

Abstract
Activation of the immune system by ANG II contributes to the pathogenesis of hypertension, and pharmacological suppression of lymphocyte responses can ameliorate hypertensive end-organ damage. Therefore, to examine the mechanisms through which lymphocytes mediate blood pressure elevation, we studied ANG II-dependent hypertension in scid mice lacking lymphocyte responses and wild-type controls. Scid mice had a blunted hypertensive response to chronic ANG II infusion and accordingly developed less cardiac hypertrophy. Moreover, lymphocyte deficiency led to significant reductions in heart and kidney injury following 4 wk of angiotensin. The muted hypertensive response in the scid mice was associated with increased sodium excretion, urine volumes, and weight loss beginning on day 5 of angiotensin infusion. To explore the mechanisms underlying alterations in blood pressure and renal sodium handling, we measured gene expression for vasoactive mediators in the kidney after 4 wk of ANG II administration. Scid mice and controls had similar renal expression for interferon-γ, interleukin-1β, and interleukin-6. By contrast, lymphocyte deficiency (i.e., scid mice) during ANG II infusion led to upregulation of tumor necrosis factor-α, endothelial nitric oxide synthase (eNOS), and cyclooxygenase-2 (COX-2) in the kidney. In turn, this enhanced eNOS and COX-2 expression in the scid kidneys was associated with exaggerated renal generation of nitric oxide, prostaglandin E2, and prostacyclin, all of which promote natriuresis. Thus, the absence of lymphocyte activity protects from hypertension by allowing blood pressure-induced sodium excretion, possibly via stimulation of eNOS- and COX-2-dependent pathways.
Keywords: inflammation, kidney diseases, T lymphocytes
the renin-angiotensin system (RAS) is a critical regulator of blood pressure and fluid homeostasis. The principal effector molecule of this system, angiotensin II (ANG II) raises blood pressure primarily through activation of type 1 angiotensin (AT1) receptors (11). The important role of AT1 receptors in the pathogenesis of hypertension is illustrated by clinical trials that show the impressive efficacy of AT1 receptor blockers (ARBs) in ameliorating hypertension and its complications, including chronic kidney disease (CKD) and cardiac hypertrophy (3, 13, 37).
In these trials, RAS inhibition appears to protect from end-organ damage to a greater degree than can be explained solely by blood pressure reduction (3, 63). Blockade of proinflammatory cellular effects of ANG II represents one blood pressure-independent mechanism through which ARB therapy could protect against target organ injury. For example, ANG II stimulates NF-κB activation and interferon-γ expression in the kidney, and immunosuppression can reverse these effects (8, 45, 46). Moreover, ANG II can drive lymphocyte proliferation (48), and mice that are deficient in lymphocyte populations have a blunted hypertensive response to ANG II (25).
Despite substantial recent progress in understanding the role of the adaptive immune system in the pathogenesis of ANG II-dependent hypertension (25, 46), several questions remain. First, the mechanisms through which lymphocytes influence the hypertensive response to ANG II are unclear. Second, whether the absence of lymphocytes can protect the heart and kidney from hypertensive injury has not been addressed. Therefore, in the present studies, we employ scid mice that are deficient in lymphocyte activity and are susceptible to both heart and kidney damage to explore the mechanisms through which lymphocytes modulate blood pressure and tissue injury in ANG II-dependent hypertension.
MATERIALS AND METHODS
Animals.
Wild-type (WT) C3H/HeSnJ (C3H) mice and C3SnSmn. CB17-PrkdcScid/J (C3H scid) were purchased from Jackson Laboratories and were maintained in the animal facility of the Durham Veterans Affairs Medical Center. All animal studies were approved by the Institutional Animal Care and Use Committee, Durham Veterans Affairs Medical Center (Durham, NC) and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
To prevent development of infection or spontaneous immunity in the scid mice, all experimental mice were maintained in sterile barrier conditions. These studies used 2- to 4-mo-old male mice.
Model of ANG II-induced hypertension.
At the initiation of the protocol, C3H WT and scid mice (n ≥ 7 mice/group) underwent left nephrectomy followed 1 wk later by implantation of a pressure-sensing catheter (TA11PA-C10; Transoma Medical) via the left common carotid artery as previously described (10). After allowing 7 days for reestablishment of diurnal blood pressure variation, baseline blood pressure and heart rate measurements were recorded for 3 days continuously by radiotelemetry (Transoma) in conscious unrestrained animals. Next, an osmotic minipump (Alzet model 2004; DURECT) was implanted to infuse ANG II (1,000 ng·kg−1·min−1; Sigma-Aldrich) or vehicle (0.9% NaCl, n = 5 in saline groups) continuously for 28 days as previously described (9). Blood pressure and heart rate measurements continued for 3 wk of ANG II infusion as previously described (9). On day 25, the mice were placed in metabolic cages, and urine was collected for 24 h. Urinary concentrations of albumin, prostaglandin E2 (PGE2), and prostacyclin (PGI2) were measured in individual samples using specific ELISAs for mouse albumin (Exocell), prostaglandin E metabolite (Cayman Chemical), and prostaglandin F1α (Cayman), respectively, as previously described (19). Creatinine (Cr) concentrations were measured with a picric acid-based method using a kit (Exocell). Albumin excretion is expressed as micrograms per milligram Cr.
Histopathological analysis.
Following 28 days of ANG II infusion, hearts and kidneys were harvested, weighed, and fixed in formalin, sectioned, and stained with Masson trichrome. All of the tissues were examined by a pathologist (Ruiz) without knowledge of the genotypes or treatment groups. The pathological abnormalities in the kidney were graded based on the presence and severity of component abnormalities, including glomerulosclerosis, mesangial expansion, chronic inflammation, tubular atrophy or casts, fibrosis, and vascular injury. Grading for each component was performed using a semiquantitative scale as previously described (58, 59) where zero was no abnormality and where one, two, three, and four represented mild, moderate, moderately severe, and severe abnormalities, respectively. In the heart, the component abnormalities scored included myocyte injury, interstitial inflammation, fibrosis, and chronic vascular injury with severity ranging from zero (no injury) to three (severe). The total injury score for each kidney or heart was a summation of these component injury scores.
Quantification of mRNA expression.
Hearts and kidneys were harvested, and total RNA was isolated by using an RNeasy Fibrous Tissue Mini Kit (for the heart) or a standard RNeasy Mini Kit (for the kidney) per the manufacturer's instructions (Qiagen, Valencia, CA). The gene expression levels of β-myosin heavy chain (MHC) and α-MHC in cardiac tissue were determined by real-time quantitative RT-PCR as previously described (32). Gene expression levels in the kidney were similarly determined for interferon-γ, interleukin-1β, interleukin-6, 5-lipoxygenase, endothelin-1, endothelial nitric oxide synthase (eNOS), tumor necrosis factor (TNF)-α, cyclooxygenase (COX)-1, and COX-2 using Taqman primers (Applied Biosystems). To separately confirm mRNA expression levels for TNF-α in the kidney, we employed RNase protection assay as previously described (8).
Metabolic studies.
In a separate experiment, seven mice from the WT and SCID groups were placed in specially designed metabolic cages (19), and balance studies were conducted as previously described (9). Briefly, the mice were fed 10 g/day gelled normal (0.25% NaCl) diet that contained all nutrients and water (Nutra-gel; Bio-Serv). After 1 wk of baseline collections, the animals were implanted with osmotic minipumps infusing ANG II as described above and were returned to the metabolic cages for two more weeks. Urinary sodium content was determined by using an IL943 Automatic Flame photometer per the manufacturer's instructions (Instrumentation Laboratory). Urinary nitrite and nitrate levels were measured using an ELISA kit (Cayman) per the manufacturer's instructions.
Statistical methods.
The values for each parameter within a group were expressed as means ± SE. For comparisons between groups with normally distributed data, statistical significance was assessed using ANOVA followed by unpaired t-test. For comparisons between groups with nonnormally distributed data, the Mann Whitney U-test was employed. For comparisons within groups, normally distributed variables were analyzed by a paired t-test, whereas nonnormally distributed variables were analyzed by the Wilcoxon signed rank test.
RESULTS
Absence of lymphocyte activity leads to blunted hypertensive response to ANG II.
To study the role of lymphocyte responses in ANG II-dependent hypertension, we used radiotelemetry to measure intra-arterial blood pressure in uninephrectomized C3H WT and C3H scid (SCID) mice at baseline and during 4 wk of chronic ANG II infusion. At baseline, mean arterial pressures (MAPs) in the SCID group were slightly but significantly lower than in the WT controls (105 ± 1 vs. 109 ± 1 mmHg; P < 0.04). As shown in Fig. 1A, ANG II induced a vigorous hypertensive response in both groups. However, following day 5 of ANG II, MAPs in the SCID group moved progressively lower such that, over the whole ANG II infusion period, average SCID MAPs (138 ± 3 mmHg) were significantly lower than in WT controls (151 ± 1 mmHg; P = 0.001). As shown in Fig. 1, B and C, systolic blood pressures (SBPs) and diastolic blood pressures (DBPs) followed a pattern similar to the MAPs such that, averaged over the ANG II infusion period, SCID SBPs were 12 mmHg lower than WT SBPs (153 ± 4 vs. 165 ± 1 mmHg; P < 0.006), and SCID DBPs were 13 mmHg lower than WT DBPs (121 ± 3 vs. 134 ± 2 mmHg; P = 0.001). The blood pressure increase following the initiation of ANG II infusion was associated with a significant decrement in heart rate in both the WT (P < 0.007; Fig. 1D) and the SCID (P = 0.001) groups, and overall during ANG infusion heart rates in the WT and SCID groups were similar [579 ± 23 vs. 596 ± 19 beats/min; P = not significant (NS)]. However, following day 5 of ANG II infusion when the blood pressures in the two groups separated because of the decrement in the SCID blood pressures, the heart rate initially rose only in the SCID group (P < 0.04).
Fig. 1.
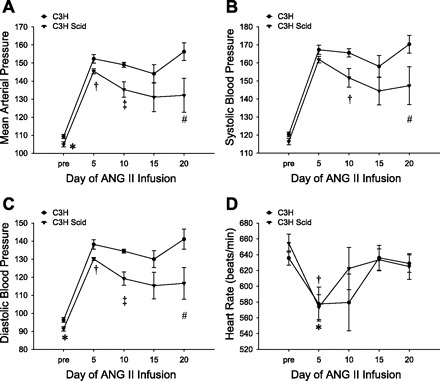
C3H SCID mice have a blunted chronic hypertensive response to ANG II. Hemodynamic parameters were measured by radiotelemetry in the experimental groups at baseline (“pre”) and during 3 wk of ANG II infusion. Values during ANG II are averaged over 5-day periods. A: mean arterial pressures. *P < 0.04 vs. C3H. †P = 0.03 vs. C3H. ‡P < 0.005 vs. C3H. #P = 0.05 vs. C3H. B: systolic blood pressures. †P = 0.02 vs. C3H. #P < 0.08 vs. C3H. C: diastolic blood pressures. *P < 0.02 vs. C3H. †P = 0.02 vs. C3H. ‡P = 0.0006 vs. C3H. #P < 0.05 vs. C3H. D: heart rates. *P < 0.04 vs. C3H Scid baseline, P < 0.04 vs. C3H Scid day 10. †P < 0.007 vs. C3H baseline. n ≥ 7 mice/group.
SCID mice have diminished cardiac damage in ANG II-dependent hypertension.
As shown in Fig. 2A, 4 wk of ANG II infusion caused marked cardiac hypertrophy in the WT group compared with saline-infused WT controls (7.2 ± 0.2 vs. 4.7 ± 0.2 mg/g; P < 0.0001). ANG II also induced significant cardiac hypertrophy in the SCID relative to saline-infused scid controls (6.1 ± 0.2 vs. 4.6 ± 0.1 mg/g; P < 0.0002). However, consistent with the lower blood pressures in the ANG II-infused SCID mice, this group had ∼40% less cardiac hypertrophy than the ANG II-infused WT group (P = 0.0007). To determine if the reduced cardiac hypertrophy in the SCID group was associated with less severe cardiac injury, hearts from the experimental groups were scored for evidence of histopathological damage following 28 days of ANG II infusion. The degree of cardiac pathology was mild in the WT group [3.3 ± 0.5 arbitrary units (AU); Fig. 2B, Table 1] with sparse myocardial injury, interstitial inflammation, fibrosis, and vascular injury. Nevertheless, as with cardiac hypertrophy, cardiac pathological changes were markedly reduced in the ANG II-infused SCID group (1.8 ± 0.4 AU; P = 0.04; Fig. 2C, Table 1) with significant protection from cardiac fibrosis (0.38 ± 0.14 vs. 0.83 ± 0.17 AU; P < 0.05; Table 1) and a trend toward protection from vascular injury in the heart (0.31 ± 0.13 vs. 0.75 ± 0.18 AU; P < 0.06; Table 1). During the development of cardiac hypertrophy, gene expression in the heart recapitulates a fetal pattern with upregulation of β-MHC and downregulation of α-MHC (14, 30). In our experiments, after 4 wk of ANG II, the β-MHC-to-α-MHC ratio in the SCID group (4.3 ± 0.7 AU) was >50% lower than in the WT controls (10.2 ± 2.9 AU; P = 0.04), reflecting the blunted cardiac hypertrophy in the lymphocyte-deficient mice. Thus, lymphocyte activity enhances the chronic hypertensive response to ANG II and accordingly exaggerates the degree of ANG II-induced cardiac enlargement and damage.
Fig. 2.

Reduced cardiac hypertrophy and injury in SCID mice infused with ANG II. A: heart weight-to-body weight ratios (mg/g) in the experimental groups following 28 days of ANG II infusion or saline control. *P < 0.0001 vs. C3H saline. †P < 0.0002 vs. C3H Scid saline and P = 0.0007 vs. C3H ANG II. B: representative heart section from C3H ANG II group showing mild myocardial injury, mononuclear cell infiltrates, fibrosis, and chronic vascular damage. C: representative heart section from C3H Scid ANG II group (×20).
Table 1.
SCID mice have reduced degree of cardiac injury induced by ANG II
| Cardiac Injury Scores, AU | Myocyte Injury | Interstitial Inflammation | Fibrosis | Chronic Vascular Injury | Total Injury |
|---|---|---|---|---|---|
| C3H WT | 0.6 ± 0.1 | 1.1 ± 0.1 | 0.8 ± 0.2 | 0.8 ± 0.2 | 3.3 ± 0.5 |
| C3H SCID | 0.3 ± 0.1 | 0.8 ± 0.2 | 0.4 ± 0.1* | 0.3 ± 0.1† | 1.8 ± 0.4‡ |
Values are means ± SE; n ≥ 12 mice/group. AU, arbitrary units; WT, wild type.
P < 0.05 vs. C3H WT.
P < 0.06 vs. C3H WT.
P = 0.04 vs. C3H WT.
Lymphocyte deficiency protects from ANG II-induced kidney injury.
To determine if the lower blood pressures in the ANG II-infused SCID group were also associated with less injury to the kidney, we measured urinary albumin excretion in the experimental groups following 4 wk of ANG II infusion. As shown in Fig. 3A, ANG II caused a dramatic increase in albuminuria in the SCID mice (1,238 ± 316 μg/mg Cr) and the WT group (1,980 ± 321 μg/mg Cr) compared with saline-infused scid (120 ± 47 μg/mg Cr; P < 0.05) and WT controls (88 ± 23 μg/mg Cr; P = 0.002), respectively. However, following ANG II, albuminuria was ∼40% lower in the SCID group than in the WT mice (P = 0.02), suggesting a role for lymphocytes in ANG II-dependent kidney injury.
Fig. 3.

Lymphocyte deficiency in SCID mice reduces hypertensive kidney injury. A: urinary albumin excretion (μg/mg creatinine) in the experimental groups following 25 days of ANG II infusion or saline control. *P = 0.002 vs. C3H saline. †P < 0.05 vs. C3H Scid saline, P = 0.02 vs. C3H ANG II. B: representative kidney section from C3H group showing mild mesangial expansion, sparse mononuclear cell infiltrates, and fibrosis. C: representative kidney section from C3H Scid group (×20).
Following 4 wk of ANG II infusion, kidneys from the WT group displayed moderate levels of pathology characterized by mild mesangial expansion and sclerosis in the glomeruli, sparse interstitial inflammation, and rare fibrosis (Fig. 3B, Table 2). Lymphocyte deficiency in the SCID group afforded protection from ANG II-induced renal damage, reducing the score for the severity of overall pathology by 30% from 9.3 ± 0.7 to 6.3 ± 1.1 AU (P < 0.03; Fig. 3C, Table 2). The amelioration in kidney damage in the SCID group was most prominent within the glomerulus (4.7 ± 0.8 vs. 7.0 ± 0.6 AU; P < 0.03). However, we also noted a trend toward a reduction in tubulointerstitial disease in the SCID mice (1.5 ± 0.3 vs. 2.2 ± 0.2 AU; P = 0.06). Thus, lymphocyte deficiency substantially limited the extent of both functional and histological renal injury in ANG II-dependent hypertension.
Table 2.
Hypertensive kidney injury is ameliorated in lymphocyte-deficient SCID mice
| Kidney Injury Scores, AU | Glomerular Injury | Interstitial Inflammation | Vascular Damage | Total Injury |
|---|---|---|---|---|
| C3H WT | 7.0 ± 0.6 | 2.2 ± 0.2 | 0.2 ± 0.1 | 9.3 ± 0.7 |
| C3H SCID | 4.7 ± 0.8* | 1.5 ± 0.3† | 0.2 ± 0.1 | 6.3 ± 1.1‡ |
Values are means ± SE; n ≥ 12 mice/group.
P < 0.03 vs. C3H WT.
P = 0.06 vs. C3H WT.
P < 0.03 vs. C3H WT.
SCID mice have exaggerated sodium excretion during ANG II infusion.
We reasoned that the lower blood pressures in the SCID mice over the whole ANG II infusion period may have served to protect them from tissue injury. Therefore, to determine if differences in renal sodium handling and intravascular volume might explain the blood pressure differences between the experimental groups, WT and SCID mice were placed in metabolic cages for 1 wk before and 2 wk following the initiation of ANG II infusion. While in the metabolic cages, the animals were fed a gelled preparation containing adequate nutrients and water to minimize differences in intake. Throughout this period, daily weights, urine volume, and sodium excretion were monitored. Overall, sodium excretion was similar between the groups as were total body weights at the beginning and end of the study (data not shown). However, beginning on day 5 of ANG II infusion when blood pressures peaked in both groups and continuing over the next 4 days during which blood pressures fell sharply in the SCID group, cumulative sodium excretion was significantly enhanced in the SCID mice (1.12 ± 0.06 mmol) compared with WT controls (0.93 ± 0.04 mmol; P = 0.02). Over this same period, urine volumes were exaggerated in the SCID group (15.4 ± 0.6 vs. 12.8 ± 0.6 ml; P = 0.01), and total body weight, a sensitive gauge of intravascular volume, declined significantly only in the SCID mice (−2.5 ± 0.08 g; P = 0.02 for paired analysis of day 5 vs. day 9 ANG II). These data suggest that differences in renal sodium handling contributed to the lower blood pressures in the ANG II-infused SCID group vs. controls.
SCID mice have enhanced renal nitric oxide production and eNOS expression.
Nitric oxide (NO) in the kidney potentiates pressure natriuresis, and NO production can be regulated by inflammatory mediators (40, 41). We therefore quantitated urinary nitrate/nitrite excretion as a measure of renal NO production on day 5 of ANG II infusion when the SCID and WT blood pressures initially diverged (Fig. 4A). We found that urinary nitrate/nitrate levels in the SCID group (1,044 ± 204 nmol/24 h) were more than twice those of WT controls (471 ± 115 nmol/24 h; P < 0.04), suggesting that the SCID natriuresis may be mediated through a NO-dependent pathway. eNOS is an important mediator of NO production. Moreover, eNOS protects against hypertension (28, 55) and can be suppressed by tissue inflammation (66). We therefore examined renal expression for eNOS by real-time PCR following saline or ANG II infusion (Fig. 4B). Following saline infusion, eNOS expression in the kidney was virtually equivalent in the WT and SCID groups (1.00 ± 0.11 vs. 1.03 ± 0.24 AU; P = NS). ANG II suppressed renal expression of eNOS in the WT group numerically but not significantly (0.77 ± 0.06 AU; P = NS vs. WT saline). By contrast, ANG II infusion caused a threefold upregulation of renal eNOS expression in the SCID group such that it greatly exceeded eNOS expression in the ANG II-infused WT group (3.11 ± 1.05 AU; P = 0.03). Thus lymphocyte deficiency permits an induction of eNOS by ANG II, leading to enhanced renal NO production and natriuresis.
Fig. 4.

Nitric oxide (NO) production is exaggerated in ANG II-infused SCID mice. A: urinary excretion of nitrate/nitrite metabolites of NO on day 5 of ANG II infusion. *P < 0.04 vs. C3H. B: real-time PCR for endothelial nitric oxide synthase (eNOS) mRNA expression in kidney following 28 days of saline or ANG II infusion. *P < 0.03 vs. C3H ANG II.
The absence of lymphocyte activity leads to alterations in gene expression for TNF-α in the kidney during ANG II infusion.
Because scid mice are deficient in lymphocyte activity, we posited that differential expression of inflammatory mediators previously linked to blood pressure regulation or kidney injury might contribute to the differences in blood pressure in our experimental groups. We therefore used real-time PCR to profile gene expression for some of these mediators in the kidneys from the WT and SCID groups after 28 days of ANG II infusion. Renal expression of interferon-γ, interleukin-1β, interleukin-6, 5-lipoxygenase, and endothelin-1 was similar between the experimental groups (Table 3). By contrast, we found surprisingly that renal expression of TNF-α was exaggerated in the SCID group compared with WT controls (1.00 ± 0.10 vs. 1.57 ± 0.28 AU; P = 0.06). To confirm this unexpected finding, we used RNase protection assay as an alternative approach to measure renal gene expression. With this method, we found that TNF-α expression in the SCID kidneys markedly exceeded that of controls (P = 0.01; Fig. 5, A and B). Thus, despite lower levels of renal injury, TNF-α expression in the kidney is augmented in the setting of lymphocyte deficiency.
Table 3.
Profile of renal mRNA expression for inflammatory mediators in C3H WT and scid mice infused with ANG II
| Gene Product | WT, AU | SCID, AU |
|---|---|---|
| Interferon-γ | 1.00 ± 0.20 | 0.89 ± 0.14 |
| Interleukin-1β | 1.00 ± 0.19 | 0.97 ± 0.12 |
| Interleukin-6 | 1.00 ± 0.32 | 1.07 ± 0.51 |
| 5-Lipoxygenase | 1.00 ± 0.09 | 0.85 ± 0.14 |
| Endothelin-1 | 1.00 ± 0.22 | 1.50 ± 0.35 |
Values are means ± SE; n ≥ 7 mice/group.
Fig. 5.

Tumor necrosis factor (TNF)-α expression is augmented in SCID kidneys following ANG II infusion. A: levels of mRNA for TNF-α and housekeeping gene L32 assessed by RNase protection assay in kidney following 28 days of ANG II infusion. B: plot of the corresponding analysis by densitometry; the lanes were run on the same gel but were noncontiguous. *P = 0.01 vs. C3H.
COX-2 expression and activity is enhanced in ANG II-infused SCID mice.
ANG II, NO, and TNF-α can influence generation of COX-2 in the kidney (7, 16, 53, 54). We therefore examined the level of renal expression for COX-2 following 4 wk of saline or ANG II infusion. COX-2 expression in the kidney following saline infusion was comparable in the WT and SCID groups (0.43 ± 0.10 vs. 0.54 ± 0.12 AU; P = NS). Compared with saline, ANG II caused a significant induction of renal COX-2 expression in both the WT (P = 0.02) and the SCID (P = 0.02) groups, but the levels of COX-2 expression in the kidneys from the ANG II-infused SCID mice (1.52 ± 0.19 AU) exceeded those in the ANG II-infused WT controls by 50% (1.00 ± 0.14 AU; P = 0.05; Fig. 6A). To determine whether lymphocyte deficiency influenced COX-1 generation in this model, we measured renal expression for COX-1 following ANG II infusion and found it to be similar in the WT and SCID groups (1.00 ± 0.18 vs. 1.13 ± 0.24 AU; P = NS; Fig. 6A).
Fig. 6.

SCID mice have enhanced renal cyclooxygenase (COX)-2 expression and urinary excretion of vasodilatory prostaglandins in ANG II-dependent hypertension. A: relative expression in kidney of COX-1 and COX-2 after 28 days of ANG II infusion. *P = 0.05 vs. C3H wild type (WT). B and C: urinary excretion of metabolites for prostaglandin E2 (B) and prostacyclin (prostaglandin I2) (C) measured by ELISA following 25 days of ANG II infusion. *P = 0.05 vs. C3H WT.
COX-2 catalyzes the generation of PGE2 and PGI2 in the kidney (57). PGE2 and PGI2, in turn, both inhibit sodium reabsorption in the nephron (6, 22, 26, 27). Because the SCID group had enhanced renal COX-2 expression and exaggerated sodium excretion in response to ANG II, we used ELISA to measure urinary excretion of PGE2 and PGI2 following 4 wk of ANG II infusion. As shown in Fig. 6B, PGE2 excretion was significantly enhanced in the SCID group (P = 0.05), and there was a trend toward increased PGI2 excretion in the SCID group as well (P < 0.07; Fig. 6C). Thus, in ANG II-dependent hypertension, lymphocyte deficiency permits an augmentation of COX-2 expression in the kidney that may drive sodium excretion through enhanced generation of vasodilatory prostaglandins.
DISCUSSION
Activation of the RAS promotes blood pressure elevation and the progression of cardiovascular and kidney disease (4, 5, 20). The role of RAS activation in the pathogenesis of hypertension is highlighted by clinical trials in which ARB and angiotensin-converting enzyme inhibitor therapy effectively controls blood pressure and ameliorates hypertensive heart disease (13, 62) and CKD (3, 37). Although activation of AT1 receptors in the kidney is clearly paramount in the hypertensive response to ANG II (9, 24, 39), several studies have now shown that activation of the immune system by ANG II can contribute to blood pressure elevation and/or tissue damage (8, 25, 45, 46). For example, pharmacological suppression of inflammatory responses in ANG II-dependent hypertension protects the heart and kidney from injury and, in some models, reduces the magnitude of blood pressure elevation (8, 45, 52). Nevertheless, the mechanisms through which immunosuppression ameliorates hypertensive end-organ damage have not been fully elucidated.
In the current studies, we examined the role of lymphocyte responses in the pathogenesis of ANG II-dependent hypertension and its associated complications of heart and kidney disease. In our model, hypertension was induced by chronic infusion of ANG II in WT mice and SCID mice, which are deficient in lymphocyte activity (12). To make the kidney more susceptible to hypertensive injury, one native kidney was removed before the induction of hypertension. In addition, we selected the C3H mouse strain for these experiments both because the development of “leaky” spontaneous immunity that circumvents lymphocyte deficiency is rare on this strain and because the C3H strain is susceptible to progressive kidney injury (34). Using this approach, we were able to directly assess the role of lymphocytes not only in blood pressure elevation but also in heart and kidney damage during ANG II-dependent hypertension.
In our model, the SCID animals lacking lymphocyte activity have a blunted chronic hypertensive response to ANG II. This finding is consistent with that of Guzik and Harrison (25) using the Rag1-deficient model even though theirs is a two-kidney model of hypertension, and suggests that on susceptible strains lymphocytes can play a significant role in potentiating the blood pressure elevation induced by ANG II. In addition, our studies show that, in the absence of functional lymphocytes, blood pressures peak during the 1st wk of ANG II infusion and thereafter regress. This diminution in blood pressure coincides with enhanced sodium excretion, suggesting that actions of lymphocytes exaggerate the hypertensive response at least in part by promoting salt reabsorption in the nephron. By contrast, in the animal with an intact adaptive immune system, the blood pressure elevation persists such that the average blood pressures during ANG II infusion are markedly higher than in the lymphocyte-deficient animal.
In several studies, cardiac enlargement correlates closely with the degree of blood pressure elevation (9). Thus, the reduced level of ANG II-induced cardiac hypertrophy in the SCID mice is consistent with their muted hypertensive response. The degree of cardiac injury caused by ANG II in the C3H strain was mild with infrequent myocardial injury and fibrosis. Moreover, vascular damage was present in <10% of the coronary vessels examined. Nevertheless, even against this background of mild hypertensive injury noted in the WT heart, lymphocyte deficiency led to a numerical decrement in all injury components examined and a significant reduction in the severity of overall cardiac pathology. The reduced cardiac injury in the SCID group during ANG II infusion is likely related to their lower blood pressures (9) and is consistent with the finding that suppression of inflammatory responses can concomitantly limit both blood pressure elevation and cardiac injury induced by ANG II (45). Nevertheless, these findings demonstrate for the first time a direct role for lymphocytes in mediating ANG II-induced cardiac hypertrophy and injury.
In the current model, ANG II also caused significant damage to the kidney both in the glomerulus and the interstitium. However, injury to the renal vasculature was negligible. On the C3H background strain, the glomerular disease was characterized by mesangial expansion rather than frank glomerulosclerosis. Accordingly, the level of albuminuria, a functional measure of glomerular damage, was substantial but not as dramatic as that seen in the 129/SvEv mouse strain, which is susceptible to glomerular injury, particularly in the setting of severe hypertension (8, 29). In the present studies, the SCID group had a >30% reduction in both albuminuria and severity of renal pathology. Lymphocyte deficiency protected both the glomerulus and the renal interstitium from hypertensive injury. Thus, although previous studies have shown that immunosuppression can limit ANG II-induced kidney damage (8, 46), the current experiments are the first to our knowledge demonstrating a direct role for lymphocytes in promoting renal injury in ANG II-dependent hypertension.
Because hypertensive damage to both the heart and the kidney tracks closely with the degree of blood pressure elevation (9, 21), the protection from tissue damage in the absence of functional lymphocytes is likely related to a blunted hypertensive response to ANG II. We therefore explored the mechanism through which lymphocyte activity promotes blood pressure elevation. Although our scid model and the Rag1-deficient model both illustrate the importance of lymphocyte activity in ANG II-dependent hypertension, the lack of substantial vascular injury in our C3H mouse model contrasts sharply with the Rag1-deficient model using C57Bl/6 mice in which vascular hypertrophy and dysfunction were prominent features (25). In that study, vascular disease induced by lymphocytes in response to ANG II was posited to augment the hypertensive response. By contrast, in our scid model, the protection from sustained hypertension is more likely related to a renal mechanism in which lymphocyte deficiency permits enhanced sodium excretion following an early peak in blood pressure. Such a finding would be consistent with the hypothesis advanced by Guyton et al. (23, 24) in which the kidney modulates intravascular volume to discourage persistent elevations in blood pressure. In this regard, based on our gene expression analysis, the absence of lymphocytes during ANG II hypertension may facilitate sodium excretion through enhanced renal expression of eNOS and COX-2.
eNOS deficiency leads to hypertension (28, 55). The preservation of flow-induced vasodilatory responses in peripheral vessels in eNOS−/− mice points to a renal mechanism for this hypertension (61), and eNOS deficiency causes dysregulation of sodium handling in the medullary thick ascending limb that is not corrected by activity of the other nitric oxide synthases (50). As seen in our WT group, eNOS is typically downregulated in the setting of inflammation and oxidative stress associated with RAS activation (38, 66, 67). However, in our SCID mice, the absence of functional lymphocytes during ANG II infusion leads to augmented renal expression of eNOS, which acts through NO to promote sodium excretion and thereby mitigate the hypertensive response (35, 40, 44). Accordingly, we have examined urinary NO excretion early during the ANG II infusion period when the WT and SCID blood pressures diverged and found NO production to be exaggerated in the SCID group. Local generation of NO also stimulates COX-2 activity in the kidney (53, 54), and the absence of lymphocytes in our SCID animals potentiated the induction of renal COX-2 expression by ANG II. The effects of COX-2 in hypertension are admittedly complex with most but not all studies indicating that actions of COX-2 blunt the hypertensive response by promoting natriuresis (2, 51, 64). However, one salient effect in our model is COX-2-mediated stimulation of PGE2 and PGI2 (57), both of which inhibit sodium reabsorption from the renal tubule (6, 22, 26, 27), and thereby facilitate natriuresis in response to ANG II-dependent hypertension. As such, deficiency of these hormones is permissive to salt-sensitive hypertension (18, 42, 47).
We posited that lymphocyte deficiency might permit induction of eNOS and COX-2 during ANG II-dependent hypertension by altering the local cytokine milieu in the kidney. Nonetheless, the renal expression of several inflammatory mediators linked to blood pressure elevation or vascular reactivity, including interferon-γ, interleukin-1β, interleukin-6, 5-lipoxygenase, and endothelin-1 (1, 15, 33, 36, 49), was not altered by the absence of lymphocyte activity in our model. However, we found that lymphocyte-deficient mice had augmented expression of TNF-α in the kidney during ANG II infusion. We speculate that the enhanced TNF expression in the SCID animals may be related to dysregulation of macrophage functions or upregulation of innate immune responses in the absence of adaptive immunity. The enhanced TNF expression in the SCID kidneys was particularly surprising because TNF-α has been implicated in hypertensive kidney injury (46), and global TNF deficiency protects from ANG II-dependent hypertension (60). We therefore confirmed this paradoxical finding with two separate methods and obtained consistent results. Thus, our data suggest that actions of TNF in hypertension may be governed by its localization within the kidney such that alterations of renal TNF expression due to infiltrating lymphocytes may have effects that are incongruent with those of generalized TNF blockade or deletion. With regard to the current model, we note that TNF can stimulate eNOS activity (31, 65) and COX-2 expression (16, 17), particularly in the presence of ANG II (43) and may therefore have contributed to the induction of NO and vasodilatory prostaglandins in the setting of lymphocyte deficiency.
Perspectives and Significance
Hypertension associated with RAS activation leads to substantial cardiovascular morbidity and mortality (4, 56). Blood pressure elevation is a major determinant of heart and kidney injury in the setting of hypertension (21). Recent studies have demonstrated that stimulation of inflammatory responses by ANG II can potentiate both blood pressure elevation and end-organ damage (8, 45). In our experiments, the absence of lymphocyte activity during ANG II-dependent hypertension significantly limits the degree of blood pressure elevation at least in part by enhancing sodium excretion. Thus, lymphocyte activation contributes to the pattern of sodium retention induced by ANG II, possibly through regulation of actions of eNOS and COX-2 in the kidney. Moreover, by enhancing the chronic hypertensive response to ANG II, lymphocytes aggravate hypertensive cardiac and renal damage. Therapies to manipulate lymphocyte function may therefore represent a novel and promising intervention for hypertensive patients with progressive end-organ injury despite the use of conventional pharmacological approaches.
GRANTS
This work was supported by funding from the Medical Research Service of the Veterans Administration.
DISCLOSURES
None.
ACKNOWLEDGMENTS
We acknowledge outstanding administrative support from Brooke Flythe.
REFERENCES
- 1. Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, Kohan DE. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest 114: 504–511, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aw TJ, Haas SJ, Liew D, Krum H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch Intern Med 165: 490–496, 2005. [DOI] [PubMed] [Google Scholar]
- 3. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001. [DOI] [PubMed] [Google Scholar]
- 4. Brunner HR. Experimental and clinical evidence that angiotensin II is an independent risk factor for cardiovascular disease. Am J Cardiol 87: 3C–9C, 2001. [DOI] [PubMed] [Google Scholar]
- 5. Brunner HR, Laragh JH, Baer L, Newton MA, Goodwin FT, Krakoff LR, Bard RH, Buhler FR. Essential hypertension: renin and aldosterone, heart attack and stroke. N Engl J Med 286: 441–449, 1972. [DOI] [PubMed] [Google Scholar]
- 6. Chen J, Zhao M, He W, Milne GL, Howard JRH, Morrow J, Hebert RL, Breyer RM, Chen J, Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol 295: F818–F825, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng HF, Wang JL, Zhang MZ, Miyazaki Y, Ichikawa I, McKanna JA, Harris RC. Angiotensin II attenuates renal cortical cyclooxygenase-2 expression. J Clin Invest 103: 953–961, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, Coffman TM. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol 295: F515–F524, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci 103: 17985–17990, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 115: 1092–1099, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crowley SD, Tharaux PL, Audoly LP, Coffman TM. Exploring type I angiotensin (AT1) receptor functions through gene targeting. Acta Physiol Scand 181: 561–570, 2004. [DOI] [PubMed] [Google Scholar]
- 12. Custer RP, Bosma GC, Bosma MJ. Severe combined immunodeficiency (SCID) in the mouse. Pathology, reconstitution, neoplasms. Am J Pathol 120: 464–477, 1985. [PMC free article] [PubMed] [Google Scholar]
- 13. Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359: 995–1003, 2002. [DOI] [PubMed] [Google Scholar]
- 14. D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., II Transgenic Galpha q overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA 94: 8121–8126, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dorrance AM. Interleukin 1-beta (IL-1beta) enhances contractile responses in endothelium-denuded aorta from hypertensive, but not normotensive, rats. Vascul Pharmacol 47: 160–165, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J Clin Invest 95: 1669–1675, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferreri NR, An SJ, McGiff JC. Cyclooxygenase-2 expression and function in the medullary thick ascending limb. Am J Physiol Renal Physiol 277: F360–F368, 1999. [DOI] [PubMed] [Google Scholar]
- 18. Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, Rockman HA, Fitzgerald GA, Koller BH, Coffman TM. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab 2: 201–207, 2005. [DOI] [PubMed] [Google Scholar]
- 19. Francois H, Athirakul K, Mao L, Rockman H, Coffman TM. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension 43: 364–369, 2004. [DOI] [PubMed] [Google Scholar]
- 20. Gavras H, Lever AF, Brown JJ, Macadam RF, Robertson JI. Acute renal failure, tubular necrosis, and myocardial infarction induced in the rabbit by intravenous angiotensin II. Lancet 2: 19–22, 1971. [DOI] [PubMed] [Google Scholar]
- 21. Griffin KA, Bidani AK. Progression of renal disease: renoprotective specificity of renin-angiotensin system blockade. Clin J Am Soc Nephrol 1: 1054–1065, 2006. [DOI] [PubMed] [Google Scholar]
- 22. Guan Y, Zhang Y, Breyer RM, Fowler B, Davis L, Hebert RL, Breyer MD. Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest 102: 194–201, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guyton AC. Blood pressure control–special role of the kidneys and body fluids. Science 252: 1813–1816, 1991. [DOI] [PubMed] [Google Scholar]
- 24. Guyton AC, Coleman TG, Cowley AV, Jr, Scheel KW, Manning RD, Jr, Norman RA., Jr Arterial pressure regulation Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52: 584–594, 1972. [DOI] [PubMed] [Google Scholar]
- 25. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hilchey SD, Bell-Quilley CP. Association between the natriuretic action of angiotensin-(1–7) and selective stimulation of renal prostaglandin I2 release. Hypertension 25: 1238–1244, 1995. [DOI] [PubMed] [Google Scholar]
- 27. Hill TW, Moncada S. The renal haemodynamic and excretory actions of prostacyclin and 6-oxo-PGF1 alpha in anaesthetized dogs. Prostaglandins 17: 87–98, 1979. [DOI] [PubMed] [Google Scholar]
- 28. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377: 239–242, 1995. [DOI] [PubMed] [Google Scholar]
- 29. Ishola DA, Jr, van der Giezen DM, Hahnel B, Goldschmeding R, Kriz W, Koomans HA, Joles JA. In mice, proteinuria and renal inflammatory responses to albumin overload are strain-dependent. Nephrol Dial Transplant 21: 591–597, 2006. [DOI] [PubMed] [Google Scholar]
- 30. Jones WK, Grupp IL, Doetschman T, Grupp G, Osinska H, Hewett TE, Boivin G, Gulick J, Ng WA, Robbins J. Ablation of the murine alpha myosin heavy chain gene leads to dosage effects and functional deficits in the heart. J Clin Invest 98: 1906–1917, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim F, Gallis B, Corson MA. TNF-α inhibits flow and insulin signaling leading to NO production in aortic endothelial cells. Am J Physiol Cell Physiol 280: C1057–C1065, 2001. [DOI] [PubMed] [Google Scholar]
- 32. Kim HS, Lee G, John SWM, Maeda N, Smithies O. Molecular phenotyping for analyzing subtle genetic effects in mice: application to an angiotensinogen gene titration. Proc Natl Acad Sci USA 99: 4602–4607, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koh KP, Wang Y, Yi T, Shiao SL, Lorber MI, Sessa WC, Tellides G, Pober JS. T cell-mediated vascular dysfunction of human allografts results from IFN-gamma dysregulation of NO synthase. J Clin Invest 114: 846–856, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuin A, Kruse JJ, Stewart FA. Proteinuria and vascular changes after renal irradiation: the role of reactive oxygen species (ROS) and vascular endothelial growth factor (Vegf). Radiat Res 159: 174–181, 2003. [DOI] [PubMed] [Google Scholar]
- 35. Lahera V, Salom MG, Miranda-Guardiola F, Moncada S, Romero JC. Effects of N G-nitro-l-arginine methyl ester on renal function and blood pressure. Am J Physiol Renal Fluid Electrolyte Physiol 261: F1033–F1037, 1991. [DOI] [PubMed] [Google Scholar]
- 36. Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. [DOI] [PubMed] [Google Scholar]
- 37. Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345: 851–860, 2001. [DOI] [PubMed] [Google Scholar]
- 38. Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt- short-term exposure to angiotensin II. Hypertension 33: 1013–1019, 1999. [DOI] [PubMed] [Google Scholar]
- 39. Lu S, Mattson DL, Cowley AW., Jr Renal medullary captopril delivery lowers blood pressure in spontaneously hypertensive rats. Hypertension 23: 337–345, 1994. [DOI] [PubMed] [Google Scholar]
- 40. Majid DS, Williams A, Navar LG. Inhibition of nitric oxide synthesis attenuates pressure-induced natriuretic responses in anesthetized dogs. Am J Physiol Renal Fluid Electrolyte Physiol 264: F79–F87, 1993. [DOI] [PubMed] [Google Scholar]
- 41. Markewitz BA, Michael JR, Kohan DE. Cytokine-induced expression of a nitric oxide synthase in rat renal tubule cells. J Clin Invest 91: 2138–2143, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martineau A, Robillard M, Falardeau P. Defective synthesis of vasodilator prostaglandins in the spontaneously hypertensive rat. Hypertension 6: I161–I165, 1984. [DOI] [PubMed] [Google Scholar]
- 43. Matsuzuka T, Miller K, Pickel L, Doi C, Ayuzawa R, Tamura M. The synergistic induction of cyclooxygenase-2 in lung fibroblasts by angiotensin II and pro-inflammatory cytokines. Mol Cell Biochem 320: 163–171, 2009. [DOI] [PubMed] [Google Scholar]
- 44. Mattson D, Roman R, Cowley A., Jr Role of nitric oxide in renal papillary blood flow and sodium excretion. Hypertension 19: 766–769, 1992. [DOI] [PubMed] [Google Scholar]
- 45. Muller DN, Dechend R, Mervaala EMA, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35: 193–201, 2000. [DOI] [PubMed] [Google Scholar]
- 46. Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol 161: 1679–1693, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nasjletti Arthur C A. Corcoran Memorial Lecture. The role of eicosanoids in angiotensin-dependent hypertension. Hypertension 31: 194–200, 1998. [DOI] [PubMed] [Google Scholar]
- 48. Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 104: 1693–1701, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Natarajan R, Rosdahl J, Gonzales N, Bai W. Regulation of 12-lipoxygenase by cytokines in vascular smooth muscle cells. Hypertension 30: 873–879, 1997. [DOI] [PubMed] [Google Scholar]
- 50. Plato CF, Shesely EG, Garvin JL. eNOS mediates l-arginine-induced inhibition of thick ascending limb chloride flux. Hypertension 35: 319–323, 2000. [DOI] [PubMed] [Google Scholar]
- 51. Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest 110: 61–69, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001. [DOI] [PubMed] [Google Scholar]
- 53. Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA 90: 7240–7244, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salvemini D, Seibert K, Masferrer JL, Misko TP, Currie MG, Needleman P. Endogenous nitric oxide enhances prostaglandin production in a model of renal inflammation. J Clin Invest 93: 1940–1947, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93: 13176–13181, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Simon N, Franklin SS, Bleifer KH, Maxwell MH. Clinical characteristics of renovascular hypertension. J Am Med Assoc 220: 1209–1218, 1972. [PubMed] [Google Scholar]
- 57. Smith WL, Marnett LJ, DeWitt DL. Prostaglandin and thromboxane biosynthesis. Pharmacol Ther 49: 153–179, 1991. [DOI] [PubMed] [Google Scholar]
- 58. Spurney RF, Ibrahim S, Butterly D, Klotman PE, Sanfilippo F, Coffman TM. Leukotrienes in renal transplant rejection in rats. Distinct roles for leukotriene B4 and peptidoleukotrienes in the pathogenesis of allograft injury. J Immunol 152: 867–876, 1994. [PubMed] [Google Scholar]
- 59. Spurney RF, Ruiz P, Pisetsky DS, Coffman TM. Enhanced renal leukotriene production in murine lupus: role of lipoxygenase metabolites. Kidney Int 39: 95–102, 1991. [DOI] [PubMed] [Google Scholar]
- 60. Sriramula S, Haque M, Majid DSA, Francis J. Involvement of tumor necrosis factor-α in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sun D, Huang A, Smith CJ, Stackpole CJ, Connetta JA, Shesely EG, Koller A, Kaley G. Enhanced release of prostaglandins contributes to flow-induced arteriolar dilation in eNOS knockout mice. Circ Res 85: 288–293, 1999. [DOI] [PubMed] [Google Scholar]
- 62. The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 342: 145–153, 2000. [DOI] [PubMed] [Google Scholar]
- 63. Viberti G, Wheeldon NM. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 106: 672–678, 2002. [DOI] [PubMed] [Google Scholar]
- 64. Wu R, Laplante MA, de Champlain J. Cyclooxygenase-2 inhibitors attenuate angiotensin II-induced oxidative stress, hypertension, and cardiac hypertrophy in rats. Hypertension 45: 1139–1144, 2005. [DOI] [PubMed] [Google Scholar]
- 65. Yang B, Rizzo V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 292: H954–H962, 2007. [DOI] [PubMed] [Google Scholar]
- 66. Zhang J, Patel JM, Li YD, Block ER. Proinflammatory cytokines downregulate gene expression and activity of constitutive nitric oxide synthase in porcine pulmonary artery endothelial cells. Res Commun Mol Pathol Pharmacol 96: 71–87, 1997. [PubMed] [Google Scholar]
- 67. Zhou MS, Adam AG, Jaimes EA, Raij L. In salt-sensitive hypertension, increased superoxide production is linked to functional upregulation of angiotensin II. Hypertension 42: 945–951, 2003. [DOI] [PubMed] [Google Scholar]