

Abstract
Iron oxygenases generate elusive transient oxygen species to catalyze substrate oxygenation in a wide range of metabolic processes. Here we resolve the reaction sequence and structures of such intermediates for the archetypal non-heme FeII and α-ketoglutarate-dependent dioxygenase TauD. Time-resolved Raman spectra of the initial species with 16O18O oxygen unequivocally establish the FeIV═O structure. 1H/2H substitution reveals direct interaction between the oxo group and the C1 proton of substrate taurine. Two new transient species were resolved following FeIV═O; one is assigned to the νFeO mode of an FeIII─O(H) species, and a second is likely to arise from the vibration of a metal-coordinated oxygenated product, such as FeII─O─C1 or FeII─OOCR. These results provide direct insight into the mechanism of substrate oxygenation and suggest an alternative to the hydroxyl radical rebinding paradigm.
Keywords: ferric-oxo, ferryl, non-heme iron, oxygenation, transient Raman
Interest in the mechanism of iron oxygenases arises from their roles in an array of critical biological functions ranging from bacterial biodegradation of xenobiotics and recalcitrant compounds to human drug metabolism and cellular regulation. Many heme and non-heme iron oxygenases are believed to share highly oxidized iron-oxo species as central elements in their reaction mechanisms (1, 2). Several transient oxygen intermediates have been studied extensively in heme enzymes, including compound I-type species of cytochrome P450s (3), but the intermediates occurring during substrate oxygenation have not been directly observed. Even less is known about the transient species in the non-heme Fe oxygenases. An FeIV-oxo intermediate was observed in the FeII and α-ketoglutarate-dependent dioxygenase TauD (4–6), the archetype of this enzyme family (7), and in the related prolyl 4-hydroxylase (8) or the chlorinating enzymes CytC3 and SyrB2 (9, 10). The Fe-oxygen vibration in TauD at 821 cm-1 was assigned to an FeIV═O stretching mode, whereas an additional oxygen vibration at 583 cm-1 was not assigned (6). Here, through the use of substrate and media isotopes and varying reaction times, we resolve vibrations associated with three distinct transient oxygen-containing species during TauD catalysis that lead us to propose an alternative to the hydroxyl radical rebinding step that is central to the traditionally accepted mechanism (Fig. 1).
Fig. 1.

Postulated mechanism of taurine oxygenation by TauD. States A, B, C, and F have been experimentally observed; state D is consistent with the chemistry of biomimetic model compounds; the radical rebinding mechanism of state G is inferred from studies of heme-containing oxygenases.
Results
Transient Oxygen Species Detected by Difference Raman Spectroscopy.
The time dependence of the TauD reaction with 1H- and 2H- taurine was examined by cryogenic continuous-flow Raman spectroscopy (Fig. 2 and Fig. S1). The previously reported oxygen vibrations (6) can be seen as shifts at 825/788 and 578/555 cm-1 between the 16O and 18O derivatives (for 1H-taurine, Fig. 2A). Intensities of the isotopic shifts diminished rapidly at longer delay times, although the decay of both species was significantly slower and overall intensities were greater with 2H-taurine.
Fig. 2.

Resolution of transient oxygen species of TauD. (A) Raman spectra with 1H- and 2H-taurine at -36 °C are shown as  isotope differences (Gray), which reveal changes in oxygen vibrations as pairs of inverted bands (Fig. S2). Black traces show simulated spectra. Frequencies of a laser plasma line (▴) and a major ethylene glycol peak (♦) are indicated. Previously reported data for 1H-taurine at 0.22 s (6) are shown for comparison. (B) Intensity profiles of individual F4, F3, and FX species in simulated
isotope differences (Gray), which reveal changes in oxygen vibrations as pairs of inverted bands (Fig. S2). Black traces show simulated spectra. Frequencies of a laser plasma line (▴) and a major ethylene glycol peak (♦) are indicated. Previously reported data for 1H-taurine at 0.22 s (6) are shown for comparison. (B) Intensity profiles of individual F4, F3, and FX species in simulated  difference spectra. The ∼800 cm-1 shift was modeled as (i) separate but overlapped species, F4 and FX (Solid Line), or (ii) as F4 alone (Dashed Line).
difference spectra. The ∼800 cm-1 shift was modeled as (i) separate but overlapped species, F4 and FX (Solid Line), or (ii) as F4 alone (Dashed Line).
The 16O/18O difference spectra around 800 cm-1 at the shortest delay times (0.8 s for 2H- and 0.4 s for 1H-taurine) could be satisfactorily described by a single 1H/2H-sensitive isotopic shift at 825/788 and 828/791 cm-1, although the features were somewhat asymmetrical with both substrates. For 2H-taurine the 18O mode exhibited a shoulder at the lower frequency side and lower intensity than the better-defined 16O mode. For 1H-taurine both oxygen isotope modes were of similar intensity and width, but a shoulder was observed around 815 cm-1. As the intensity of the 16O mode decreased with time, a pronounced downshift of the positive bands from 825 and 828 to ∼815 cm-1 was observed with both substrates. The  band (negative) did not show a frequency shift over time. We interpret the clear shift of the 16O mode as arising from a third oxygen isotope-sensitive species with a 16O frequency of ∼815 cm-1 and an isotopic shift of ∼25–30 cm-1. The three oxygen isotope-sensitive vibrations with 16O modes at ∼825, 578, and ∼815 cm-1 are termed here the F4, F3, and FX species, respectively.
band (negative) did not show a frequency shift over time. We interpret the clear shift of the 16O mode as arising from a third oxygen isotope-sensitive species with a 16O frequency of ∼815 cm-1 and an isotopic shift of ∼25–30 cm-1. The three oxygen isotope-sensitive vibrations with 16O modes at ∼825, 578, and ∼815 cm-1 are termed here the F4, F3, and FX species, respectively.
Modeling of the Transient Spectra.
Temporal intensity profiles and accurate frequencies of the F4, F3, and FX species observed for 1H- and 2H-taurine were obtained by spectral global regression analysis (Fig. 2B). Whereas individual early spectra in the 800 cm-1 region were well described as a single F4 species, global simulation of the complete dataset (Fig. 2B, dashed line) yielded a low fidelity of fit because of a shift from 826 to 815 cm-1 over time (Fig. S3). To model the observed frequency changes it was necessary to use two independent but overlapping species F4 plus FX (solid lines in Fig. 2B; Fig. S3). Difference spectra at 0.22 s for 1H-taurine and 0.75 s for 2H-taurine provided a good estimate for frequencies and width of F4. For the dual-shift model, the spectrum of the 2H-taurine derivative at 3 s provided an estimate for the FX species. These values were used as initial guesses in the subsequent global regression analysis of the dual-shift model. Comparison of the regression results for models of F4 only or overlapping F4 and FX shifts are provided in SI Text and Fig. S3. The 16O frequency of the FX species was 815 cm-1 (Fig. 2A) with a  isotopic shift of ∼29 cm-1. Unlike the shift of the 16O mode, the broadening of the 18O mode with 2H-taurine showed no clear time dependence and was modeled as a vibrational energy transfer common in proteins (11, 12), as described in SI Text.
isotopic shift of ∼29 cm-1. Unlike the shift of the 16O mode, the broadening of the 18O mode with 2H-taurine showed no clear time dependence and was modeled as a vibrational energy transfer common in proteins (11, 12), as described in SI Text.
In contrast to the 800 cm-1 region, the spectra in the ∼600 cm-1 region were modeled as a single F3 species. Individual (Fig. 3) and time-dependent (Fig. 2) spectra of the 578 cm-1 species with 1H- and 2H-taurine isotopes were satisfactorily modeled by using a single, symmetrical frequency shift with a half-width of 16 cm-1. This mode in the 1H-taurine data from the earlier study (0.22 s) was not included in the analysis because of its broadening due to the lower resolution (see SI Text).
Fig. 3.

Sensitivity of TauD intermediates to 1H/2H substitution. Data are shown as 16O - 18O isotopic differences observed at 1.5 s (Gray) along with simulated spectra (Black). Individual contributions of F4 (Red), FX (Blue), and baselines (Dashed Line) are shown. Intensities of individual spectra in both regions were adjusted for clarity. Experimental conditions are the same as in Fig. 2.
The temporal dependencies of the intensities for F4, F3, and FX obtained during global regression of the complete dataset are compared in Fig. 2B. We conclude that all three profiles are noncoincident, with F4 exhibiting the fastest, exponential decay. F3 was delayed in comparison to F4, particularly with 2H-taurine. FX showed a biphasic profile, developing early in the reaction with 2H-taurine, and exhibited the longest lifetime among the three species. 2H-taurine led to an expected increase in the lifetime and intensity of the F4 species (13) and, importantly, had similar effects on the F3 and FX species.
Isotopic Sensitivity.
Visual comparison of transient spectra showed a 3–4 cm-1 increase in the frequency of F4 from ∼825 to 828 cm-1 upon 1H/2H-taurine substitution. Global regression to an F4-only model yielded shifts of +5.1 ± 1.1 (1H/2H-taurine) and -33.1 ± 5.2 cm-1 ( ) for the 820.4 ± 1.0 cm-1 mode. The small amplitude and large error in the
) for the 820.4 ± 1.0 cm-1 mode. The small amplitude and large error in the  shift were caused by the downshift of the 16O mode at later times. Corresponding shifts in the dual-species F4/FX model confirmed the 1H/2H sensitivity of F4 but provided more stringent values of +1.8 ± 1.8 and -37.0 ± 0.7 cm-1 for the 824.6 ± 1.8 cm-1 mode of F4, respectively (Fig. 3). A part of the observed shift is clearly seen in Fig. 2, and the increased uncertainty in the position and the 1H/2H sensitivity of F4 are attributed to the spectral overlap with FX. The F3 species showed -23.2 ± 0.6 (
shift were caused by the downshift of the 16O mode at later times. Corresponding shifts in the dual-species F4/FX model confirmed the 1H/2H sensitivity of F4 but provided more stringent values of +1.8 ± 1.8 and -37.0 ± 0.7 cm-1 for the 824.6 ± 1.8 cm-1 mode of F4, respectively (Fig. 3). A part of the observed shift is clearly seen in Fig. 2, and the increased uncertainty in the position and the 1H/2H sensitivity of F4 are attributed to the spectral overlap with FX. The F3 species showed -23.2 ± 0.6 ( ) and +1.4 ± 0.7 cm-1 (1H/2H-taurine) shifts of the 578.3 ± 0.7 cm-1 mode. Corresponding shifts of the 815.5 ± 1.8 cm-1 mode of FX were -28.5 ± 1.8 and -2.9 ± 1.7 cm-1. Media 1H/2H substitution for the 2H-taurine sample caused additional shifts of -1.0 ± 1.1, -0.4 ± 0.4, and -1.4 ± 0.8 cm-1 for F4, F3, and FX, respectively, which are insignificant considering the indirect frequency calibration used for 2H media because of solvent frequency shifts.
) and +1.4 ± 0.7 cm-1 (1H/2H-taurine) shifts of the 578.3 ± 0.7 cm-1 mode. Corresponding shifts of the 815.5 ± 1.8 cm-1 mode of FX were -28.5 ± 1.8 and -2.9 ± 1.7 cm-1. Media 1H/2H substitution for the 2H-taurine sample caused additional shifts of -1.0 ± 1.1, -0.4 ± 0.4, and -1.4 ± 0.8 cm-1 for F4, F3, and FX, respectively, which are insignificant considering the indirect frequency calibration used for 2H media because of solvent frequency shifts.
Mixed Oxygen Isotope Studies.
To ascertain the number of oxygen atoms associated with each of the three transient oxygen species, cryogenic continuous-flow Raman studies were carried out by using the mixed oxygen isotope 16O18O (see description in SI Text). The  and
and  difference spectra yielded frequencies identical to each other and to those observed for the
difference spectra yielded frequencies identical to each other and to those observed for the  difference (Fig. 4). Normalized intensities of the isotopic shifts observed with 16O18O were half of those observed for the
difference (Fig. 4). Normalized intensities of the isotopic shifts observed with 16O18O were half of those observed for the  difference. The difference between the 16O18O derivative and the average of the
difference. The difference between the 16O18O derivative and the average of the  and
and  derivatives yielded no new isotopic shifts. The feature-rich double difference spectra characteristic of dioxygen compounds (Fig. S2, Bottom, Right) were not observed for any of the TauD species described in this study, ruling out the presence of an intact O─O bond in any of these intermediates. The featureless
derivatives yielded no new isotopic shifts. The feature-rich double difference spectra characteristic of dioxygen compounds (Fig. S2, Bottom, Right) were not observed for any of the TauD species described in this study, ruling out the presence of an intact O─O bond in any of these intermediates. The featureless  difference spectrum shows that all intermediates contain a single atom derived from O2.
difference spectrum shows that all intermediates contain a single atom derived from O2.
Fig. 4.

Sensitivity of TauD intermediates to 16O18O substitution. Intensities of isotopic difference spectra (Gray) between symmetrically ( or
or  ) and asymmetrically (16O18O) labeled derivatives were normalized by using an internal standard, and the simulated spectra (Black) were obtained by using the results in Fig. 2. Experimental conditions are the same as in Fig. 3 except for an increased spectral resolution.
) and asymmetrically (16O18O) labeled derivatives were normalized by using an internal standard, and the simulated spectra (Black) were obtained by using the results in Fig. 2. Experimental conditions are the same as in Fig. 3 except for an increased spectral resolution.
Oxygen-Labeled Model Compounds.
To assist in the structural identification of the unique oxygen-sensitive species, we examined the magnitudes of 16O/18O isotopic shifts for compounds that resemble potential oxygenation products—isopropanol, propionic acid, and their salts—as shown in Fig. 5 and Figs. S4 and S5.
Fig. 5.

Effect of metal binding on oxygen vibrations of secondary alcohol derivatives. Infrared absorption spectra of (a) isopropanol, (b) Na+ isopropoxide, (c) Zn2+ diisopropoxide, and (d) Zn2+ monoisopropoxide complexes are shown as 16O - 18O isotopic difference. Assignment of O─C─C symmetrical and asymmetrical stretching modes is shown by arrows. The νOCCsm region in traces b and c is magnified for clarity.
Although the O─C─C symmetrical stretching mode of isopropanol (νOCCsm = 818/807 cm-1, Fig. 5) is very close in frequency to FX (816 cm-1), the IR and Raman isotopic difference spectra showed a much smaller 16O/18O isotopic shift (Δν = 11 cm-1, trace a). Formation of the Na+ isopropoxide complex caused significant changes in oxygen-sensitive vibrations in the νOCCsm and νOCCas regions where multiple narrow shifts were observed (trace b). When Na+ was replaced with a limited amount of Zn2+ to form a highly insoluble diisopropoxide complex, vibrational changes were partially reversed and the observed 16O - 18O difference spectrum (trace c) was closer to that of isopropanol (trace a) except for the appearance of two narrow shifts in the νOCCsm region. Binding of additional Zn2+ to form a THF-soluble monoisopropoxide complex resulted in a further downshift of oxygen-sensitive vibrations, a change in their relative intensities, and a significantly larger Δν = 19 cm-1 of a single mode in the νOCCsm region (trace d). This large shift was observed in the absolute spectra of Zn2+ monoisopropoxide derivatives (Fig. S4, trace b) excluding the possibility that it is the result of two small overlapping shifts such as observed in the case of Zn2+ diisopropoxide derivatives (Fig. S4, trace d).
To identify the 16O/18O shift of the OCO scissoring mode (δOCO) of singly labeled propionic acid, a 50% 18O-enriched sample was prepared that is expected to yield a 1∶2∶1 ratio of unlabeled, singly, and doubly labeled isotopes as opposed to mostly doubly labeled isotope in a 90% 18O-enriched sample. A broad Raman vibration was observed at 844 and 827 cm-1 for unlabeled and 90% 18O-enriched propionic acid, respectively; vibrations were not resolved for the 50% enriched sample. Three different well-resolved vibrations were observed at 867, 862, and 850 cm-1 in the solid state Raman spectrum of the 50% enriched Na propionate (Fig. S5) with relative intensities of 1∶2∶1 corresponding to 0/2, 1/2, and 2/2 exchanged oxygen atoms. The same pattern and isotopic shifts were observed for Zn2+ propionate at 906, 894, and 881 cm-1 (Fig. S5). The 16O18O mode observed for both salts showed no evidence of broadening or splitting indicative of asymmetry in binding for both propionate salts.
Discussion
Our cryogenic continuous-flow Raman studies have allowed us to observe three oxygen isotope-sensitive modes with noncoincident temporal profiles. The distinct timing for each feature demonstrates that no two modes arise from the same species (e.g., stretching and bending modes) and the different species are not in rapid equilibrium, such as observed for intermediates in CytC3 and SyrB2 (9, 10). Thus, we conclude that three separate detectable species exist in this reaction, which progress in the order of F4 → F3 → FX (Fig 2B).
Our earlier assignment of νFeIV═O in F4 was based on the frequency and amplitude of the  isotope shift (6); however, this mode falls close to typical values of the νO─O of iron-peroxo complexes. The 578 cm-1 mode is similarly close to typical values for the νFe─O and δFe─O─O modes of peroxo and superoxo complexes. We utilized asymmetrically labeled 16O18O oxygen (Fig. 4) to distinguish unequivocally the number of oxygen atoms associated with F4 and the other two species (14). We observed no new frequencies in the isotopic difference or double difference spectra involving the 16O18O derivative of TauD and conclude that a single oxygen atom is present in all three species.
isotope shift (6); however, this mode falls close to typical values of the νO─O of iron-peroxo complexes. The 578 cm-1 mode is similarly close to typical values for the νFe─O and δFe─O─O modes of peroxo and superoxo complexes. We utilized asymmetrically labeled 16O18O oxygen (Fig. 4) to distinguish unequivocally the number of oxygen atoms associated with F4 and the other two species (14). We observed no new frequencies in the isotopic difference or double difference spectra involving the 16O18O derivative of TauD and conclude that a single oxygen atom is present in all three species.
The frequency, 16O/18O isotope sensitivity, and 1H/2H-taurine kinetics of F4 agree with the FeIV═O intermediate previously examined by Mössbauer effect, x-ray absorption, and stopped-flow UV-visible spectroscopies (4, 5, 13). 2H-taurine labeling caused a 2 cm-1 increase in the frequency of F4 (Fig. 3). Solvent 1H/2H substitution had no discernible effect (< 2 cm-1) on the observed frequencies (Fig. 3), indicating that no exchangeable protons interact directly with the oxygen atom in F4. Whereas a longer lifetime for the FeIV═O species with 2H-taurine was expected, the effect of deuteration on the νFe═O mode provides direct evidence for interactions between the oxo group and substrate hydrogen (15–18).
A single oxygen atom associated with the 578 cm-1 mode of F3 suggests an Fe─O(H) structure, but F3 showed no sensitivity to substrate or bulk solvent 1H/2H substitution (Fig. 3). This mode is consistent with the calculated νFe─O of tripodal FeIII─O- (19) but is lower than the νFe─O (671 cm-1) of the only reported non-heme FeIII─O- model complex (20). This frequency is at the high end of calculated νFe─OH values (19) and distinct from those of most experimentally measured hydroxyl modes, but it is very close to the νFe(IV)─OH mode of protonated compound II of chloroperoxidase (CPO) at 565 cm-1 (21) and the νFe(III)─OH modes of di-iron hydroxomethemerythrin at 565 cm-1 (22) and a synthetic FeIII─OH model at 574 cm-1 (23). The 16O/18O shifts are similar between all of these species and F3. Unlike F3, however, CPO and hydroxomethemerythrin exhibit characteristic 1H/2H downshifts of 13 and 5 cm-1, respectively. Such downshifts are well documented for the Fe─OH(2) stretching mode of ferric aqua- and hydroxy-hemes (typically 5–20 cm-1). A few heme ferric-hydroxy complexes lack 1H/2H sensitivity or even exhibit an upshift with deuteration. This unusually small sensitivity of the νFe─OH mode to deuteration was rationalized by (i) a highly bent geometry of the hydroxyl ligand and (ii) strong hydrogen bonding that leads to an upshift of νFe─O upon deuteration (16–18). Indeed, the crystal structure of the model FeIII─OH revealed a highly bent hydroxide (∠Fe─O─H = 93°) because of interactions with the iron ligand carboxylate, as well as strong hydrogen bonding between the oxygen and three other caging ligands (23). One could argue that a similar configuration may take place in F3, particularly if iron-bound D101 could rotate to form a hydrogen bond with the hydroxo group. On the other hand, the insensitivity of F3 to bulk media deuteration clearly argues against additional hydrogen bonding to the hydroxo oxygen, without which hydrogen bonding with the carboxylate would weaken. Whereas small rearrangements relative to the static crystal structure could take place, there is no evidence for other steric interference in the active site that could significantly distort the Fe─O─H angle. Although the protonation state of the oxygen in F3 cannot be assigned unambiguously, the structure of the active site and the lack of detectable media 1H/2H shifts raise a significant probability that the oxo group in F3 is deprotonated.
Although the intensity of FX is low under the present conditions, it is sufficient to discern clear time-dependent spectral changes. Accounting for FX in spectral modeling resulted in a noticeably better fit to the experimental spectra (Fig. S3), and the Δν16O/18O = -37 cm-1 observed for F4 in the F4/FX model is a better match for the expected Δν = -36 cm-1 than the Δν = -33 cm-1 observed for the F4-only model. Our data indicate a small (∼3 cm-1) decrease in the observed frequency of FX with 2H-taurine to the extent that its low intensity with 1H-taurine allows. The slower decay of FX with 2H-taurine implies the kinetic involvement of a substrate proton(s) in the subsequent step. FX must contain a single oxygen atom from O2, and yet it cannot be assigned to known Fe-oxygen compounds: The 29 cm-1 16O/18O shift is smaller than the ΔνFe(IV)═O = ∼ 36 cm-1, and the 815 cm-1 frequency is too high for an Fe─OX stretching mode (νtyp. < 600 cm-1) (6), suggesting that FX originates from an oxygenated product of the reaction.
The δOCO mode of succinate, which incorporates one atom from O2 (Fig. 6A) (24), is not likely to give rise to FX because the Δδ = ∼ 13 cm-1 of the symmetrical salt of the 16OC18O carboxylate is too small compared to that of FX (Fig. S5). This shift may increase in a monodentate Fe-carboxylate complex similar to Zn2+ alkoxide (Fig. 5), but the intensity profile of FX with 2H-taurine (Fig. 2B) further argues against its assignment to succinate, which should be formed concurrently with F4. The second atom from O2 forms the product aldehyde (Figs. 1 and 6) via a transient C1 alcohol or alkoxide of taurine. An aldehyde would not coordinate to FeII, and its oxygen vibrations appear at significantly higher frequencies. The νOCCsm of isopropanol (Fig. 5), the simplest secondary alcohol, is very close to FX in frequency but has a much smaller 16O/18O shift (Δν = 11 cm-1). This isotopic shift almost doubled in one of the Zn2+-isopropoxide complexes to Δν = 19 cm-1 (Fig. 5). Whereas it is still smaller than the shift observed for FX, our results show that the geometry of the alkoxide complex had greater effect on oxygen vibrations than the nature of the metal. Thus, it is vibrationally feasible that the formation of a transient FeII-alkoxide complex can give rise to the transient FX species. The amplitude of the isotopic shift in TauD can be further affected by the geometry of the complex, the mass and electronic effect of a sulfur atom, and the vibrational interactions at the active site.
Fig. 6.

Possible mechanisms of taurine oxygenation by TauD. The structure of the active site following O─O bond cleavage (A) is the FeIV═O species. According to the standard pathway, species A oxidizes substrate to form the Fe─OH complex (B), which is followed by radical rebinding to form an alcohol (C) and decomposition into products (D). Alternative pathways involve formation of an alkoxide (C′) upon concerted (B → C′) or stepwise (B → B′ → C′ deprotonation via a transient FeIII─O- species. Atoms originating from O2 are grayed; α-ketoglutarate (R1) and taurine (R2) are partially abbreviated.
Resonance enhancement of Raman spectra requires electronic absorption near the excitation wavelength (364 nm). Stopped-flow absorption spectroscopy of the TauD reaction provided no evidence of multiple distinctive optical species in the near-UV region at higher temperatures (4, 25, 26), nor were such species seen during in situ optical measurements under cryogenic continuous-flow conditions (6). Thus, either the absorption changes at 300–400 nm are contributed by several unresolved species with broad absorption spectra or the new species do not absorb in this region. Enhancement of F4 may occur via pre- or near-resonance with its 318-nm absorption (4), whereas excitation of FX may be even less favorable implying either a larger energy difference from the electronic transition or a weaker chromophore. Although the FeII alkoxo complex should not exhibit charge transfer transitions similar to the FeII-αKG-TauD complex (27), the low symmetry of the FeII site in TauD may favor spin-forbidden mid-UV transitions below 300 nm, thus allowing for a weak enhancement of oxygen vibrations. In any case, the rapid isotope-dependent disappearance of FX shows that it originates from a transient substrate-derived species.
Possible oxygenation mechanisms following the FeIV═O species in the reaction of TauD are depicted in Fig. 6. Binding of α-ketoglutarate and taurine at the active site allows for O2 binding, O─O bond cleavage, and concomitant formation of CO2, succinate, and the highly oxidized FeIV═O species (F4) (Fig. 1A–F). The oxo group of F4 is poised to oxidize the substrate (Fig. 6) via direct interactions with a proton on the C1 carbon as revealed by its substrate isotope sensitivity, similar to heme FeIV═O species (16–18). A short Fe─C1 distance in TauD agrees with crystallographic data of the enzyme-substrate complex (28, 29) and with electron spin-echo envelop modulation spectroscopy results that place a substrate C1 deuteron 1.8 Å from the FeII─NO center (30).
According to the generally described mechanism for this enzyme family (7, 31, 32), taurine oxidation proceeds via direct hydrogen atom transfer from C1 of the substrate to the FeIV═O group yielding a FeIII─OH and a substrate radical (Fig. 6, A → B), supported by a large taurine 1H/2H kinetic isotope effect on the decay of the FeIV species. Our results indicate that a metastable deprotonated FeIII─O- species (F3) may be formed as the FeIV═O decays. F3 could be formed by rapid proton transfer from FeIII─OH to a nearby base immediately following hydrogen atom transfer (B → B′). A less likely direct route (not depicted) may involve an A → B′ transition via a concerted electron and proton transfer from the C1 carbon to the FeIV═O and a base, respectively. Both pathways will lead to the 1H/2H isotope-sensitive (33) formation of an FeIII─O- species and protonation of a nearby base contingent on the relative pKas of the hydroxide and the base. In the absence of the base only B is expected.
The classical radical rebinding mechanism applied to TauD posits the transfer of OH• from FeIII─OH onto  to form the alcohol (B → C), deprotonation, and cleavage of the C1─S bond with the formation of sulfite, aminoacetaldehyde, and regeneration of FeII (C → D). A neutral alcohol will not bind to a metal and is expected to exhibit a much narrower 16O/18O shift than we observe; therefore, structure C is not likely to give rise to the FX species, which is vibrationally distinct from an alcohol. We propose that an FeII─O─C1 alkoxy complex of taurine (C′) is formed instead of an alcohol, tentatively accounting for the observed FX. The formation of C′ from FeIII─OH (B → C′) would require concerted deprotonation, because metal alkoxides undergo rapid hydrolysis. If the oxygen is already deprotonated in F3, formation of FX could occur directly (B′ → C′). Both routes would likely be reversible until the C1─S bond is cleaved (C′ → D). The substrate proton can subsequently protonate the sulfonate oxygen to maintain charge neutrality, thus explaining the distinctly longer lifetimes observed for F3 and FX with 2H-taurine.
to form the alcohol (B → C), deprotonation, and cleavage of the C1─S bond with the formation of sulfite, aminoacetaldehyde, and regeneration of FeII (C → D). A neutral alcohol will not bind to a metal and is expected to exhibit a much narrower 16O/18O shift than we observe; therefore, structure C is not likely to give rise to the FX species, which is vibrationally distinct from an alcohol. We propose that an FeII─O─C1 alkoxy complex of taurine (C′) is formed instead of an alcohol, tentatively accounting for the observed FX. The formation of C′ from FeIII─OH (B → C′) would require concerted deprotonation, because metal alkoxides undergo rapid hydrolysis. If the oxygen is already deprotonated in F3, formation of FX could occur directly (B′ → C′). Both routes would likely be reversible until the C1─S bond is cleaved (C′ → D). The substrate proton can subsequently protonate the sulfonate oxygen to maintain charge neutrality, thus explaining the distinctly longer lifetimes observed for F3 and FX with 2H-taurine.
The possible formation of ferric oxide as F3 (B → B′ or A → B′) requires a base that is stronger than the FeIII─OH group. The [FeIIIH3buea(OH)]- complex has a pKa of 25 and substitution of MnIII for FeIII increased the pKa of the terminal oxo ligand to 28.3 (34). Oxidation of [MnIIIH3buea(OH)]- to [MnIVH3buea(OH)]0 decreased the pKa from 28.3 to ∼15 (35). A pKa of > 15 was also estimated for porphyrinoid [(TBP8Cz)MnIV(OH)]0 (36) indicating that the macrocycle does not affect the MnIV─OH pKa value. These observations suggest that the FeIV─OH in FeIVH3buea(OH)]0 or porphyrin analogs will be basic under similar conditions (pKa≥12). On the other hand, the porphyrin FeIV─OH complex in most histidine-ligated peroxidases is significantly more acidic (pKa < 4) (19). Therefore, heme and non-heme FeIII─OH complexes in proteins are also likely to be significantly more acidic than in corresponding model compounds. The structure of FeII-αKG-TauD suggests that H99 and H255 are neutral and, thus, the FeIII─OH complex should be neutral (Fig. 6, B′), similar to most ferric histidine-ligated hemes. In contrast, a net negative charge on [FeIIIH3buea(OH)]- and cysteine-ligated hemes (37) is likely to contribute to the higher proton affinity of their oxo complexes (19, 38). The paramount role of protein ligands and local charges in determining the pKa of metal-bound oxygen is further exemplified by siderophores, where the pKa1 of bound water can shift by as much as 8 pH units (39). Greater variability is expected in the low dielectric environment of a protein.
Direct assessment of the pKa for an FeIII─OH is of great significance, especially for iron oxygenases, because the proton affinity of the reduced state has been correlated with the ability of the high valent species to abstract hydrogen in model systems (40), as well as in heme enzymes (19). The deprotonation of FeIII─OH has not been observed experimentally in any biological process, although FeIII─O- states have been generated cryogenically (41). Moreover, the protonation of oxo groups in globins occurs relatively slowly even at T > 160 K in spite of a strong coupling to 1H, as opposed to peroxidases where protonation occurs at 77 K. Such a difference in proton affinities of FeIII─O(H) states correlates with the relative basicity of cysteine-ligated FeIV─OH in CPO (19, 38) and the “push” of electron density toward oxygen in imidazolate-ligated peroxidases (37) as opposed to structurally similar hemes in globins. The effect of axial ligands illustrates the latitude of ligand-mediated protein control over the reactivity of heme complexes, and an even greater role of metal ligands may be expected in structurally diverse non-heme enzymes.
Conclusions
This study presents the direct observation of three transient iron species occurring during substrate oxygenation by TauD. Resolution of two unique intermediates in addition to characterization of the FeIV═O species allowed us to develop an alternative mechanism for catalysis by the FeII/αKG-dependent oxygenases. Ferric-oxo and metal-substrate alkoxo complexes similar to those proposed here also may occur transiently in a broader range of Fe oxygenases. Chemical models and molecular dynamics calculations for the observed species will further our understanding of biological oxygen catalysis with implications for a diverse array of related enzymes.
Materials and Methods
Enzyme Purification.
TauD was purified as an apoprotein from Escherichia coli BL21(DE3) containing pME4141 as previously described (42, 43). The isolated enzyme was assayed by reaction of the released sulfite with Ellman’s reagent, providing a specific activity ranging from 2.4 to 3.6 μmol min-1 mg-1. Because of the nature of the experiment, large quantities of protein are required; an estimated 17 g of purified TauD was consumed while generating the results reported here.
Isotopic Substitution.
1H-taurine (Sigma), 2H-taurine (99.5%, C/D/N Isotopes, Inc.), and  (≥99%, Isotec Laboratories, Inc.) were used as acquired. Asymmetrically labeled oxygen (16O18O) was synthesized as described in SI Text. Bulk solvent 2H substitution of the 50% ethylene glycol plus 25 mM Tris in
(≥99%, Isotec Laboratories, Inc.) were used as acquired. Asymmetrically labeled oxygen (16O18O) was synthesized as described in SI Text. Bulk solvent 2H substitution of the 50% ethylene glycol plus 25 mM Tris in 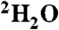 buffer was carried out by alternating evaporation and dilution with 50% volumes of
buffer was carried out by alternating evaporation and dilution with 50% volumes of  (99%, Cambridge Isotopes) up to five times to achieve a final exchangeable 2H enrichment > 95%. This substitution resulted in shifting of most of the ethylene glycol Raman vibrations. The pH of the resulting buffer was adjusted while correcting for 2H activity (p2H = 0.4 pH units greater than the pH meter reading). Isotopically substituted model compounds were prepared by oxygen exchange with H218O as described in SI Text.
(99%, Cambridge Isotopes) up to five times to achieve a final exchangeable 2H enrichment > 95%. This substitution resulted in shifting of most of the ethylene glycol Raman vibrations. The pH of the resulting buffer was adjusted while correcting for 2H activity (p2H = 0.4 pH units greater than the pH meter reading). Isotopically substituted model compounds were prepared by oxygen exchange with H218O as described in SI Text.
Sample Preparation.
Anaerobic solutions of 0.5 mM TauD in 25 mM Tris, pH 8.0, 50% (vol/vol) ethylene glycol were prepared by 10 cycles between mild vacuum and Ar gas. Anaerobic solutions of 0.5 mM ferrous ammonium sulfate, 2.0 mM 1H- or 2H-labeled taurine, 2.0 mM Na α-ketoglutarate, and 0.1 mM Na ascorbate were added to anaerobic TauD and formation of the ternary complex was confirmed optically (43). Sample integrity during Raman measurements was confirmed by recording the visible absorption spectrum before and after the experiment by using a 1-cm flow cuvette and an optical fiber spectrophotometer (model S2000; Ocean Optics).
Cryogenic Continuous-Flow Raman Spectroscopy.
The continuous-flow approach with active mixing used in this study was similar to that implemented earlier (6) but with major improvements of the experimental setup (see SI Text and Fig. S1). Temperature at the coldest point (mixer outlet, -36 ± 1 °C) was controlled by a model 34 temperature controller (Cryogenic Control Systems, Inc.) via a stream of cold N2 gas and was reproducible to ± 0.1 °C.
During the measurements, TauD samples and oxygenated buffers were continuously mixed in equal volumes, delivered into a rectangular quartz flow cuvette (25 mm long, cross-section 0.25 × 0.8 mm), and probed at ∼5 mm from the mixer outlet with the dead volume of ∼2 μL. Sample syringes were driven by computer-controlled high pressure syringe pump modules (Harvard Apparatus) at constant flow rates of 20–160 μL/ min (after mixing), which resulted in delay times of 0.75–6 s for the current dead volume.
Raman scattering was excited with the 363.8-nm line of an argon ion laser (Coherent, model 70), as described previously (6), with laser power of 60 mW at the point of sampling. The scattered light was collected at 90° geometry by using an F = 0.6 aspherical lens and analyzed by using a single polychromator (model TRIAX 550; Jobin Yvon) equipped with an imaging CCD detector (model 5000; Jobin Yvon). The typical spectral slit width was 8 cm-1, except for the 16O18O and  measurements where the spectral slit width was reduced to 5 cm-1; the spectra were recorded at 0.36 cm-1/pixel and subsequently smoothed by using a binomial algorithm with the half-width of 2 cm-1 to preserve experimental resolution. Rayleigh scattering was rejected by using a notch filter (Kaiser Optical Systems). To minimize the effect of velocity distribution in laminar flow along the cuvette walls, the laser beam was focused to 0.15 mm diameter in a 0.25-mm-wide cuvette and only scattering from the central 80% height of the cuvette (0.8 mm) was collected. A two-step calibration of Raman shifts against external and in situ standards was used to increase reproducibility to ± 0.3 cm-1. For further details on sample excitation and detailed calibration procedures, see SI Text.
measurements where the spectral slit width was reduced to 5 cm-1; the spectra were recorded at 0.36 cm-1/pixel and subsequently smoothed by using a binomial algorithm with the half-width of 2 cm-1 to preserve experimental resolution. Rayleigh scattering was rejected by using a notch filter (Kaiser Optical Systems). To minimize the effect of velocity distribution in laminar flow along the cuvette walls, the laser beam was focused to 0.15 mm diameter in a 0.25-mm-wide cuvette and only scattering from the central 80% height of the cuvette (0.8 mm) was collected. A two-step calibration of Raman shifts against external and in situ standards was used to increase reproducibility to ± 0.3 cm-1. For further details on sample excitation and detailed calibration procedures, see SI Text.
Spectral Analysis.
Complete time-resolved datasets of 16O/18O difference spectra with 1H- and 2H-taurine were analyzed simultaneously for frequencies and amplitudes of 16O/18O and 1H/2H-taurine isotopic shifts by using global nonlinear regression analysis using custom routines for Igor Pro (WaveMetrics, Inc.). Reported frequency and amplitude errors represent ± 1 SD of the value obtained during regression analysis. See SI Text for further details.
Supplementary Material
Acknowledgments.
This work was supported by the National Institutes of Health Grants GM063584 (to R.P.H.) and GM070544 (to D.A.P.). Synthesis of 16O18O was generously supported by Drs. John L. Lipscomb and the late Gerald T. Babcock and was carried out under the auspices of the Office of Basic Energy Sciences, U.S. Department of Energy.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911565107/DCSupplemental.
References
- 1.Kovaleva EG, Lipscomb JD. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 3.Makris TM, Davydov R, Denisov IG, Hoffman BM, Sligar SG. Mechanistic enzymology of oxygen activation by the cytochromes P450. Drug Metab Rev. 2002;34:691–708. doi: 10.1081/dmr-120015691. [DOI] [PubMed] [Google Scholar]
- 4.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 5.Riggs-Gelasco PJ, et al. EXAFS spectroscopic evidence for an FeO unit in the Fe(IV) intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 6.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/α-ketoglutarate dioxygenase. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 7.Hausinger RP. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 8.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc Natl Acad Sci USA. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 10.Matthews MM, et al. Substrate-triggered formation and remarkable stability of the C-H-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hashimoto S, Tatusuno Y, Kitagawa T. Resonance Raman evidence for oxygen exchange between FeIV═O heme and bulk water during enzymic catalysis of horseradish peroxidase and its relation with the heme-linked ionization. Proc Natl Acad Sci USA. 1986;83:2417–2421. doi: 10.1073/pnas.83.8.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizutani Y, Kitagawa T. Direct observation of cooling of heme upon photodissociation of carbonmonoxy myoglobin. Science. 1997;278:443–446. doi: 10.1126/science.278.5337.443. [DOI] [PubMed] [Google Scholar]
- 13.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD) J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 14.Kitagawa T, Ogura T. Oxygen activation mechanism at the binuclear site of heme-copper oxidase superfamily as revealed by time-resolved resonance Raman spectroscopy. Prog Inorg Chem. 1997;45:431–479. [Google Scholar]
- 15.Feis A, Marzocchi MP, Paoli M, Smulevich G. Spin state and axial ligand bonding in the hydroxide complexes of metmyoglobin, methemoglobin, and horseradish peroxidase at room and low temperatures. Biochemistry. 1994;33:4577–4583. doi: 10.1021/bi00181a019. [DOI] [PubMed] [Google Scholar]
- 16.Sitter AJ, Reczek CM, Terner J. Heme-linked ionization of horseradish peroxidase compound II monitored by the resonance Ramn Fe(IV)═O stretching vibration. J Biol Chem. 1985;260:7515–7522. [PubMed] [Google Scholar]
- 17.Hashimoto S, Teraoka J, Inubushi T, Yonetani T, Kitagawa T. Resonance Raman study on cytochrome c peroxidase and its intermediate. Presence of the Fe(IV)═O bond in compound ES and heme-linked ionization. J Biol Chem. 1986;261:1110–1118. [PubMed] [Google Scholar]
- 18.Proshlyakov DA, et al. Selective resonance Raman observation of the “607 nm” form generated in the reaction of oxidized cytochrome c oxidase with hydrogen peroxide. J Biol Chem. 1994;269:29385–29388. [PubMed] [Google Scholar]
- 19.Green MT. Application of Badger’s rule to heme and non-heme iron-oxygen bonds: an examination of ferryl protonation states. J Am Chem Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 20.MacBeth CE, et al. O2 activation by nonheme iron complexes: a monomeric Fe(III)-oxo complex derived from O2. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 21.Stone KL, Behan RK, Green MT. Resonance Raman spectroscopy of chloroperoxidase compound II provides direct evidence for the existence of an iron(IV)-hydroxide. Proc Natl Acad Sci USA. 2006;103:12307–12310. doi: 10.1073/pnas.0603159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiemke AK, Loehr TM, Sanders-Loehr J. Resonance Raman study of oxyhemerythrin and hydroxomethemerythrin. Evidence for hydrogen bonding of ligands to the Fe-O-Fe center. J Am Chem Soc. 1986;108:2437–2443. doi: 10.1021/ja00269a050. [DOI] [PubMed] [Google Scholar]
- 23.Ogo S, et al. Structural and spectroscopic features of a cis (hydroxo)-FeIII-(carboxylato) configuration as an active site model for lipoxygenases. Inorg Chem. 2002;41:5513–5520. doi: 10.1021/ic0200040. [DOI] [PubMed] [Google Scholar]
- 24.Welford RW, et al. Incorporation of oxygen into the succinate co-product of iron(II) and 2-oxoglutarate dependent oxygenases from bacteria, plants and humans. FEBS Lett. 2005;579:5170–5174. doi: 10.1016/j.febslet.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 25.Grzyska PK, et al. Steady-state and transient kinetic analyses of taurine/α-ketoglutarate dioxygenase: effects of oxygen concentration, alternative sulfonates, and active site variants on the Fe(IV) intermediate. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- 26.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM., Jr Kinetic dissection of the catalytic mechanism of taurine:α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 27.Solomon EI, Decker A, Lehnert N. Non-heme iron enzymes: Contrasts to heme catalysis. Proc Natl Acad Sci USA. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elkins JM, et al. X-ray crystal structure of Escherichia coli taurine/α-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry. 2002;41:5185–5192. doi: 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- 29.O’Brien JR, Schuller DJ, Yang VS, Dillard BD, Lanzilotta WN. Substrate-induced conformational changes in Escherichia coli taurine/α-ketoglutarate dioxygenase and insight into the oligomeric structure. Biochemistry. 2003;42:5547–5554. doi: 10.1021/bi0341096. [DOI] [PubMed] [Google Scholar]
- 30.Muthakumaran RB, Grzyska PK, Hausinger RP, McCracken J. Probing the Fe-substrate orientation for taurine/α-ketoglutarate dioxygenase using deuterium ESEEM spectroscopy. Biochemistry. 2007;46:5951–5959. doi: 10.1021/bi700562t. [DOI] [PubMed] [Google Scholar]
- 31.Purpero VM, Moran GR. The diverse and pervasive chemistries of the α-keto acid dependent enzymes. J Biol Inorg Chem. 2007;12:587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]
- 32.Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr Non-heme Fe(IV)-oxo intermediates. Accounts Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohen A, Limbach H-H. Isotope Effects in Chemistry and Biology. Boca Raton, FL: CRC; 2005. [Google Scholar]
- 34.Gupta R, Borovik AS. Monomeric MnIII/II and FeIII/II complexes with terminal hydroxo and oxo ligands: Probing reactivity via O-H bond dissociation energies. J Am Chem Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]
- 35.Parsell TH, Yang M-Y, Borovik AS. C-H bond cleavage with reductants: Re-investigating the reactivity of monomeric MnIII/IV-oxo complexes and the role of oxo ligand basicity. J Am Chem Soc. 2009;131:2762–2763. doi: 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lansky DE, Goldberg DP. Hydrogen atom abstraction by a high-valent manganese(V)-oxo corrolazine. Inorg Chem. 2006;45:5119–5125. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]
- 37.Dawson JD. Probing structure-function relations in heme-containing oxygenases and peroxidase. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 38.Green MT, Dawson JD, Gray HB. Oxoiron(IV) in chloroperoxidase compound II is basic: Implications for P450 chemistry. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 39.Harris WR, Carrano CJ, Raymond KN. Coordination chemistry of microbial iron transport compounds. 16. Isolation, characterization, and formation constants of ferric aerobactin. J Am Chem Soc. 1979;101:2722–2727. [Google Scholar]
- 40.Mayer JM. Hydrogen atom abstraction by metal-oxo complexes: Understanding the analogy with organic reactions. Accounts Chem Res. 1998;31:441–450. [Google Scholar]
- 41.Davydov R, Osborne RL, Kim SH, Dawson JD, Hoffman BM. EPR and ENDOR studies of cryoreduced compound II of peroxidases and myoglobin proton-coupled electron transfer and protonation status of ferryl hemes. Biochemistry. 2008;47:5147–5155. doi: 10.1021/bi702514d. [DOI] [PubMed] [Google Scholar]
- 42.Ryle MJ, et al. O2- and α-ketoglutarate-dependent tyrosyl radical formation in TauD, an α-keto acid-dependent non-heme iron dioxygenase. Biochemistry. 2003;42:1854–1862. doi: 10.1021/bi026832m. [DOI] [PubMed] [Google Scholar]
- 43.Ryle MJ, Padmakumar R, Hausinger RP. Stopped-flow kinetic analysis of Escherichia coli taurine/α-ketoglutarate dioxygenase: interactions with α-ketoglutarate, taurine, and oxygen. Biochemistry. 1999;38:15278–15286. doi: 10.1021/bi9912746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.