomes has functional significance, these results imply that hDaxx may be an important factor in regulating the functions of ATRX. This hypothesis forms the basis of the remainder of this paper.
FIG. 3.

ATRX is not recruited to sites associated with ICP0-null mutant HSV-1 genomes in hDaxx-depleted HepaRG cells. (A) hDaxx depletion. Cell extracts harvested from normal HepaRG, shLuci-expressing HALL, and shDaxx-expressing HALD2 cells and were resolved on a 7.5% polyacrylamide gel, and hDaxx expression was detected by Western blotting. Actin is a loading control. (B) Confocal immunofluorescence images demonstrating simultaneous detection of hDaxx, ATRX, and PML in HALL and HALD2 cells, using rabbit anti-Daxx (07-471), mouse anti-PML (5E10), and goat anti-ATRX (D19) antibodies. The secondary antibodies were FITC-conjugated anti-mouse, Alexa 647-conjugated anti-rabbit, and Alexa 555-conjugated anti-goat IgGs. (C) Immunofluorescence analysis of ATRX cells in developing ICP0-null mutant HSV-1 plaques in HALL and HALD2 cells. Cells were infected at an MOI of 0.5 (HALL) or 0.1 (HALD2) and costained the following day for ATRX (H300, anti-rabbit Alexa 488) and ICP4 (58S, anti-mouse Cy3).
Reconstitution of hDaxx expression in hDaxx-depleted cells.
In order to confirm the importance of hDaxx in ATRX recruitment to sites associated with HSV-1 genomes, we reintroduced hDaxx expression into HALD2 cells by constructing a lentiviral vector that encodes EYFP-tagged hDaxx (Fig. 4A). The hDaxx mRNA transcribed from this vector was modified to contain silent mutations that render it noncomplementary to the shRNAs expressed in the hDaxx-depleted cells. To allow stable expression of the EYFP-hDaxx fusion protein, the vector included a neomycin resistance gene, and the resulting cell lines were maintained in double selection of puromycin (for the original shRNA vector) and neomycin (see Table 2). The other key feature of the vector system is the use of a weak promoter (that of the HSV-1 glycoprotein gD gene) to avoid excessive expression of the reintroduced protein. Cells expressing EYFP-hDaxx fusion proteins were enriched by FACS to establish lines in which close to 100% of the cells expressed the reintroduced proteins. Control cell lines expressing EYFP only were also produced to control for any effects of EYFP expression on the subsequent assays. Western blot analysis showed that the reintroduced hDaxx fusion protein was expressed at close-to-endogenous levels (Fig. 4B), with the EYFP-hDaxx band (approximately 140 kDa) corresponding to the size of endogenous hDaxx plus the EYFP tag of approximately 25 kDa. Cell line HL-ED expressed both endogenous hDaxx and reintroduced EYFP-hDaxx but was not used in any further experiments. Cell line HD-E (HepaRG cells expressing shDaxx2 and EYFP) was used as a control for hDaxx depletion.
FIG. 4.
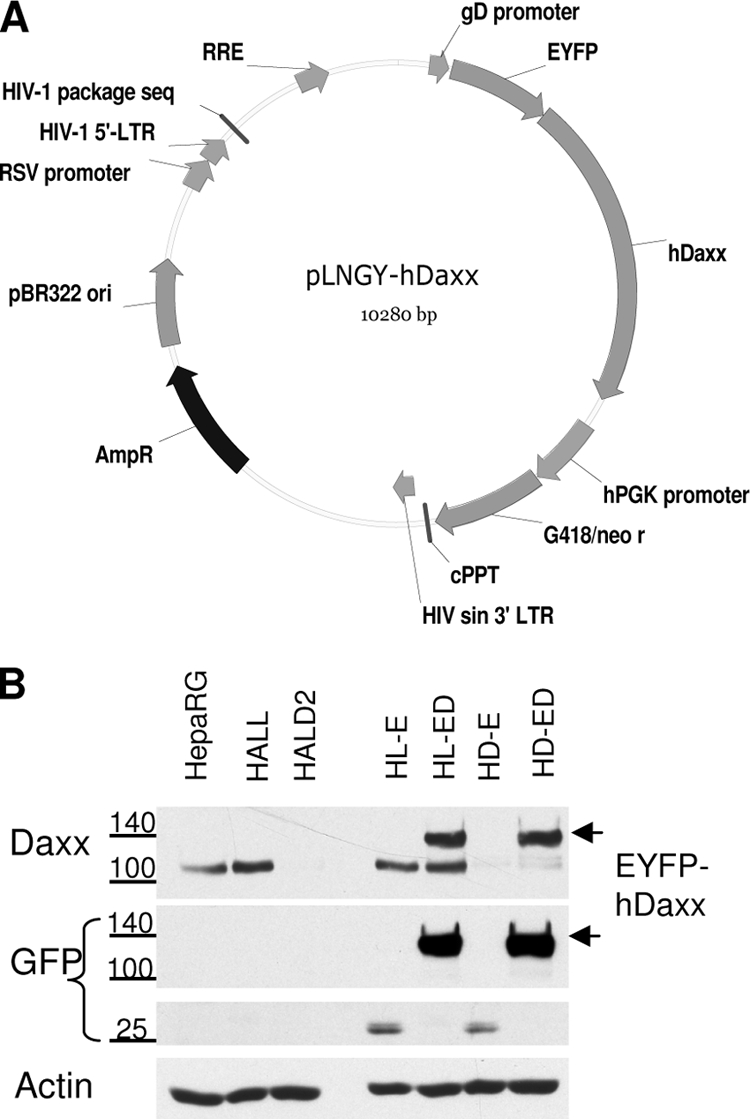
Reintroduction of hDaxx into hDaxx-depleted cells. (A) Map of the pLNGY-hDaxx vector construct (Table 1). RRE, rev response element; hPGK promoter, human phosphoglycerate kinase promoter; cPPT, polypurine tract; LTR, long terminal repeats; sin, self-inactivating; RSV promoter, Rous sarcoma virus promoter; AmpR, ampicillin resistance gene; G418/neo, neomycin resistance gene. (B) Western blot analysis of hDaxx expression in the generated cell lines. The reintroduced fusion protein EYFP-hDaxx in HD-ED cells is indicated by arrowheads on the right. Antibodies used for detection of the proteins are indicated on the left: hDaxx and EYFP expression levels were detected by anti-hDaxx antibody (D7810) and anti-GFP antibody (ab290), respectively. The positions of relevant size markers are indicated.
Immunofluorescence analysis of HD-ED cells demonstrated that reintroduced wt EYFP-hDaxx colocalizes with PML at ND10 (Fig. 5, row F) and, more importantly, relocalizes ATRX into these structures (Fig. 5, row C). In HD-E cells, however, the pattern of ATRX dispersal is consistent with that seen in HALD2 cells, expressing shDaxx only (Fig. 5, row B). Cells were also stained with anti-hDaxx antibody to confirm depletion of endogenous hDaxx and expression of the reintroduced fusion protein (Fig. 5, rows E and F). Figure 5, rows A and D, show control examples of the parent HepaRG cells.
FIG. 5.

Reintroduced hDaxx relocates ATRX to ND10 and to sites associated with incoming ICP0-null mutant HSV-1 genomes. Immunofluorescence analysis of PML, ATRX, and hDaxx distribution in uninfected (rows A to F) and ICP0-null HSV-1-infected (rows G to I) HepaRG, HD-E, and HD-ED cells (as indicated on the relevant panels). Cells on coverslips were costained for PML (r8) and ATRX (39F) (A to C) or PML (5E10) and hDaxx (07-471) (D to F). For analysis of the redistribution of the proteins during ICP0-null mutant virus infection, cells were infected at an MOI of 0.1 (HD-E) or 0.5 (HepaRG and HD-ED) and processed for immunofluorescence analysis the following day. Cells were costained for ICP4 (58S) and ATRX (H300). The secondary antibodies used throughout the experiment were Cy5-conjugated anti-rabbit (blue) and Alexa 555-conjugated anti-mouse (red) IgGs. Green fluorescence resulted from the autofluorescence of the EYFP tag.
During infection of hDaxx-reconstituted cells (HD-ED) with ICP0-null mutant HSV-1, ATRX recruitment to the sites associated with viral genomes and early replication compartments was restored to a pattern similar to that in infected HepaRG cells (Fig. 5, rows G and I). ATRX remained dispersed in HD-E cells infected with ICP0-null mutant virus (Fig. 5, row H), consistent with data shown in Fig. 3C. Expression of EYFP-hDaxx did not affect recruitment of PML in the ICP0-null mutant virus-infected cells (data not shown). These observations confirm the requirement of hDaxx for ATRX recruitment to sites associated with HSV-1 genomes at the early stages of infection.
Depletion of ATRX or hDaxx supports increased efficiency of ICP0-null mutant HSV-1 infection.
Previously, we found that depletion of either PML or Sp100 improves the efficiency of ICP0-null mutant HSV-1 infection and that simultaneous depletion of both improves infection yet further. Both proteins are recruited to sites closely associated with ICP0-null mutant HSV-1 genomes at the early stages of infection, which correlates with reduced gene expression (28, 30). By analogy with these findings, we set out to test whether recruitment of ATRX and hDaxx may also contribute to cell-mediated repression of ICP0-null HSV-1 gene expression, particularly since ATRX and hDaxx are components of a chromatin-associated complex with repressive properties. In addition to the hDaxx-depleted cell line described above, we generated ATRX-depleted HepaRG cells, achieving a greater efficiency of depletion and a lower rate of recovery of ATRX expression than in our previous study using HFs (65). Depletion of both major and truncated isoforms of ATRX in HAA cells was confirmed by Western blotting (Fig. 6A).
FIG. 6.

Efficiency of HSV-1 infection in ATRX-depleted cells. (A) Detection of ATRX expression by Western blot analysis using 39F antibody, showing expression of both ATRX isoforms in HALL cells and barely detectable levels of expression in HAA cells. Samples were resolved on a 6% polyacrylamide gel. USP7 was used as a loading control. (B) Kinetics of viral protein expression in ATRX-depleted cells. HALL and HAA cells were infected with wt or ICP0-null HSV-1 at an MOI of 2, and samples were harvested for Western blot analysis at 0, 4, 6, and 8 h postinfection (hpi). Samples were resolved on a 7.5% polyacrylamide gel, and membranes were probed for ICP4 and UL42, using antibodies 58S and Z1F11, respectively. Actin was a loading control. (C) Efficiency of plaque formation of HSV-1 in ATRX-depleted cells. Cells were infected with wt HSV-1 strain in1863 and ICP0-null HSV-1 strain dl1403/CMVlacZ at sequential dilutions. After 24 h of infection, cells were fixed and stained for β-galactosidase expression. The plots represent the mean relative efficiencies of plaque formation as the numbers of plaques determined to occur in HAA cells in relation to the level for HALL cells from three independent experiments. Error bars represent standard error values.
The effect of depleting either ATRX or hDaxx on the efficiency of HSV-1 infection was investigated by measuring viral gene expression and plaque-forming efficiency. Depletion of neither ATRX nor hDaxx affected the expression of typical IE and early proteins (ICP4 and UL42, respectively) during wt HSV-1 infection (Fig. 6B, top panel, and 7A). When the cells were infected with ICP0-null mutant HSV-1, however, ATRX depletion resulted in a slight but reproducible increase in UL42 expression (Fig. 6B, bottom panel). Depletion of hDaxx resulted in a more prominent increase in UL42 expression in ICP0-null HSV-1 infections (Fig. 7B). The increases in UL42 expression were always much more substantial than any effect on ICP4 in both ATRX- and hDaxx-depleted cells. This may reflect the fact that ICP4 expression levels are higher in ICP0-null mutant infections than might be expected on the basis of the plaque-forming defect (7, 19).
FIG. 7.

Efficiency of HSV-1 infection in hDaxx-depleted cells. (A and B) Kinetics of ICP4 and UL42 expression in hDaxx-depleted and hDaxx-reconstituted cells. HepaRG, HALL, HD-E, and HD-ED cells were infected with wt (A) or ICP0-null mutant (B) HSV-1 at an MOI of 2, and samples were harvested for Western blot analysis at 0, 4, 6, and 8 h postinfection (hpi). Samples were resolved on a 7.5% polyacrylamide gel, and membranes were probed for ICP4, UL42, and hDaxx using antibodies 58S, Z1F11, and D7810, respectively. Actin was a loading control. (C and D) Efficiency of plaque formation of HSV-1 in hDaxx-depleted and hDaxx-reconstituted cells. Cells seeded into 24-well plates were infected with wt HSV-1 strain in1863 (C) or ICP0-null HSV-1 dl1403/CMVlacZ (D) at sequential dilutions and stained for β-galactosidase expression 24 h later. The plots represent mean values for plaque numbers relative to those in HepaRG cells, obtained from 4 independent experiments. Error bars represent standard error values.
Depletion of neither ATRX nor hDaxx had any substantial effect on wt HSV-1 plaque formation (Fig. 6C and 7C). In contrast, we observed 5- to 10-fold increases in ICP0-null mutant plaque formation in ATRX- and hDaxx-depleted cells, compared to the levels for control HALL and naïve HepaRG cells (Fig. 6D and 7D). Similar results were obtained using HALD2 cells that express shDaxx2 only (data not shown), implying that EYFP expression in HD-E cells did not interfere with the assay. Importantly, these increases in ICP0-null mutant virus gene expression and plaque formation in hDaxx-depleted cells were reversed upon reintroduction of the wt EYFP-hDaxx fusion protein (Fig. 7B and D), providing strong evidence for a role for hDaxx in regulating ICP0-null mutant HSV-1 infection.
The ATRX interaction region of hDaxx is required for fully efficient repression of ICP0-null mutant HSV-1 infection and gene expression.
With the above data summarized, it has been shown that (i) both ATRX and hDaxx contribute to efficient repression of ICP0-null mutant HSV-1 infection and (ii) hDaxx is required for recruitment of ATRX to ICP0-null mutant HSV-1 genomes, but not vice versa. These observations prompted the question as to whether ATRX recruitment is dependent on its interaction with hDaxx. Tang et al. (94) identified an hDaxx domain that is essential for its interaction with ATRX, called PAH1 (paired amphipathic helices) (Fig. 8A). Using the lentiviral expression system, we constructed HD.ΔPAH cells that are depleted of endogenous hDaxx and express an EYFP-hDaxx fusion protein that lacks 73 amino acids spanning the PAH1 domain. Western blot analysis indicated that the expressed EYFP-hDaxx.ΔPAH1 fusion protein was smaller than EYFP-hDaxx present in HD-ED cells and was expressed at levels similar to those of the endogenous hDaxx protein (Fig. 8B).
FIG. 8.

Deletion of the ATRX interaction domain of hDaxx. (A) Schematic representation of wt hDaxx (based on that in reference 102), the EYFP-hDaxx construct, and the EYFP-hDaxxΔPAH1 mutant, from which amino acids 48 to 120 spanning the ATRX interacting domain PAH1 were removed by PCR-based splicing mutagenesis. PAH, paired amphipathic helices; D/E, Asp/Glu-rich motif; S/P/T, Ser/Pro/Thr-rich domain. (B) Western blot analysis of expression of hDaxx proteins in HepaRG, HALL, HD-E, HD-ED, and HD.ΔPAH cells. The membrane was probed with the indicated antibodies. Samples were resolved on a 7.5% polyacrylamide gel. (C) Immunoprecipitation analysis of ATRX and hDaxx interaction in HD-ED cells and HD.ΔPAH cells. Lanes 1 and 6 represent 2.5% input of the total extract sample incubated in each immunoprecipitation reaction. HD-ED and HD.ΔPAH cells extracts were immunoprecipitated with either anti-ATRX (antibody H300; lanes 4 and 9), anti-Daxx (antibody 07-471; lanes 5 and 10), control rabbit serum 201 raised against USP7 (lanes 3 and 8), or no antibody (lanes 2 and 7). Samples were analyzed on a 6% polyacrylamide gel and probed with anti-ATRX 39F and anti-hDaxx D7810 antibodies.
Coimmunoprecipitation (Co-IP) assays were conducted to test whether the ΔPAH1 deletion had successfully eliminated the interaction between ATRX and hDaxx (Fig. 8C). Bands corresponding to both ATRX and hDaxx were detected in immune precipitates prepared from extracts of HD-ED cells using either anti-ATRX or anti-Daxx antibodies, respectively (Fig. 8C, lanes 4 and 5), confirming the interaction between the two proteins. However, the reciprocal coimmune precipitation was not observed when extracts from HD.ΔPAH were used, although both proteins were precipitated independently by their respective antibodies (lanes 9 and 10). This result was reproducible and indicated that the interaction between ATRX and hDaxx in HD.ΔPAH cells had been abolished. Co-IP of the ATRX/hDaxx complex using the anti-Daxx antibody (lane 5) was not as efficient as that with the anti-ATRX antibody (lane 4), and this efficiency varied slightly among independent experiments (Fig. 12). An anti-USP7 rabbit serum was used as an irrelevant antibody control in Co-IP reactions (Fig. 8C, lanes 3 and 8). An interaction between hDaxx and USP7 reported previously (93) was not detected in our Co-IP assays.
FIG. 12.

ICP0 neither interacts with nor disrupts the ATRX/hDaxx complex. HepaRG cells were infected with wt HSV-1 at an MOI of 5, and cells were harvested in IP lysis buffer at 4 h postinfection. Immunoprecipitation was carried out using no antibody or anti-ICP0 190, anti-ATRX H300, anti-Daxx 07-471, and anti-USP7 210 antibodies, as indicated. Input lanes contained 2.5% of the sample used for immunoprecipitation. Western blot analysis was carried out using mouse anti-ATRX 39F, anti-Daxx D7810, and anti-ICP0 11060 antibodies.
Immunofluorescence analysis of uninfected HD.ΔPAH cells (Fig. 9A and B) revealed that the ATRX interaction domain of hDaxx is required for ATRX localization to ND10 (consistent with previous reports) (49, 94). As with its PAH1-dependent localization to ND10 in uninfected cells, ATRX remained dispersed in HD.ΔPAH cells that had been infected with ICP0-null mutant HSV-1, even though the mutant hDaxx protein was efficiently recruited to sites associated with viral genomes (Fig. 9C and D). These results confirm that the interaction between ATRX and hDaxx is required for ATRX recruitment viral genome-associated sites and identify the PAH1 domain as being important for this process.
FIG. 9.

The ATRX interaction domain of hDaxx is essential for ATRX localization to ND10 and recruitment to HSV-1 early replication foci in the absence of ICP0. Panels A and B show uninfected HD.ΔPAH cells stained for the indicated proteins. Panels C and D show HD.ΔPAH cells located at the periphery of a developing plaque after infection with ICP0-null mutant HSV-1 at an MOI of 0.5 PFU/cell and after a 24-h infection period. ATRX, hDaxxΔPAH, PML, and ICP4 were detected with antibodies H300, 07-471, 5E10, and 58S, respectively.
We then analyzed the plaque-forming efficiency and protein expression levels of ICP0-null mutant virus in HD.ΔPAH cells, in comparison with the levels for hDaxx-reintroduced and hDaxx-depleted cells (Fig. 10). Deletion of the hDaxx-ATRX interaction domain resulted in increased plaque-forming efficiency in ICP0-null virus and increases in ICP4 and UL42 production levels, which were comparable to those observed in hDaxx-depleted cells (Fig. 10B). As expected, wt HSV-1 gene expression and plaque formation were identical in this set of cells (Fig. 10A and C). The increase in ICP0-null mutant plaque formation in hDaxx-depleted HD-E cells could be reversed by reintroduction of wt hDaxx in HD-ED cells but not by introduction of ATRX binding deficient hDaxx in HD.ΔPAH cells (Fig. 10D). Hence, a functional ATRX/hDaxx complex is required for their role in the repression of ICP0-null mutant HSV-1 infection. This requirement is likely to be related to the chromatin-associated functions of the ATRX/hDaxx complex.
FIG. 10.

The ATRX interaction region of hDaxx is required for fully efficient hDaxx-mediated repression of ICP0-null mutant HSV-1 gene expression (A and B) and plaque formation (C and D). (A and B) Cells were infected with the indicated viruses at an MOI of 2.0 PFU/cell, and samples were harvested at the indicated time points after infection. Samples were resolved on a 7.5% polyacrylamide gel. Membranes were probed for the expression of ICP4, UL42, and hDaxx using antibodies 58S, Z1F11, and D7810, respectively. Actin was a loading control. (C and D) Cells seeded into 24-well plates were infected with wt HSV-1 strain in1863 (C) or ICP0-null HSV-1 dl1403/CMVlacZ (D) at sequential dilutions and stained for β-galactosidase activity the following day to reveal the plaques. The plots represent mean values for plaque numbers relative to those in HepaRG cells, obtained from 3 independent experiments. Error bars represent standard error values.
The efficiency of establishment of HSV-1 quiescence is influenced by hDaxx and its interaction with ATRX.
ICP0 efficiently stimulates reactivation or derepression of HSV-1 in cultured cell models of quiescent infection (39, 79, 81, 88, 96); hence, in the absence of ICP0, the virus has a higher probability of establishing a quiescent infection. In view of the roles of ATRX and hDaxx in the repression of ICP0-null HSV-1 lytic infection, we investigated whether the ATRX/hDaxx complex influences the efficiency of establishment of quiescent infections. These assays use virus strain in1374, an ICP0-mutant HSV-1 with a temperature sensitive mutation within ICP4, a mutation within the VP16 activation domain, and a marker β-galactosidase gene (80). Expression from in1374 genomes is rapidly repressed even after infection at a relatively high MOI, allowing the production of cell cultures in which a high proportion of cells maintain in1374 genomes in a repressed state at 18 to 24 h after infection. The proportion of virus genome-positive cells can be assessed by coinfection with HSV-1 strain tsK, which expresses ICP0 and stimulates marker gene expression (80). These control samples indicated that the majority of cells in these populations had been infected with in1374 (data not shown). Changes in the establishment of quiescence in hDaxx-depleted and reconstituted cells were detected by the proportion of β-galactosidase-positive cells at 24 h after in1374 infection (Fig. 11A). Depletion of hDaxx resulted in a substantially increased number of β-galactosidase-positive cells compared to that in control HALL cells. This increase was completely reversed in wt hDaxx but not in EYFP-hDaxx.ΔPAH1 reconstituted cells (Fig. 11). These results indicate that a small but significant proportion of cells in which the hDaxx-ATRX interaction fails to occur have a reduced capacity to repress in1374 gene expression. Therefore, the ATRX/hDaxx complex may have a role in the efficiency of establishment of a quiescent HSV-1 infection in a cultured cell model.
FIG. 11.

Establishment of quiescent infections in hDaxx-depleted, EYFP-hDaxx-reconstituted, and EYFP-hDaxxΔPAH1-introduced cells. Cells were infected with HSV-1 strain in1374, incubated at the restrictive temperature of 38.5°C overnight, and processed for β-galactosidase staining the following day. (A) Images of HALL, HD-E, HD-ED, and HD.ΔPAH cells at 24 h after infection with in1374. (B) Relative cell numbers of β-galactosidase-expressing cells in the above-mentioned cell types, compared to those obtained in parallel in1374-infected HepaRG cells. The plots represent mean values obtained from 3 independent experiments. Error bars are standard error values.
ICP0 does not disrupt the ATRX/hDaxx complex.
Previous evidence reported that ICP0 associates with histone deacetylase (HDAC) enzymes (64) and interacts with and disrupts the REST/CoREST/HDAC1 chromatin repressor complex (34). By analogy with the above-mentioned model, we therefore considered whether dispersal of ATRX and hDaxx during the early stages of wt HSV-1 infection could be due to ICP0-mediated disruption of the ATRX/hDaxx complex. However, Co-IP assays revealed no differences in the efficiency of ATRX/hDaxx Co-IP from extracts of cells infected with wt HSV-1 for 4 h (Fig. 12), a time at which both proteins are completely dispersed from ND10 (data not shown). ICP0 was not detected in the immune precipitates, whereas the interaction between ICP0 and USP7 was successfully detected. These data indicate that HSV-1 ICP0 interacts directly with neither ATRX nor hDaxx and that it does not affect their functionalities by disrupting the complex between them.
Evidence for increased phosphorylation of hDaxx during HSV-1 infection.
If ICP0 does not disrupt the ATRX/hDaxx complex, a different mechanism by which ICP0 overcomes their repressive properties must be utilized. During the course of the experiments represented in Fig. 7, it was noted that hDaxx is modified during HSV-1 infection, seemingly in an ICP0-dependent fashion. We analyzed hDaxx expression by Western blotting over 10 h of infection with wt or ICP0-null mutant viruses at an MOI of 5. As the wt virus infection progressed from the 3-h time point onwards, the lower band of the hDaxx doublet decreased in mobility and merged with the higher-band isoforms (Fig. 13A). This shift did not occur in the parallel ICP0-null mutant virus infection, even at late times in this experiment when UL42 expression was abundant (Fig. 13B). A similar pattern of Daxx migration was observed in previous studies when HIPK1, which is responsible for Daxx phosphorylation in mouse cells, was overexpressed (16). Therefore, we investigated whether the hDaxx band shift during wt virus infection is a result of hyperphosphorylation. Phosphatase treatment of mock-infected samples resulted in loss on the slower-mobility bands and an increase in the intensity of the highest-mobility form, suggesting that hDaxx is normally subject to phosphorylation (Fig. 13C and D). Analysis of the wt-HSV-1-infected samples at an MOI of 5.0 harvested at 4 and 7 h postinfection indicated that the infection-specific band shift was also due to phosphorylation (Fig. 13C) since phosphatase treatment of aliquots of the same infected samples caused loss of the lower-mobility forms and the appearance of a single, more intense band of higher mobility (Fig. 13C). The ICP0-null-mutant-infected samples gave results that were indistinguishable from those given by the mock-infected samples in this assay (Fig. 13D).
FIG. 13.

Phosphorylation of hDaxx during wt HSV-1 infection. (A and B) Cells were seeded into 24-well plates at a density of 1 × 105 cells per well and infected with wt (A and C) or ICP0-null (B and D) HSV-1 at an MOI of 5. Samples were harvested at the indicated time points (A and B) and resolved on 7.5% gels for ICP4, UL42, and actin and on 6% gels for hDaxx. For panels C and D, samples were harvested in IP lysis buffer (without EDTA), and then aliquots of the same samples were mock treated (left-hand sets of 3 lanes) or treated with λ-phosphatase (right-hand sets of 3 lanes) prior to Western blot analysis. M, mock.
We observed some variability in the gel migration of hDaxx and its modified forms in replicate experiments (compare Fig. 13A and C). In some experiments, the higher-mobility forms decreased in intensity as the wt HSV-1 infection progressed, with a concomitant increase in a lower-mobility band (Fig. 13A), while in others, all the hDaxx bands decreased in intensity, resulting in a smear of reduced mobility forms (Fig. 13C). We interpret these differences as an issue of resolution of the phosphorylated forms on individual gels, and where the bands become less intense, it is because of multiple modified species smearing up the gel. It is not due to any loss of hDaxx, because the same sample after phosphatase treatment gives a single intense band of higher mobility (Fig. 13C).
These results imply that hDaxx function may be regulated by phosphorylation during HSV-1 infection. Since there is no evidence that ICP0 is capable of directly phosphorylating substrates, we suggest that the increased phosphorylation of hDaxx is an indirect consequence of ICP0 expression.
DISCUSSION
A concept has emerged in recent years that cells constitutively express a number of proteins that, in addition to their normal roles in cellular pathways, have properties that inhibit virus infection. This process has been dubbed intrinsic antiviral resistance or intrinsic defense, and in the cases of HCMV and HSV-1, this relates to cell-mediated repression of viral gene expression. In turn, the viruses encode functions that counteract this defense in order to ensure efficient initiation of lytic infection. One arm of cellular intrinsic antiviral resistance is provided by components of ND10 structures within the infected cell nucleus. ICP0-induced degradation of the core ND10 component PML and selected isoforms of Sp100 (12, 21) provides a means of inactivating one aspect of cellular intrinsic resistance to HSV-1 infection. Both PML and Sp100 have been demonstrated to contribute to intrinsic resistance to HSV-1 infection (28, 30, 57, 73), but prior to this study, the effects of hDaxx and ATRX on HSV-1 infection had not been investigated in any detail. We found that both proteins are rapidly recruited to sites associated with incoming HSV-1 genomes and that depletion of either of them increases the efficiency of ICP0-null mutant HSV-1 infection. This increase can be reversed by reintroduction of wt hDaxx but not by a mutant unable to interact with ATRX. On the basis of these findings, we propose that the ATRX/hDaxx complex takes part in intrinsic cellular defense against HSV-1 infection. The possible mechanisms by which this might occur are discussed below.
Since hDaxx interacts with HDAC II (42, 60), it is attractive to suggest that hDaxx influences the efficiency of transcription from parental HSV-1 genomes via HDAC-dependent chromatin remodeling, especially as inhibition of HDACs by trichostatin A (TSA) decreases hDaxx transcriptional repressor activity (60). This model is supported by analogy with the role of hDaxx in regulating HCMV infection. The HCMV tegument protein pp71 interacts with hDaxx (41, 48) and in some reports induces its degradation (45, 86). Since repression of IE gene expression of pp71-null mutant virus can be overcome by RNAi-mediated depletion of hDaxx, it follows that a major role of pp71 is to overcome the repressive effects of hDaxx (9, 82, 86, 98). Repression of HCMV gene expression in the absence of pp71 can also be overcome by TSA treatment (86), while hDaxx-mediated repression of the HCMV IE promoter correlates with assembly of a chromatin structure with decreased levels of histone acetylation (98). Taken together, the data are consistent with a model that, in the case of HCMV, invokes HDAC activity as an important component of hDaxx-mediated intrinsic cellular resistance to infection.
There is conflicting evidence concerning the role of HDAC-related mechanisms in regulating ICP0-null mutant gene expression. The role of chromatin-related mechanisms in regulating HSV-1 infection at various levels has been reviewed recently (53, 56), so for simplicity, the following discussion relates only to the specific instance of ICP0 activity during the early stages of lytic infection of cultured cells. In favor of a central role for HDACs in repressing ICP0-null mutant HSV-1 gene expression are the following pieces of evidence: (i) HDAC inhibitors can improve ICP0-null mutant replication in some cell lines (78, 96), (ii) ICP0 interacts with a number of HDAC enzymes when both are highly expressed (64), (iii) there is increased assembly of nucleosomes containing deacetylated histones on the ICP0-null mutant compared to the level for wt HSV-1 genomes (13), and (iv) ICP0 interacts with CoREST and dissociates HDAC1/2 from the REST/CoREST/HDAC1/2 chromatin repressor complex (34-36). On the other hand, a number of observations are not consistent with a simple hypothesis invoking HDAC activity as a dominant factor in regulating ICP0-null mutant infections. These include the following: (i) HDAC inhibitors do not improve ICP0-null mutant replication in HFs or HepaRG cells (28); (ii) unlike TSA, high-level ICP0 expression does not lead to a general increase in acetylated histone H4 (64); and (iii) the proposed interaction of ICP0 with the CoREST complex is not essential for significant ICP0 function in lytic and derepression assays (29). The first of these pieces of evidence, in particular, is inconsistent with the hypothesis that repression of ICP0-null mutant replication by hDaxx and ATRX involves an absolute requirement for HDAC activity. Therefore, alternative possibilities must be considered.
Alternative models for hDaxx-mediated repression have been proposed in other systems. These mechanisms include sequestration of SUMO-modified transcriptional activators through the hDaxx SUMO interaction motif (89) and interaction with DNA methyltransferase DNMT1 leading to methylation of certain target promoters (83). These mechanisms seem unlikely in the case of HSV-1 infection because, in the first case, hDaxx appears to be acting at the sites of viral genomes rather than sequestering other factors at remote sites and, in the second case, there is no evidence of regulation of HSV-1 through DNA methylation (54). An attractive model for repression mediated by hDaxx and ATRX stems from their interaction and presence in a chromatin-remodeling complex (99). The complex has ATPase activity that can be stimulated by DNA or nucleosomes, and it alters nucleosome structure at a nucleosome entry site in an in vitro system, without altering nucleosome phasing (99). ATRX includes near its N terminus a so-called ADD domain that contains a GATA-like zinc finger and a PHD finger, which is also present in certain DNA methyltransferases (2). Many of the defects that cause the ATR-X syndrome are within the ADD domain (2). In the DNA methyltransferase examples, the ADD domain has been shown to interact with unmodified histone H3 tails (75), providing a mechanism (by analogy) by which ATRX might interact with chromatin. The mouse homologue of ATRX was found to interact with HP1 via its PHD finger domain (59), and indeed, ATRX localizes to heterochromatic foci (49), possibly by associating with HP1 (68) or methyl-CpG-binding protein MeCP2 (72). Although speculative, the above information opens the possibility that the ATRX/hDaxx complex could associate with any nucleosomes that are assembled on viral DNA and allow recruitment of other proteins to create a repressive environment. This model is compatible with the relatively low density of nucleosomes found on HSV-1 genomes (13, 74), whereas HDAC-mediated repression models imply compaction of chromatin with a high nucleosome density.
Whatever the mechanism of hDaxx/ATRX-mediated repression, ICP0 must in some way inactivate it. ICP0 completely inhibits the recruitment of the two proteins to sites associated with incoming HSV-1 genomes, and this may provide a primary method of overcoming their repressive effects, especially in view of the correlation between ICP0 biological activity and the ability of ICP0 to inhibit recruitment of these and other ND10 proteins (29). However, the observation that hDaxx becomes hyperphosphorylated during wt HSV-1 infection of HepaRG cells (Fig. 13) provides another potential mechanism for inactivation of the repressive activities of hDaxx. It may be relevant that the mouse homologue of hDaxx is a direct substrate for phosphorylation by HIPK1, resulting in a reduction in Daxx-mediated repression and, intriguingly, the displacement of Daxx from ND10 (16). Furthermore, it is the nonphosphorylated form of hDaxx that is found in complex with HDAC II and core histones (42) and also with the transcription factor Pax3 (a complex which results in the repression of Pax3-stimulated genes) (43). We have no evidence that ICP0 is involved directly in hDaxx phosphorylation, but this phosphorylation could be an indirect consequence of ICP0-induced disruption of ND10. It would be of interest to define the kinases that are responsible for hDaxx phosphorylation during wt HSV-1 infection and to investigate the consequence of their inhibition.
In conclusion, we report here that both hDaxx and ATRX contribute to the repression of HSV-1 gene expression and infection that occurs in the absence of ICP0 and that the domain of hDaxx that interacts with ATRX is required for hDaxx-mediated repression. These observations support the hypotheses that ND10 plays a role in intrinsic cellular resistance to HSV-1 infection and that multiple ND10 components, perhaps using distinct pathways, are involved in this process.
Acknowledgments
This work was supported by a Medical Research Council studentship held by V.L. and through Medical Research Council funding of the laboratory of R.D.E.
We thank Chris Preston and Chris Boutell for helpful comments on the manuscript, Tom Gilbey for FACS service, and Richard Gibbons, John Sinclair, Philippe Gripon, and Roel van Driel for reagents.
Footnotes
Published ahead of print on 10 February 2010.
REFERENCES
- 1.Amin, H. M., S. Saeed, and S. Alkan. 2001. Histone deacetylase inhibitors induce caspase-dependent apoptosis and downregulation of daxx in acute promyelocytic leukaemia with t(15;17). Br. J. Haematol. 115:287-297. [DOI] [PubMed] [Google Scholar]
- 2.Argentaro, A., J. C. Yang, L. Chapman, M. S. Kowalczyk, R. J. Gibbons, D. R. Higgs, D. Neuhaus, and D. Rhodes. 2007. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc. Natl. Acad. Sci. U. S. A. 104:11939-11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumann, C., A. Schmidtmann, K. Muegge, and R. De La Fuente. 2008. Association of ATRX with pericentric heterochromatin and the Y chromosome of neonatal mouse spermatogonia. BMC Mol. Biol. 9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boddy, M. N., K. Howe, L. D. Etkin, E. Solomon, and P. S. Freemont. 1996. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 13:971-982. [PubMed] [Google Scholar]
- 5.Boutell, C., A. Orr, and R. D. Everett. 2003. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 77:8686-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai, W., and P. A. Schaffer. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 66:2904-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canning, M., C. Boutell, J. Parkinson, and R. D. Everett. 2004. A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279:38160-38168. [DOI] [PubMed] [Google Scholar]
- 9.Cantrell, S. R., and W. A. Bresnahan. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80:6188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantrell, S. R., and W. A. Bresnahan. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 79:7792-7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang, H. Y., H. Nishitoh, X. Yang, H. Ichijo, and D. Baltimore. 1998. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281:1860-1863. [DOI] [PubMed] [Google Scholar]
- 12.Chelbi-Alix, M. K., and H. de The. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935-941. [DOI] [PubMed] [Google Scholar]
- 13.Cliffe, A. R., and D. M. Knipe. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82:12030-12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham, A. L., R. J. Diefenbach, M. Miranda-Saksena, L. Bosnjak, M. Kim, C. Jones, and M. W. Douglas. 2006. The cycle of human herpes simplex virus infection: virus transport and immune control. J. Infect. Dis. 194(Suppl. 1):S11-S18. [DOI] [PubMed] [Google Scholar]
- 15.De La Fuente, R., M. M. Viveiros, K. Wigglesworth, and J. J. Eppig. 2004. ATRX, a member of the SNF2 family of helicase/ATPases, is required for chromosome alignment and meiotic spindle organization in metaphase II stage mouse oocytes. Dev. Biol. 272:1-14. [DOI] [PubMed] [Google Scholar]
- 16.Ecsedy, J. A., J. S. Michaelson, and P. Leder. 2003. Homeodomain-interacting protein kinase 1 modulates Daxx localization, phosphorylation, and transcriptional activity. Mol. Cell. Biol. 23:950-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Efstathiou, S., and C. M. Preston. 2005. Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res. 111:108-119. [DOI] [PubMed] [Google Scholar]
- 18.Everett, R. D. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761-770. [DOI] [PubMed] [Google Scholar]
- 19.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett, R. D., W. C. Earnshaw, J. Findlay, and P. Lomonte. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18:1526-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Everett, R. D., P. Freemont, H. Saitoh, M. Dasso, A. Orr, M. Kathoria, and J. Parkinson. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everett, R. D., and G. G. Maul. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Everett, R. D., M. Meredith, and A. Orr. 1999. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 73:417-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Everett, R. D., M. Meredith, A. Orr, A. Cross, M. Kathoria, and J. Parkinson. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16:1519-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everett, R. D., and J. Murray. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79:5078-5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Everett, R. D., J. Murray, A. Orr, and C. M. Preston. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81:10991-11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Everett, R. D., A. Orr, and C. M. Preston. 1998. A viral activator of gene expression functions via the ubiquitin-proteasome pathway. EMBO J. 17:7161-7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everett, R. D., C. Parada, P. Gripon, H. Sirma, and A. Orr. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82:2661-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everett, R. D., M. L. Parsy, and A. Orr. 2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol. 83:4963-4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everett, R. D., S. Rechter, P. Papior, N. Tavalai, T. Stamminger, and A. Orr. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett, R. D., and A. Zafiropoulos. 2004. Visualization by live-cell microscopy of disruption of ND10 during herpes simplex virus type 1 infection. J. Virol. 78:11411-11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibbons, R. J., D. J. Picketts, L. Villard, and D. R. Higgs. 1995. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 80:837-845. [DOI] [PubMed] [Google Scholar]
- 33.Gripon, P., S. Rumin, S. Urban, J. Le Seyec, D. Glaise, I. Cannie, C. Guyomard, J. Lucas, C. Trepo, and C. Guguen-Guillouzo. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 99:15655-15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu, H., Y. Liang, G. Mandel, and B. Roizman. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:7571-7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu, H., and B. Roizman. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134-17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu, H., and B. Roizman. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 83:181-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 75:3240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris, R. A., R. D. Everett, X. X. Zhu, S. Silverstein, and C. M. Preston. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 63:3513-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heckman, K. L., and L. R. Pease. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2:924-932. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann, H., H. Sindre, and T. Stamminger. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollenbach, A. D., C. J. McPherson, E. J. Mientjes, R. Iyengar, and G. Grosveld. 2002. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 115:3319-3330. [DOI] [PubMed] [Google Scholar]
- 43.Hollenbach, A. D., J. E. Sublett, C. J. McPherson, and G. Grosveld. 1999. The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. EMBO J. 18:3702-3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmstrom, S. R., S. Chupreta, A. Y. So, and J. A. Iniguez-Lluhi. 2008. SUMO-mediated inhibition of glucocorticoid receptor synergistic activity depends on stable assembly at the promoter but not on DAXX. Mol. Endocrinol. 22:2061-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang, J., and R. F. Kalejta. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334-338. [DOI] [PubMed] [Google Scholar]
- 46.Ishov, A. M., and G. G. Maul. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134:815-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishov, A. M., A. G. Sotnikov, D. Negorev, O. V. Vladimirova, N. Neff, T. Kamitani, E. T. Yeh, J. F. Strauss III, and G. G. Maul. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147:221-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishov, A. M., O. V. Vladimirova, and G. G. Maul. 2002. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 76:7705-7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishov, A. M., O. V. Vladimirova, and G. G. Maul. 2004. Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J. Cell Sci. 117:3807-3820. [DOI] [PubMed] [Google Scholar]
- 50.Jamieson, D. R., L. H. Robinson, J. I. Daksis, M. J. Nicholl, and C. M. Preston. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J. Gen. Virol. 76:1417-1431. [DOI] [PubMed] [Google Scholar]
- 51.Jang, M. S., S. W. Ryu, and E. Kim. 2002. Modification of Daxx by small ubiquitin-related modifier-1. Biochem. Biophys. Res. Commun. 295:495-500. [DOI] [PubMed] [Google Scholar]
- 52.Khelifi, A. F., M. S. D'Alcontres, and P. Salomoni. 2005. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 12:724-733. [DOI] [PubMed] [Google Scholar]
- 53.Knipe, D. M., and A. Cliffe. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6:211-221. [DOI] [PubMed] [Google Scholar]
- 54.Kubat, N. J., R. K. Tran, P. McAnany, and D. C. Bloom. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 78:1139-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuo, H. Y., C. C. Chang, J. C. Jeng, H. M. Hu, D. Y. Lin, G. G. Maul, R. P. Kwok, and H. M. Shih. 2005. SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of Daxx. Proc. Natl. Acad. Sci. U. S. A. 102:16973-16978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kutluay, S. B., and S. J. Triezenberg. 2009. Role of chromatin during herpesvirus infections. Biochim. Biophys. Acta 1790:456-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kyratsous, C. A., and S. J. Silverstein. 2009. Components of nuclear domain 10 bodies regulate varicella-zoster virus replication. J. Virol. 83:4262-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lechner, M. S., D. C. Schultz, D. Negorev, G. G. Maul, and F. J. Rauscher III. 2005. The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem. Biophys. Res. Commun. 331:929-937. [DOI] [PubMed] [Google Scholar]
- 59.Le Douarin, B., A. L. Nielsen, J. M. Garnier, H. Ichinose, F. Jeanmougin, R. Losson, and P. Chambon. 1996. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 15:6701-6715. [PMC free article] [PubMed] [Google Scholar]
- 60.Li, H., C. Leo, J. Zhu, X. Wu, J. O'Neil, E. J. Park, and J. D. Chen. 2000. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 20:1784-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li, R., H. Pei, D. K. Watson, and T. S. Papas. 2000. EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene 19:745-753. [DOI] [PubMed] [Google Scholar]
- 62.Lin, D. Y., H. I. Fang, A. H. Ma, Y. S. Huang, Y. S. Pu, G. Jenster, H. J. Kung, and H. M. Shih. 2004. Negative modulation of androgen receptor transcriptional activity by Daxx. Mol. Cell. Biol. 24:10529-10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin, D. Y., Y. S. Huang, J. C. Jeng, H. Y. Kuo, C. C. Chang, T. T. Chao, C. C. Ho, Y. C. Chen, T. P. Lin, H. I. Fang, C. C. Hung, C. S. Suen, M. J. Hwang, K. S. Chang, G. G. Maul, and H. M. Shih. 2006. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 24:341-354. [DOI] [PubMed] [Google Scholar]
- 64.Lomonte, P., J. Thomas, P. Texier, C. Caron, S. Khochbin, and A. L. Epstein. 2004. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 78:6744-6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lukashchuk, V., S. McFarlane, R. D. Everett, and C. M. Preston. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543-12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 74:2679-2690. [DOI] [PubMed] [Google Scholar]
- 67.Maul, G. G., A. M. Ishov, and R. D. Everett. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67-75. [DOI] [PubMed] [Google Scholar]
- 68.McDowell, T. L., R. J. Gibbons, H. Sutherland, D. M. O'Rourke, W. A. Bickmore, A. Pombo, H. Turley, K. Gatter, D. J. Picketts, V. J. Buckle, L. Chapman, D. Rhodes, and D. R. Higgs. 1999. Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc. Natl. Acad. Sci. U. S. A. 96:13983-13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meinecke, I., A. Cinski, A. Baier, M. A. Peters, B. Dankbar, A. Wille, A. Drynda, H. Mendoza, R. E. Gay, R. T. Hay, B. Ink, S. Gay, and T. Pap. 2007. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 104:5073-5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Michaelson, J. S., and P. Leder. 2003. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 116:345-352. [DOI] [PubMed] [Google Scholar]
- 71.Morozov, V. M., N. A. Massoll, O. V. Vladimirova, G. G. Maul, and A. M. Ishov. 2008. Regulation of c-met expression by transcription repressor Daxx. Oncogene 27:2177-2186. [DOI] [PubMed] [Google Scholar]
- 72.Nan, X., J. Hou, A. Maclean, J. Nasir, M. J. Lafuente, X. Shu, S. Kriaucionis, and A. Bird. 2007. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. U. S. A. 104:2709-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Negorev, D. G., O. V. Vladimirova, A. Ivanov, F. Rauscher III, and G. G. Maul. 2006. Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J. Virol. 80:8019-8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oh, J., and N. W. Fraser. 2008. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 82:3530-3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Otani, J., T. Nankumo, K. Arita, S. Inamoto, M. Ariyoshi, and M. Shirakawa. 2009. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 10:1235-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Picketts, D. J., D. R. Higgs, S. Bachoo, D. J. Blake, O. W. Quarrell, and R. J. Gibbons. 1996. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum. Mol. Genet. 5:1899-1907. [DOI] [PubMed] [Google Scholar]
- 77.Pluta, A. F., W. C. Earnshaw, and I. G. Goldberg. 1998. Interphase-specific association of intrinsic centromere protein CENP-C with HDaxx, a death domain-binding protein implicated in Fas-mediated cell death. J. Cell Sci. 111:2029-2041. [DOI] [PubMed] [Google Scholar]
- 78.Poon, A. P., Y. Liang, and B. Roizman. 2003. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J. Virol. 77:12671-12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Preston, C. M. 2007. Reactivation of expression from quiescent herpes simplex virus type 1 genomes in the absence of immediate-early protein ICP0. J. Virol. 81:11781-11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Preston, C. M., and M. J. Nicholl. 2005. Human cytomegalovirus tegument protein pp71 directs long-term gene expression from quiescent herpes simplex virus genomes. J. Virol. 79:525-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Preston, C. M., and M. J. Nicholl. 1997. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 71:7807-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Preston, C. M., and M. J. Nicholl. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 87:1113-1121. [DOI] [PubMed] [Google Scholar]
- 83.Puto, L. A., and J. C. Reed. 2008. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 22:998-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Russell, J., N. D. Stow, E. C. Stow, and C. M. Preston. 1987. Herpes simplex virus genes involved in latency in vitro. J. Gen. Virol. 68:3009-3018. [DOI] [PubMed] [Google Scholar]
- 85.Ryo, A., A. Hirai, M. Nishi, Y. C. Liou, K. Perrem, S. C. Lin, H. Hirano, S. W. Lee, and I. Aoki. 2007. A suppressive role of the prolyl isomerase Pin1 in cellular apoptosis mediated by the death-associated protein Daxx. J. Biol. Chem. 282:36671-36681. [DOI] [PubMed] [Google Scholar]
- 86.Saffert, R. T., and R. F. Kalejta. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863-3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saffert, R. T., and R. F. Kalejta. 2008. Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both? Future Virol. 3:265-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Samaniego, L. A., L. Neiderhiser, and N. A. DeLuca. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shih, H. M., C. C. Chang, H. Y. Kuo, and D. Y. Lin. 2007. Daxx mediates SUMO-dependent transcriptional control and subnuclear compartmentalization. Biochem. Soc. Trans. 35:1397-1400. [DOI] [PubMed] [Google Scholar]
- 90.Song, J. J., and Y. J. Lee. 2004. Tryptophan 621 and serine 667 residues of Daxx regulate its nuclear export during glucose deprivation. J. Biol. Chem. 279:30573-30578. [DOI] [PubMed] [Google Scholar]
- 91.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 92.Stuurman, N., A. de Graaf, A. Floore, A. Josso, B. Humbel, L. de Jong, and R. van Driel. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 101:773-784. [DOI] [PubMed] [Google Scholar]
- 93.Tang, J., L. K. Qu, J. Zhang, W. Wang, J. S. Michaelson, Y. Y. Degenhardt, W. S. El-Deiry, and X. Yang. 2006. Critical role for Daxx in regulating Mdm2. Nat. Cell Biol. 8:855-862. [DOI] [PubMed] [Google Scholar]
- 94.Tang, J., S. Wu, H. Liu, R. Stratt, O. G. Barak, R. Shiekhattar, D. J. Picketts, and X. Yang. 2004. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 279:20369-20377. [DOI] [PubMed] [Google Scholar]
- 95.Tavalai, N., and T. Stamminger. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 1783:2207-2221. [DOI] [PubMed] [Google Scholar]
- 96.Terry-Allison, T., C. A. Smith, and N. A. DeLuca. 2007. Relaxed repression of herpes simplex virus type 1 genomes in murine trigeminal neurons. J. Virol. 81:12394-12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thompson, R. L., and N. M. Sawtell. 2006. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J. Virol. 80:10919-10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woodhall, D. L., I. J. Groves, M. B. Reeves, G. Wilkinson, and J. H. Sinclair. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652-37660. [DOI] [PubMed] [Google Scholar]
- 99.Xue, Y., R. Gibbons, Z. Yan, D. Yang, T. L. McDowell, S. Sechi, J. Qin, S. Zhou, D. Higgs, and W. Wang. 2003. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. U. S. A. 100:10635-10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang, X., R. Khosravi-Far, H. Y. Chang, and D. Baltimore. 1997. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89:1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yao, F., and P. A. Schaffer. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yeung, P. L., L. Y. Chen, S. C. Tsai, A. Zhang, and J. D. Chen. 2008. Daxx contains two nuclear localization signals and interacts with importin alpha3. J. Cell. Biochem. 103:456-470. [DOI] [PubMed] [Google Scholar]
- 103.Zhong, S., S. Muller, S. Ronchetti, P. S. Freemont, A. Dejean, and P. P. Pandolfi. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748-2752. [PubMed] [Google Scholar]