

Abstract
The tumor suppressor gene HYPERMETHYLATED IN CANCER 1 (HIC1), which encodes a transcriptional repressor, is epigenetically inactivated in various human cancers. Here, we show that HIC1 is a direct transcriptional repressor of the gene encoding ephrin-A1, a cell surface ligand implicated in the pathogenesis of epithelial cancers. We also show that mouse embryos lacking both Hic1 alleles manifest developmental defects spatially associated with misexpression of ephrin-A1, and that overexpression of ephrin-A1 is a feature of tumors arising in Hic1 heterozygous mice in which the remaining wild-type allele is epigenetically silenced. In breast cancer, we find that ephrin-A1 expression is common in vivo, but that in cell culture, expression of the EphA receptors is predominant. Restoration of HIC1 function in breast cancer cells leads to a reduction in tumor growth in vivo, an effect that can be partially rescued by co-overexpression of ephrin-A1. Interestingly, overexpression of ephrin-A1 in vitro triggers downregulation of EphA2 and EphA4 levels, resulting in an expression pattern similar to that seen in vivo. We conclude that Hic1 spatially restricts ephrin-A1 expression in development, and that upregulated expression of ephrin-A1 resulting from epigenetic silencing of HIC1 in cancer cells may be an important mechanism in epithelial malignancy.
Keywords: Hic1, Ephrin-A1, EphA2, EphA4
Introduction
We first identified the tumor suppressor HYPERMETHYLATED IN CANCER 1 (HIC1) in a region of chromosome 17p13 frequently targeted for allelic loss and CpG island hypermethylation in human cancer (Wales et al., 1995). HIC1 resides within the critical deletion region associated with the Miller-Dieker Syndrome (MDS), a congenital disorder characterized by severe facial and neurologic deficits (Chong et al., 1997; Hirotsune et al., 1997). Homozygous deletion of Hic1 in mice results in embryonic lethality with severe craniofacial and somatic defects similar to many of the abnormalities in MDS (Carter et al., 2000), whereas Hic1 heterozygotes develop age- and gender-dependent tumors associated with promoter hypermethylation and gene silencing of the remaining wild-type allele (Chen et al., 2003). These studies suggest that Hic1 has important roles in development and tumor suppression. However, the mechanisms behind this assumption have not been fully elucidated.
Hic1 is a sequence-specific transcriptional repressor with an N-terminal POZ domain, and five C2H2 zinc fingers at the C-terminus (Deltour et al., 1998; Deltour et al., 1999; Deltour et al., 2002). Recent studies have demonstrated four transcriptional targets of Hic1, the class III histone deacetylase Sirt1 (Chen et al., 2005), the proneural bHLH transcription factor Atoh1 (Briggs et al., 2008), the cell cycle and apoptosis regulator E2F1 (Jenal et al., 2009), and a G-protein coupled receptor CXCR7 (van Rechem et al., 2009). Importantly, deregulation of Sirt1 and Atoh1 in the absence of Hic1 function is associated with widespread effects in both development and tumor growth (Chen et al., 2005; Briggs et al., 2008). Furthermore, these data suggest that the identification of other genes regulated by Hic1 might better define the functional importance of Hic1 in development and epithelial tumor suppression.
Here, we define a role for HIC1 in the regulation of ephrin-A1, which encodes a cell surface ligand for Eph receptors. Eph receptors form the largest subfamily of receptor tyrosine kinases (RTKs). Bidirectional ephrin/Eph signaling occurs in the context of cell-cell interactions and during the establishment of morphogenic boundaries in vertebrate development (Kullander and Klein, 2002; Pasquale, 2005; Pasquale, 2008). Ephrin-A1 is a glycophosphatidylinositol (GPI) linked EphA ligand that is frequently overexpressed in epithelial malignancies (Easty et al., 1999; Ogawa et al., 2000; Nakamura et al., 2005; Herath et al., 2006). Recent studies show that overexpression of ephrin-A1 can promote tumor growth in vivo (Brantley-Sieders et al., 2006; Shi et al., 2008). Using a combination of cDNA array analysis, ephrin-A1 expression studies, and characterization of the ephrin-A1 promoter, we show here that Hic1 is a direct transcriptional repressor of ephrin-A1. In Hic1 deficient mouse embryos, widespread overexpression of ephrin-A1 is seen in epithelial tissues and the central nervous system. In breast cancer cells in which HIC1 is epigenetically silenced, in vivo growth can be blocked by restoration of HIC1 function, and then partially rescued by co-expression of ephrin-A1. Based on our findings, we propose that the misexpression of ephrin-A1 may be important in many of the morphological abnormalities seen in mice lacking Hic1, and that epigenetic silencing of HIC1 in cancer cells may constitute an important step in promoting the growth of breast cancer cells through upregulation of ephrin-A1 in vivo.
Results
Hic1 downregulates expression of ephrin-A1
To search for potential transcriptional targets of HIC1 in carcinomas, we used an adenoviral vector AdHIC1 (Chen et al., 2005; Briggs et al., 2008) to overexpress HIC1 protein in MCF-7 cells, a cell line in which HIC1 expression is epigenetically silenced by promoter hypermethylation (Wales et al., 1995). Western blot analysis of cell lysates with a HIC1 specific antibody revealed sustained, high level protein expression in response to AdHIC1 adenoviral infection over a 10-day period (Fig. 1a). Immunofluoresence analysis of infected cells revealed that overexpressed HIC1 protein was readily detectable in the nucleus (Supplementary Fig. S1). RNA from MCF-7 cells 9 hours after infection with a control virus, or AdHIC1, was analyzed using the Affymetrix platform. Highly significant expression changes of >2 fold were seen in 70 genes, 58 of which were downregulated. Genes repressed by 3 fold or more are shown in Supplementary Table S1. Although a number of important genes involved in cell growth and differentiation are represented, we pursued ephrin-A1 as the leading candidate based on its marked 23.4 fold repression, and the potential functional consequences of ephrin-A1 overexpression in both development and cancer.
Fig. 1.

Effects of HIC1 overexpression on expression of ephrin-A1. (a) Western blot analysis of MCF-7 cell lysates treated with AdHIC1 for 0–14 days. The filter was probed with HIC1 antibody. GAPDH was used as loading control. (b) Quantitative real time RT-PCR assays for the ephrin-A1/β-actin mRNA ratio from cell lines treated with control (Adβ-Gal) or HIC1 expressing (AdHIC1) adenoviruses. Total RNA was prepared at 9 hrs post viral infection, followed by cDNA synthesis and expression analysis. The graph shows mean values and standard deviations of three independent experiments. (c) Western blot analysis of HIC1 and ephrin-A1 in lysates obtained from ZR-75-1 and MDA-MB453 cells infected with Adβ-gal or AdHIC1 adenoviruses. Equal loading was confirmed by immunoblotting for GAPDH. (d) Quantitative real time RT-PCR assays for the ephrin-A1/β-actin mRNA ratio from Hic1 wild-type and knockout MEF cells.
Repression of ephrin-A1 steady-state mRNA expression by HIC1 was confirmed in separate experiments by real-time quantitative RT-PCR in MCF-7 and MDA-MB231, both of which are known to have marked hypermethylation of HIC1 (Wales et al., 1995). As shown in Fig. 1b, re-expression of HIC1 leads to 3.8- and 3.5-fold reduction of ephrin-A1 mRNA in MCF-7 and MDA-MB231 cells respectively. In cell culture, previous studies show that ephrin-A1 protein was expressed at extremely low levels in both MCF-7 and MDA-MB231 cells (Macrae et al., 2005). To investigate whether HIC1 can repress ephrin-A1 expression at the protein level, we overexpressed HIC1 in two breast cancer cell lines, ZR-75-1 and MDA-MB453, in which HIC1 is epigenetically silenced whereas ephrin-A1 is robustly expressed (Supplementary Fig. S2). In both cell lines, overexpression of HIC1 leads to marked downregulation of endogenous ephrin-A1 protein within 48 hrs (Fig. 1c). To validate ephrin-A1 as a Hic1 transcriptional target in a more physiological condition, we examined ephrin-A1 expression in mouse embryonic fibroblasts (MEFs) derived from Hic1+/+, Hic1+/−, and Hic1−/− mice. Knockout of Hic1 leads to 1.32- and 4.15-fold increase of ephrin-A1 mRNA in Hic1+/− and Hic1−/− MEF cells respectively (Fig. 1d). In addition, we used a siRNA-based strategy to knockdown HIC1 in human WI38 cells as previously described (Chen et al., 2005). We found that the mRNA levels of ephrin-A1 increased by nearly 3-fold upon HIC1 knockdown, compared to a non-target siRNA control (Fig. 2). Since aging Hic1+/− heterozygote mice develop carcinomas, sarcomas and lymphomas in which the remaining wild-type allele is silenced by promoter DNA hypermethylation (Chen et al., 2003), we asked whether loss of Hic1 in these tumors was associated with increased expression of ephrin-A1. Of 8 tumors examined, 6 demonstrated membrane and cytoplasmic expression of ephrin-A1 in the absence of Hic1 expression (Supplementary Fig. S3). By contrast, a hemangiosarcoma (Tumor 8), which was not hypermethylated at the wild-type Hic1 allele and which expressed Hic1 protein (Chen et al., 2003; Chen et al., 2004; Chen et al., 2005), lacked ephrin-A1 protein expression (Supplementary Fig. S3). Only 1 of the 8 tumors lacked both proteins. These results indicate that ephrin-A1 expression is downregulated by Hic1.
Fig. 2.
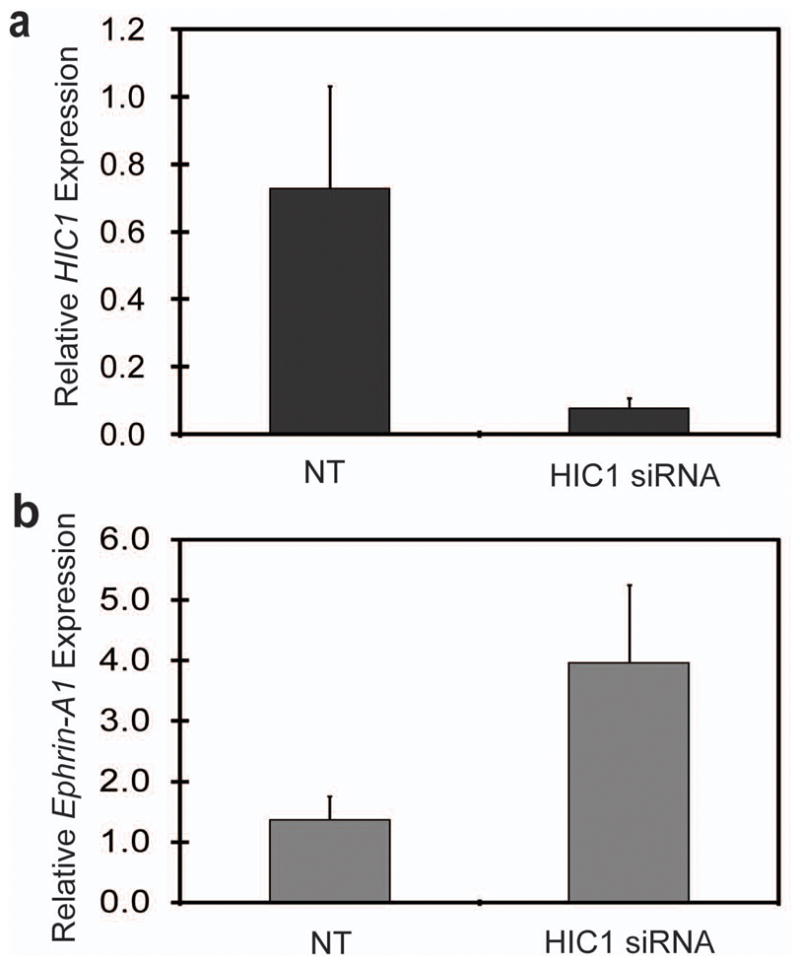
Knockdown of endogenous HIC1 in the WI38 human fibroblast cells up-regulates ephrin-A1 expression. WI38 cells were either mock-transfected, transfected with HIC1 siRNA or transfected with non-target siRNA control (NT) as previously described. Total RNA was extracted and expression levels of HIC1 (a) and ephrin-A1 (b) mRNA were assessed by real-time quantitative RT-PCR. Values were normalized to human β-actin.
Hic1 is a sequence-specific transcriptional repressor of ephrin-A1
We next analyzed 7.9 kb from transcription start site of ephrin-A1 gene for consensus DNA binding sites using the TRANSFAC search engine (Wingender et al., 2000). Numerous trans activation sequences were identified within 2 kb of the transcription start site, including consensus binding sites for NFkB and basic-helix-loop-helix transcription factors (Fig. 3a). Analysis of the HIC1 consensus sequence (Chen et al., 2005) demonstrated a total of 8 binding sites, all between −4 kb to −8 kb from the transcription start site, and they are listed in Supplementary Table S2 and shown graphically in Fig. 3a. No potential HIC1 binding sites were identified within the first 4.0 kb upstream of the transcription start site. Functional correlates of these findings were investigated using luciferase reporter assays. Four human ephrin-A1 promoter constructs (Fig. 3a) were created by subcloning the appropriate fragments into the pGL3-basic vector. Basal activity of these constructs, assessed by transient transfection in MCF-7 cells, showed that the ephrin-A1 promoter is highly active, with a 2-fold increase in activity observed with the inclusion of sequence containing the putative NFkB binding sites (Fig. 3a).
Fig. 3.
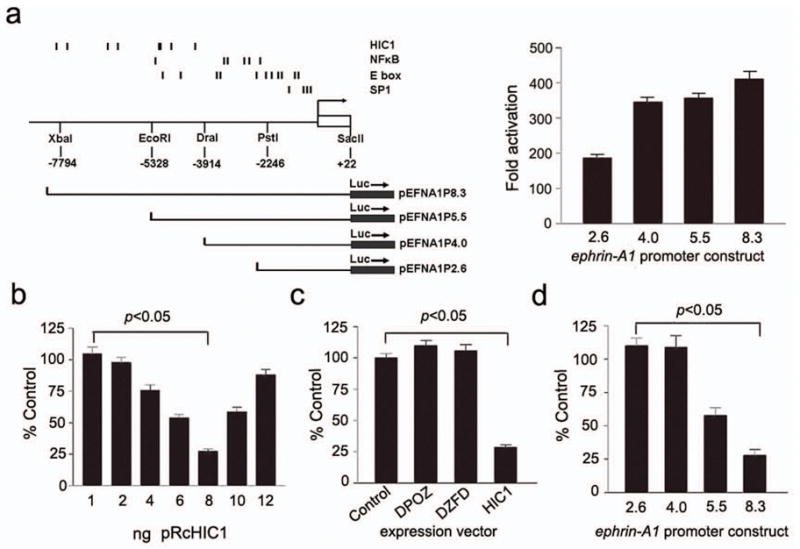
HIC1 is a direct transcriptional repressor of the ephrin-A1 promoter. (a) Left, map of the ephrin-A1 promoter region, with the positions of selected consensus binding sites indicated above. The length of the different promoter constructs used in reporter assays is shown below. Right, reporter activity in MCF-7 cells, transiently transfected with the four pEFNA1P promoter constructs as listed in left panel. Activity of each promoter construct is shown as fold activation compared with a pGL3-basic empty vector control. (b) Reporter activity in MCF-7 cells transiently co-transfected with the pEFNA1P8.3 reporter and increasing amount of the pRcHIC1 expression vector. The results are normalized to those for empty control vectors and are expressed as a relative ratio of firefly luciferase to Renilla luciferase. (c) Reporter activity in MCF-7 cells transiently co-transfected with the pEFNA1P8.3 reporter and 8ng of each of the expression constructs: pCDNA3.1 (control), a HIC1 mutant lacking the POZ domain pRcΔPOZ (DPOZ), a mutant lacking the zinc finger domain pRcΔZFM (DZFD), and pRcHIC1 (HIC1). (d) Reporter activity in MCF-7 cells transiently co-transfected with 8ng of pRcHIC1, and each of the four pEFNA1P reporter constructs listed in Fig. 3a. Results are shown as mean, SEM, N=6.
Transient co-transfection experiments using an expression vector encoding wild-type HIC1 (pRcHIC1), and the ephrin-A1 reporter construct pEFNA1P8.3, showed that transfection of increasing amounts of pRcHIC1 in ng quantities resulted in repression of reporter gene activity (Fig. 3b). Interestingly, this effect is lost with increases in the amount of HIC1 expression vector above 8ng/well. This result suggests quenching of the repressive effect above this dose. We next compared the effects of wild-type HIC1 with mutant expression vectors lacking the N-terminal POZ domain (pRcDPOZ) or the C-terminal zinc fingers (pRcDZFM). As shown in Fig. 3c, both mutant vectors failed to repress luciferase activity. These data suggest that both the C-terminal DNA binding domain and the N-terminal POZ domain are required for the biological function of HIC1 in transcriptional repression of ephrin-A1. In order to confirm that the HIC1 binding sites identified in the ephrin-A1 promoter were necessary for repression in this assay, we compared the ability of exogenous HIC1 to repress luciferase activity in the four promoter constructs depicted in Fig. 3a. Our results demonstrate that the 8 HIC1 binding sites are required for full repression, and that the two reporter constructs that do not contain HIC1 binding sites do not respond to exogenous HIC1 (Fig. 3d).
To establish that endogenous Hic1 protein is present at the ephrin-A1 promoter, we performed chromatin immunoprecipitation (ChIP) assays in NIH3T3 cells, which robustly express Hic1 protein that can be easily immunoprecipitated (Chen et al., 2005; Briggs et al., 2008). Importantly, there is high similarity between the human and mouse ephrin-A1 promoters for positions of the Hic1 binding sites as shown in Fig. 3a. The mouse promoter has 9 sites from −4.0 kb to −8.0 kb and no sites within −2.0 kb from the transcription start site (Supplementary Table S2). Primers spanning 200–300bp regions of the mouse ephrin-A1 promoter (Supplementary Table S3) were optimized for PCR using NIH3T3 genomic DNA (data not shown). PCR amplification at the predicted size occurred in several regions of the mouse ephrin-A1 promoter containing predicted Hic1 binding sites (primers C, D, E, F, G) (Fig. 4), demonstrating sequence specific occupancy of the mouse ephrin-A1 promoter by endogenous Hic1 protein. Thus, the functional Hic1 binding sites identified in the mouse promoter are located approximately between −4 kb and −8 kb upstream of the transcription start site, which is consistent with the location of HIC1 binding sites in the human ephrin-A1 promoter identified by the promoter deletion analysis. Taken together, all of the above data in this section show that Hic1 is a direct sequence specific transcriptional repressor of ephrin-A1.
Fig. 4.

ChIP analysis of Hic1 at the mouse ephrin-A1 promoter in NIH3T3 cells. The mouse ephrin-A1 promoter is depicted with the predicted Hic1 binding sites shown above, and the distance in kb from the start of exon 1 is shown below. The area of the promoter covered by each primer pair (A-L) is shown below this scale. Amplification products for each primer pair, with (+) and without (−) anti-Hic1 antibody in the immunoprecipitation are shown in the lower panel.
Misexpression of ephrin-A1 in Hic1 deficient mouse embryos
Having identified and verified ephrin-A1 as a novel direct target for transcriptional repression by the tumor suppressor Hic1, we next examined in vivo correlates between Hic1 and ephrin-A1 in our Hic1 knockout mouse model. Homozygous embryos show a variable phenotype, ranging from severe craniofacial, neural and somatic disorders to minimal macroscopic defects (Carter et al., 2000). We analyzed Hic1 knockout mice at E14.5, and compared them with wild-type littermates. In these embryos, we observed an overall increase in ephrin-A1 staining in Hic1 knockout embryos. An example of this overall increase in staining can be seen in mid gestation embryos throughout the central nervous system (CNS) (Fig. 5a). Some increased ephrin-A1 staining sites in the Hic1 knockout embryo, such as forebrain, midbrain, and skin, are correlated very well with the localization of Hic1 staining in a Hic1+/−embryo (Supplementary Fig. S4). In the developing parietal cortex of a Hic1−/− embryo (Fig. 5b), abnormal expression of ephrin-A1 was observed in the cortical plate cells staining positive for Tuj1, a marker of early cortical neurons. As can be seen by direct comparison, this domain of increased ephrin-A1 expression correlates with the region of normal Hic1 expression in wild-type embryos (Fig. 5b). In the E17.5 embryonic liver, we also observed an increase in the intensity of ephrin-A1 expression in Hic1+/− and Hic1−/− embryos (Fig. 5c). Interestingly, Hic1+/−embryos manifest an intermediate increase when compared with Hic1+/+ and Hic1−/− littermates. In summary, these data show that ephrin-A1 expression domains in mouse development are markedly expanded in mice lacking Hic1.
Fig. 5.

Abnormal expression of ephrin-A1 in Hic1 deficient mice. (a) Sagittal sections of E14.5 Hic1 wild-type (+/+) and knockout (−/−) embryos stained for ephrin-A1 by immunoperoxidase, detected by DAB and counterstained with hematoxylin. Scale bar = 500μm. (b) Dual label confocal immunofluorescence analysis of ephrin-A1 expression in axial sections of the developing telencephalon from E14.5 embryos. cp = cortical plate, v = ventricle. Upper panel, ephrin-A1 is abnormally expressed in the cortical plate of a Hic1−/− embryo. Immunolocalization of Hic1 (Green) in cortical plate (cp) neurons in an E14.5 Hic1+/+ embryo. Scale bar = 200μm. Lower panel, cortical plate neurons from a Hic1−/− embryo stained with ephrin-A1 (green) and Tuj1 (red). Scale bar = 20μm. (c) Immunostaining of ephrin-A1 expression in E17.5 livers taken from Hic1 wild-type (+/+), heterozygous (+/−), and null (−/−) mouse embryos. Scale bar = 50μm.
HIC1 inhibits tumorigencity of breast Cancer Cells partly through downregulation of ephrin-A1
Transcriptional regulation of ephrin-A1 has direct implications for HIC1 as a tumor suppressor. In cell culture, Western blot analysis of lysates from MCF-7 and MDA-MB231 cells showed that ephrin-A1 protein was expressed at extremely low levels, whereas EphA2 and EphA4 proteins were readily detectable (Fig. 6a). Strikingly, when both cell lines are grown as xenografts in nude mice, we observed a marked decrease in EphA2 and EphA4 expression with a concomitant upregulation of ephrin-A1 expression (Fig. 6a). Expression of EphA2 and EphA4 was not influenced by adenoviral infection or overexpression of HIC1 in cell culture (data not shown).
Fig. 6.

HIC1 inhibits tumorigenic behavior of human breast cancer cells in vivo. (a) Western blot analysis on ephrin-A1, EphA2 and EphA4 expression in lysates from MCF-7 and MDA-MB231 cells grown in vitro or in vivo. (b) Western blot analysis of ephrin-A1, EphA2 and EphA4 in lysates obtained from MDA-MB231 cells infected with Adβ-gal or AdEphrin-A1 adenoviruses. Equal loading was confirmed by immunoblotting for GAPDH. (c) Tumor volume of flank xenografts in athymic nude mice injected with MDA-MB231 cells infected with AdHIC1, Adβ-gal or AdEphrin-A1 adenoviruses. The black bars represent the mean values of tumor volume. Numbers below the x-axis indicate the number of mice that developed tumors. (d) Tumor volume of flank xenografts in athymic nude mice injected with MDA-MB231 cells infected with a combination of with AdHIC1 and Adβ-gal, or AdHIC1 and AdEphrin-A1 adenoviruses. The mean values of tumor volume and the number of mice that developed tumors are also indicated as in C.
In initial studies, we did not observe any changes in breast cancer cell growth or viability in response to HIC1 overexpression in vitro (Supplementary Fig. S5), suggesting that an appropriate tumor-stromal/endothelial cell interaction may be required for the tumor suppressor function of HIC1. In fact, in breast cancer, ephrin expression by epithelial cells is associated with reciprocal Eph receptor expression in adjacent vascular endothelial cells (Ogawa et al., 2000; Brantley-Sieders et al., 2006). We hypothesized that this tumor-microenvironment signaling might be affected by HIC1 expression leading to downregulation of ephrin-A1 in cancer cells and suppression of tumor growth. Most studies assessing the effects of ephrin-A1 overexpression have employed exogenous ephrin-A1-Fc. In contrast, we generated a recombinant adenovirus expressing human ephrin-A1 (AdEphrin-A1) (Supplementary Fig. S6), which allowed us to express ephrin-A1 in breast cancer cells. MDA-MB231 cells infected with AdEphrin-A1 virus showed robust expression of ephrin-A1, which led to 8.4- and 9.3-fold reduction of EphA2 proteins at 24 or 48 hrs post viral treatment respectively. By contrast, overexpression of ephrin-A1 alone resulted in 3.2- and 3.9-fold decrease in the EphA4 protein level for the same time points (Fig. 6b). Cells infected with AdHic1, Adβ-gal or AdEphrin-A1 were then injected subcutaneously into nude mice. After 5 weeks, the tumor volume analysis revealed a marked decrease in engraftment efficiency induced by overexpression of HIC1 (p<0.05), and a modest increase in tumor growth in response to ephrin-A1 overexpression (p<0.05; Fig. 6c). To test the contribution of ephrin-A1 downregulation to the tumor suppressor effect of HIC1 overexpression, we then co-infected cells with equal amounts of AdHIC1/Adβ-gal or AdHIC1/AdEphrin-A1. As shown in Fig. 6d, co-overexpression of ephrin-A1 partially restores the ability of MDA-MB231 cells to engraft nude mice, consistent with a positive role for ephrin-A1 as an important protein in engraftment and growth of breast cancer (Ogawa et al., 2000), and with observations demonstrating that ephrin-A1 siRNA can block angiogenesis and metastasis in an in vivo model of breast cancer (Brantley-Sieders et al., 2006). Together, these data support the notion that ephrin-A1 can be overexpressed in breast cancer in vivo when there is a loss of the transcriptional repression of HIC1 on the ephrin-A1 promoter.
Discussion
Using animal models, we have previously demonstrated that the transcriptional repressor Hic1 is not only a critical developmental regulator but also an epigenetically regulated tumor suppressor (Carter et al., 2000; Chen et al., 2003; Chen et al., 2004; Chen et al., 2005; Briggs et al., 2008). In this study, we identified and characterized ephrin-A1 as another transcriptional target of Hic1. This finding has two important implications. First, we have previously shown that homozygous deletion of Hic1 in mice results in embryonic lethality with severe craniofacial and somatic defects. Since the molecular mechanism underlying the developmental disorders has not been fully characterized, identification of ephrin-A1 as a new direct transcriptional target of Hic1 provides another explanation for the functional importance of Hic1 in development. Second, cancer has been traditionally viewed as a genetic disease that is driven by the sequential accumulation of mutations, resulting in the loss of function of tumor suppressor genes or the constitutive activation of proto-oncogenes (Kinzler and Vogelstein, 1996; Hanahan and Weinberg, 2000). It is now apparent, however, that epigenetic gene silencing and associated promoter CpG island DNA hypermethylation constitute an alternative mechanism to mutations that inactivate tumor suppressor genes (Jones and Baylin, 2007; Esteller, 2008). Given the high frequency of epigenetic silencing of HIC1 in most common human tumor types, and the high incidence of malignancies developed in Hic1 heterozygous mice, our data presented here reveals a potentially crucial role for HIC1 as tumor suppressor through the transcriptional regulation of ephrin-A1 signaling.
Ephrins and their receptors play a critical role in tissue patterning, morphogenesis, angiogenesis, and neural development (Poliakov et al., 2004; Pasquale, 2005; Arvanitis and Davy, 2008). Our data show that ephrin-A1 is misexpressed in Hic1 deficient mouse embryos, especially in the CNS, where abnormalities are often severe in this knockout model (Carter et al., 2000). In light of the normally restricted expression pattern of ephrin-A1, the misexpression of this ligand seen in Hic1 knockout embryos is a striking finding, and suggests that Hic1 plays an important role in coordinating development through transcriptional regulation of ephrin-A1. In addition, the broad specificity of ephrin-A1, which can bind to a wide range of EphA receptors, suggests that its misexpression could lead directly to the dramatic abnormalities in development in Hic1 deficient embryos through aberrant ephrin-A1 signaling.
Perhaps the most important direct implication of our current data regards the role for HIC1 as a tumor suppressor gene, and a potential functional link to overexpression of ephrin-A1 in epithelial malignancies such as breast cancer. In fact, previous studies have shown that HIC1 is frequently hypermethylated and transcriptionally inactivated in breast cancer cell lines and primary tumors (Wales et al., 1995; Fujii et al., 1998). Meanwhile, overexpression of both ephrins and Eph receptors has been observed in a variety of human malignancies (Easty et al., 1999; Tang et al., 1999; Takai et al., 2001) including breast cancer (Ogawa et al., 2000). Decreased protein expression of ephrin-A1 by an expression vector-based antisense RNA inhibited 3-dimensional growth of HT29 colon adenocarcinoma cells (Potla et al., 2002). Importantly, a positive role for epithelial-specific ephrin-A1 in tumor initiation in vivo has also been demonstrated in the Apcmin mouse model, in which overexpression of ephrin-A1 in enterocytes markedly enhanced the number of invasive tumors in both intestine and colon (Shi et al., 2008). Furthermore, disruption of the ephrin-A1 receptor, EphA2, leads to increased susceptibility to chemically induced carcinogenesis in mouse skin, suggesting that EphA2 has tumor suppressor functions (Guo et al., 2006). Together, these data suggest that ephrin-A1 exerts a positive role on tumorigenesis and ephrin-A1 receptor, EphA2, acts as a negative regulator of tumorigenesis. Our studies of breast cancer cells reveal a critical role of HIC1 in the regulation of tumorigenesis, at least partly through transcriptional repression of ephrin-A1. In a xenograft model, we demonstrate that restoration of HIC1 function in breast cancer cells leads to a significant reduction in tumor growth in nude mice, and importantly, co-overexpression of ephrin-A1 with HIC1 partially restores the ability of breast cancer cells to engraft nude mice, supporting a positive role of ephrin-A1 in promoting malignant cell growth in vivo. In contrast to these above data, other studies in breast cancer and prostate cancer cell lines have shown that EphA2 expression is a prominent feature in many of these systems, and that activation of EphA2 by an ephrin-A1-Fc fusion protein results in inhibition of the oncogenic Ras/MAPK signaling in several different cell types (Miao et al., 2001; Macrae et al., 2005; Miao et al., 2009).
Three important issues might explain these contradictory findings. First, data supporting a positive role for ephrin-A1, including data presented here, have been performed in vivo, suggesting that cell lines in tissue culture may not represent the potential for non-cell autonomous signaling mediated by ephrin-A1 expressed on tumor cells. It has been well established that vascular recruitment by tumors is a critical step in tumorigenesis. A growing body of evidence demonstrates that upregulation of ephrin-A1 in breast cancer cells has been directly implicated in tumor angiogenesis, neovascularization and metastasis through cancer-specific expression of ephrin-A1, and reciprocal expression of EphA receptors in tumor stromal and endothelial cells (Brantley-Sieders et al., 2004; Brantley-Sieders et al., 2006). For example, endothelial cells isolated from EphA2-deficient mice displayed defective ephrin-A1 induced migration in vitro and in vivo, and EphA2-deficient mice showed a diminished angiogenic response to ephrin-A1 in vivo (Brantley-Sieders et al., 2004). Furthermore, siRNA-mediated ephrin-A1 knockdown in metastatic mammary cancer cells leads to decreased lung metastasis in vivo (Brantley-Sieders et al., 2006). Interestingly, a recent study showed that a functional and soluble form of ephrin-A1 is released from ectopic ephrin-A1-expressing glioblastoma multiforme (GBM) cells and endogenous ephrin-A1 expressing breast cancer cells. The soluble ephrin-A1 may interact with EphA2 receptors on endothelial cells, and therefore provide a paracrine role for ephrin-A1 in stimulating angiogenesis (Wykosky et al., 2008). Here, we clearly show that in vivo, the ratio of ephrin-A1 to EphA2/4 expression is dramatically altered in favor of ephrin-A1 expression, supporting the notion that tumor-expressed ephrin-A1 interacts with EphA2 on endothelial cell to promote tumorigenesis via angiogenesis. Second, when ephrin-A1 is overexpressed by tumor cells either endogenously or ectopically, these cells exhibit downregulation of EphA2 (Macrae et al., 2005; Wykosky et al., 2008; Miao et al., 2009). We have shown that overexpression of ephrin-A1 results in reduction of EphA2 and EphA4 expression in breast cancer cells, thus reducing the capacity for these receptors to signal. Downregulation of EphA2/4 in response to ephrin-A1 overexpression in tumor cells could provide a mechanism by which the tumor cells can circumvent quenching the released ephrin-A1 before it has a chance to exert a paracrine function on the endothelial cells within the tumor environment (Wykosky and Debinski, 2008). Finally, ephrin-A1 can bind to all Ephreceptors of the A subclass, and the receptor EphA2 binds ephrin-A1 with the highest affinity (Holder and Klein, 1999). Therefore, the biological function of ephrin-A1 signaling during tumorigenesis is not limited to inhibition of Ras/MAPK signaling. For example, ephrin-A1-specific siRNA treatment can increase p21 expression in a hepatocellular carcinoma cell line (Iida et al., 2005), suggesting that the balance between ephrin-A1 and EphA expression may have cell-type specific effects.
In summary, our studies support the concept that ephrin-A1 plays a positive role in tumor growth in vivo, and are consistent with previous reports of ephrin-A1 acting through EphA receptors in the tumor microenvironment in vivo. Our data suggest a mechanism to explain how loss of Hic1 function may be a potentially important event in carcinogenesis through the activation of aberrant tumor-endothelial cell interactions mediated by ephrin-Eph signaling.
Materials and methods
Antibodies
Antibodies for immunofluorescence: ephrin-A1 (Santa Cruz, sc-911) 1:1000; Tuj1 (Covance, MMS-435P) 1:100. For immunohistochemistry: HIC1 (Rabbit polyclonal) 1: 100 as previously described (12, 13); ephrin-A1 (Santa Cruz, sc-911) 1:200. For Western blot: HIC1 1:100; ephrin-A1 (Santa Cruz, sc-20719) 1:200; EphA2 (Santa Cruz, sc-924) 1:2000; EphA4 (Upstate, 07-309) 1: 1000; and GAPDH (Upstate, MAB374) 1: 10,000.
cDNA Array Analysis
Adenoviruses were prepared as described (He et al., 1998). Total RNA was extracted from MCF-7 cells infected with either wild-type AdHIC1 or mutant HIC1 lacking POZ domain (AdΔPOZ) as a control. The transduction efficiency in MCF-7 cells was 75–80% as determined by GFP expression. Analysis of gene expression was performed according to standard protocols on an Affymetrix HGU95A chip.
RT-PCR
All RNA samples were quality controlled using a Biorad Experion Bioanalyzer, then subjected to DNase I treatment prior to reverse transcription. Real time quantitative PCR studies were performed using QuantiTect SYBR Green Kit (QIAGEN). All quantitative calculations were performed using the ΔΔc-t method. Primer sequences are listed in Supplementary Table S4.
Luciferase Reporter Assays
Reporter gene assays were performed as previously described (Zhang et al., 2008). We measured luciferase activity in a luminometer (BD Biosciences) and transfection efficiency was normalized using the paired Renilla luciferase activity by using the Dual Luciferase Reporter Assay system (Promega) according to the manufacturer’s instruction. Further experimental details can be found in Supplementary Information (SI) Methods.
Chromatin Immunoprecipitation
This was performed essentially as described (Briggs et al., 2008) using a rabbit polyclonal antibody to Hic1 to detect the presence of endogenous Hic1 at the mouse ephrin-A1 promoter. Details are provided in SI Methods.
Supplementary Material
Acknowledgments
Financial support: This work was supported by National Cancer Institute Grant R01 CA43318 (SB Baylin), and the American Cancer Society Postdoctoral Fellowship (W Zhang).
This work was supported by National Cancer Institute Grant R01 CA43318 (SB Baylin), and the American Cancer Society Postdoctoral Fellowship (W Zhang). We thank Craig D. Peacock, Leslie Meszler, Lillian Dasko-Vincent for technical assistance, Leander Van Neste for biostatistical support, and Kathy Bender for manuscript preparation and submission.
References
- Arvanitis D, Davy A. Eph/ephrin signaling: networks. Genes Dev. 2008;22:416–29. doi: 10.1101/gad.1630408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117:2037–49. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Fang WB, Hwang Y, Hicks D, Chen J. Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 2006;66:10315–24. doi: 10.1158/0008-5472.CAN-06-1560. [DOI] [PubMed] [Google Scholar]
- Briggs KJ, Corcoran-Schwartz IM, Zhang W, Harcke T, Devereux WL, Baylin SB, et al. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev. 2008;22:770–85. doi: 10.1101/gad.1640908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter MG, Johns MA, Zeng X, Zhou L, Zink MC, Mankowski JL, et al. Mice deficient in the candidate tumor suppressor gene Hic1 exhibit developmental defects of structures affected in the Miller-Dieker syndrome. Hum Mol Genet. 2000;9:413–9. doi: 10.1093/hmg/9.3.413. [DOI] [PubMed] [Google Scholar]
- Chen WY, Zeng X, Carter MG, Morrell CN, Chiu Yen RW, Esteller M, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33:197–202. doi: 10.1038/ng1077. [DOI] [PubMed] [Google Scholar]
- Chen W, Cooper TK, Zahnow CA, Overholtzer M, Zhao Z, Ladanyi M, et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell. 2004;6:387–98. doi: 10.1016/j.ccr.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–48. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith AC, et al. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet. 1997;6:147–55. doi: 10.1093/hmg/6.2.147. [DOI] [PubMed] [Google Scholar]
- Deltour S, Guerardel C, Stehelin D, Leprince D. The carboxy-terminal end of the candidate tumor suppressor gene HIC-1 is phylogenetically conserved. Biochim Biophys Acta. 1998;1443:230–2. doi: 10.1016/s0167-4781(98)00219-x. [DOI] [PubMed] [Google Scholar]
- Deltour S, Guerardel C, Leprince D. Recruitment of SMRT/N-CoR-mSin3A-HDAC-repressing complexes is not a general mechanism for BTB/POZ transcriptional repressors: the case of HIC-1 and gammaFBP-B. Proc Natl Acad Sci U S A. 1999;96:14831–6. doi: 10.1073/pnas.96.26.14831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltour S, Pinte S, Guerardel C, Wasylyk B, Leprince D. The human candidate tumor suppressor gene HIC1 recruits CtBP through a degenerate GLDLSKK motif. Mol Cell Biol. 2002;22:4890–901. doi: 10.1128/MCB.22.13.4890-4901.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easty DJ, Hill SP, Hsu MY, Fallowfield ME, Florenes VA, Herlyn M, et al. Up-regulation of ephrin-A1 during melanoma progression. Int J Cancer. 1999;84:494–501. doi: 10.1002/(sici)1097-0215(19991022)84:5<494::aid-ijc8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Fujii H, Biel MA, Zhou W, Weitzman SA, Baylin SB, Gabrielson E. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene. 1998;16:2159–64. doi: 10.1038/sj.onc.1201976. [DOI] [PubMed] [Google Scholar]
- Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, et al. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006;66:7050–8. doi: 10.1158/0008-5472.CAN-06-0004. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herath NI, Spanevello MD, Sabesan S, Newton T, Cummings M, Duffy S, et al. Over-expression of Eph and ephrin genes in advanced ovarian cancer: ephrin gene expression correlates with shortened survival. BMC Cancer. 2006;6:144. doi: 10.1186/1471-2407-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S, Pack SD, Chong SS, Robbins CM, Pavan WJ, Ledbetter DH, et al. Genomic organization of the murine Miller-Dieker/lissencephaly region: conservation of linkage with the human region. Genome Res. 1997;7:625–34. doi: 10.1101/gr.7.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder N, Klein R. Eph receptors and ephrins: effectors of morphogenesis. Development. 1999;126:2033–44. doi: 10.1242/dev.126.10.2033. [DOI] [PubMed] [Google Scholar]
- Iida H, Honda M, Kawai HF, Yamashita T, Shirota Y, Wang BC, et al. Ephrin-A1 expression contributes to the malignant characteristics of {alpha}-fetoprotein producing hepatocellular carcinoma. Gut. 2005;54:843–51. doi: 10.1136/gut.2004.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal M, Trinh E, Britschgi C, Britschgi A, Roh V, Vorburger SA, et al. The tumor suppressor gene hypermethylated in cancer 1 is transcriptionally regulated by E2F1. Mol Cancer Res. 2009;7:916–22. doi: 10.1158/1541-7786.MCR-08-0359. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–86. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- Macrae M, Neve RM, Rodriguez-Viciana P, Haqq C, Yeh J, Chen C, et al. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell. 2005;8:111–8. doi: 10.1016/j.ccr.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Miao H, Wei BR, Peehl DM, Li Q, Alexandrou T, Schelling JR, et al. Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway. Nat Cell Biol. 2001;3:527–30. doi: 10.1038/35074604. [DOI] [PubMed] [Google Scholar]
- Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R, Kataoka H, Sato N, Kanamori M, Ihara M, Igarashi H, et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci. 2005;96:42–7. doi: 10.1111/j.1349-7006.2005.00007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–52. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–75. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Poliakov A, Cotrina M, Wilkinson DG. Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev Cell. 2004;7:465–80. doi: 10.1016/j.devcel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Potla L, Boghaert ER, Armellino D, Frost P, Damle NK. Reduced expression of EphrinA1 (EFNA1) inhibits three-dimensional growth of HT29 colon carcinoma cells. Cancer Lett. 2002;175:187–95. doi: 10.1016/s0304-3835(01)00613-9. [DOI] [PubMed] [Google Scholar]
- Shi L, Itoh F, Itoh S, Takahashi S, Yamamoto M, Kato M. Ephrin-A1 promotes the malignant progression of intestinal tumors in Apc(min/+) mice. Oncogene. 2008;27:3265–73. doi: 10.1038/sj.onc.1210992. [DOI] [PubMed] [Google Scholar]
- Takai N, Miyazaki T, Fujisawa K, Nasu K, Miyakawa I. Expression of receptor tyrosine kinase EphB4 and its ligand ephrin-B2 is associated with malignant potential in endometrial cancer. Oncol Rep. 2001;8:567–73. doi: 10.3892/or.8.3.567. [DOI] [PubMed] [Google Scholar]
- Tang XX, Evans AE, Zhao H, Cnaan A, London W, Cohn SL, et al. High-level expression of EPHB6, EFNB2, and EFNB3 is associated with low tumor stage and high TrkA expression in human neuroblastomas. Clin Cancer Res. 1999;5:1491–6. [PubMed] [Google Scholar]
- van Rechem C, Rood BR, Touka M, Pinte S, Jenal M, Guerardel C, et al. The scavenger chemokine (C-X-C motif)receptor7 CXCR7 is a direct target gene of hypermethylated in cancer 1 HIC1. J Biol Chem. 2009;284:20927–35. doi: 10.1074/jbc.M109.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, Cavenee WK, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–7. doi: 10.1038/nm0695-570. [DOI] [PubMed] [Google Scholar]
- Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–9. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J, Palma E, Gibo DM, Ringler S, Turner CP, Debinski W. Soluble monomeric EphrinA1 is released from tumor cells and is a functional ligand for the EphA2 receptor. Oncogene. 2008;27:7260–73. doi: 10.1038/onc.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Glockner SC, Guo M, Machida EO, Wang DH, Easwaran H, et al. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008;68:2764–72. doi: 10.1158/0008-5472.CAN-07-6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.