

Abstract
Background
Pancreatic cancer is a disease of near uniform fatality and the overwhelming majority of patients succumb to their advanced malignancy within a few months of diagnosis. Despite considerable advances in our understanding of molecular mechanisms underlying pancreatic carcinogenesis, this knowledge has not yet been fully translated into clinically available treatment strategies that yield significant improvements in disease free or overall survival.
Objective
Cell line-based in vitro model systems provide powerful tools to identify potential molecular targets for therapeutic intervention as well as for initial pre-clinical evaluation of novel drug candidates. Here we provide a brief overview of recent literature on cell line-based model systems of pancreatic cancer and their application in the search for novel therapeutics against this vicious disease.
Conclusion
While in vitro models of pancreatic cancer are of tremendous value for genetic studies and initial functional screenings in drug discovery, they carry several imanent drawbacks and are often poor in predicting therapeutic response in humans. Therefore, in most instances they are successfully exploited to generate hypothesis and identify molecular targets for novel therapeutics, which are subsequently subject to further in-depth characterization using more advanced in vivo model systems and clinical trials.
Keywords: pancreatic cancer, cell line models, mouse models of pancreatic cancer, translational research, drug discovery
1. Introduction
Ductal adenocarcinoma of the pancreas (a.k.a. pancreatic cancer; the terms pancreatic cancer and pancreatic ductal adenocarcinoma will be used synonymously in the text) is an almost uniformly lethal malignancy, which accounts for an estimated 213,000 annual deaths worldwide and is the fourth most common cause of cancer-related mortality in the United States, with an overall median survival of less than 6 months 1, 2. The five year survival rate is below 5% when combined for all stages, and <2% for the majority of cases that present at with advanced (metastatic) disease, which are amongst the most dismal seen in any human malignancy 1.
Ongoing research efforts over the past decade have led to significant advances in our understanding of the underlying etiological and molecular mechanisms facilitating pancreatic carcinogenesis. Nevertheless, this increase in knowledge has not yet been translated into clinically tangible advances in terms of novel therapeutic strategies with marked improvement of overall patient survival 3.
Cell-line based in vitro model systems of pancreatic cancer provide powerful tools for discovery of molecular targets for novel therapeutics as well as for preclinical evaluation of drug candidates. A brief review of available in vitro models and its use and limitations in drug research is given in the following text.
2. In vitro culture of non-neoplastic pancreatic cells
The human pancreas is a complex organ consisting of several tissue compartments and as of to date we are still far from fully understanding all of the physiologic interactions underlying regulation of organ development and homeostasis, as well as those governing the development of malignant neoplasia. The question of the ‘cell of origin’ of pancreatic cancer has long been a controversial issue. While the traditional model, which is mostly based on morphologic similarities observed by light microscopy on histological specimens, suggests that pancreatic cancer arises from the ductal cell compartment, there is an opposing hypothesis suggesting that pancreatic cancer arises from transdifferentiated acinar cells 4–6. A variation of the latter theory is the concept of pancreatic cancer possibly arising from a yet to be defined population of pancreatic stem/progenitor cells, which some schools of thought believe might reside within the acinar cell compartment, or in centro-acinar cells 7–9. Establishment and in vitro culture of non-neoplastic pancreatic cells is of interest with regard to pancreatic cancer research in at least two aspects: firstly, it allows distinct examination of conditions regulating growth and differentiation of the respective distinct cell compartments in an isolated in vitro setting, as well as determination of the genetic alterations required for malignant transformation of these cells. Secondly, such non-neoplastic cells provide valuable controls in functional studies using novel experimental therapeutic approaches, specifically identifying therapeutic targets that cancer cells depend on in order to maintain a fully malignant phenotype, while exerting little, or ideally, no effects on these non-malignant cells.
2.1 Pancreatic ductal cell culture
Despite the immense relevance for pancreatic cancer research, surprisingly few cases of long-term propagated in vitro cultures of pancreatic ductal cells have been reported. This may be due to several factors, such as the relative scarcity of ductal cells in the human pancreas (<5% of the total pancreatic volume), a general lack of knowledge regarding physiologic properties regulating their growth and differentiation, thus hampering establishment of appropriate culture conditions, and the frequent occurrence of senescence in in vitro cultures of ductal cells, which often prevent successful long-term culture 10–12. Therefore, initially it has proven to be a challenge to propagate human pancreatic ductal cells in culture for more than 1–2 months 13–15.
Generation of two distinct models of epithelial cell lines that could readily be maintained in long term culture has been described, and these were generated either through immortalization by introduction of the human papillomavirus 16 gene E6E7 proteins, or by stable transfection with human telomerase reverse transcriptase (hTERT) and growth in a special culture medium containing epidermal growth factor (EGF) 16–18. Unfortunately, these immortalized lines do not represent ideal in vitro models of genuinely ‘normal’ human pancreatic ductal cells, since introduction of viral proteins abrogates the function of key tumor suppressor pathways like p53 or Rb, while the presence of EGF activates several oncogenic pathways, e.g. downstream of EGFR. Nevertheless, both of these cell lines lack key functional features of fully malignant pancreatic cancer cells, e.g. anchorage independent growth in soft agar or the ability to engraft as tumors in athymic mice, and have thus been of tremendous value as controls in functional in vitro assays. Moreover, they have been successfully used in various in vitro-transformation models 17, 19, 20 (see example in Figure 1).
Figure 1.
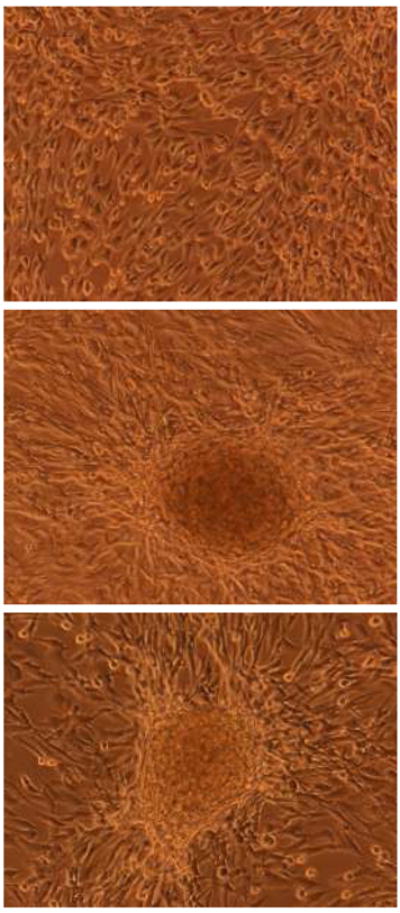
(top) Non-transformed hTERT-immortalized HPNE cells grow as a monolayer in tissue culture. (middle and bottom) Examples of transformed HPNE cells with multi-layering (“focus” formation). In this example, the gene(s) responsible for this neoplastic phenotype are not known, as transformation was induced by transient activation of a Sleeping Beauty transposon, leading to disruption and/or activation of cancer-associated genes in the HPNE genome. In vitro models of pancreatic epithelial cells can be used for such “forward” genetics experiments, as well as for more traditional reverse genetics using known combination of oncogenes or tumor suppressor genes, as discussed in the text.
2.2 Culture of acinar cells
While isolation and in vitro propagation of rat pancreatic acinar cells under special conditions has been described 21, 22, long-term culture of human or mouse pancreatic acinar cells has proven to be challenging 23. Short term (primary) explants of mouse acinar cells have been established and have been successfully utilized for a variety of ex vivo functional assays over their limited period (<7 days) of viability 24.
2.3 Culture of islet cells
Isolation of human islet cells has so far proved to be difficult and long-term survival under in vitro culture conditions has been limited 25–31. As observed with ductal cells, cultured islet cells undergo senescence in in vitro culture. However, as opposed to duct cells, immortalization by stable transfection techniques has so far been mostly unsuccessful in the case of islet cells 23.
3. Generation of pancreatic cancer cell lines
There are two general approaches for the generation of cell lines from human pancreatic cancers that are frequently used: Cell lines can either be directly established from primary patient-derived tumor tissue samples or from murine xenografts of human pancreatic cancers in athymic nude or SCID mice 32–35. The latter method carries the advantage that ex vivo passaging in mice is a relatively innocuous way to enrich the neoplastic cell compartment, while at the same time eliminating stromal components of human origin, thus enhancing the probability for successful establishment of a cancer cell line 36, 37. Addition of matrigel can further increase the take rate of xenografted tumor tissue samples 38. Disadvantages of this method include the requirement of significantly more time (propagation of a subcutaneous xenograft from primary human pancreatic cancer tissue samples typically requires some weeks up to several months), the chance of contamination with murine fibroblasts and the risk of acquiring additional genetic alterations during the period of in vivo growth as a murine xenograft.
Success rates for direct establishment of pancreatic cancer cell lines from primary tissues are generally relatively low, and appear to be only slightly higher when metastatic tumor tissues are used as starting material as opposed to primary tumor tissue samples 35, 39–47. Jaffee et al. systematically optimized a protocol for establishment of cell lines from primary pancreatic cancer tissues 48. In this study, the authors describe four key factors as being critical for successful in vitro establishment of pancreatic cancer cell lines: firstly, freshly harvested tissues have to be cut into fragments of one to five mm in diameter and digested by collagenase (300 units/mL) and hyaluronidase (200 units/mL) overnight on a shaker at 37 °C. Secondly, before initial plating stromal cells have to be removed from the digested samples as far as possible by two 20 min centrifugation steps at gravity. Plating of resuspended cells is then done at a density of 1–2 million viable cells per milliliter of fresh Panc media. Thirdly, for optimum growth of primary carcinoma cells RPMI-1640 base medium containing 15% of fetal bovine serum (FBS), 200 μM of L-glutamine, 1× non-essential amino acids solution and 1% sodium pyruvate has to be supplemented with human insulin (0.2 units/mL) and insulin-like growth factors (IGF) I and II (0.01 μg/mL each). Lastly, during the first weeks of in vitro culture stromal overgrowth has to be avoided and contaminating fibroblasts have to be eliminated by repeated differential trypsinization. Using this optimized protocol, the authors report a success rate of ~30% for the establishment of pancreatic cancer cell lines from primary tissue samples.
In our experience complete purification of a primary cancer cell line from contaminating human (in the case of primary human tumor samples as starting material) or murine fibroblasts (in the case of xenograft tissues) can often be challenging despite repetitive differential trypsinization. It is therefore tempting to speculate, whether novel, improved techniques might become available in the near future that allow for reliable and more rapid purification of primary cell lines.
4. In vitro methods for translational research in pancreatic cancer
Pancreatic cancer is a complex genetic disease and the underlying genetic and functional alterations required during the multistep pancreatic cancer progression cascade are still not fully understood 49. Isolation of patient derived pancreatic cancer cell lines provides the only means available to date to accurately mimic the entirety of genomic alterations involved in the human disease in an in vitro model system that can be maintained in culture for more than a few days 50 (see 51 for a review on molecular genetics of pancreatic cancer).
The use of cell line-based in vitro approaches for drug discovery in pancreatic cancer carries some obvious immanent shortcomings which often limit the applicability of such techniques. For example, while direct drug effects on the neoplastic cells themselves can generally be studied on cell lines in a relatively straightforward manner, the vast majority of known or supposed tumor-host interactions are difficult to impossible to accurately mimic in a purely cell culture-based system. These include - to various degrees – provision of a specific growth environment formed by the extracellular matrix of tumor stromal cells, tumor-associated neo-angiogenesis, secretion of cytokines and chemokines and possibly other growth-modulating factors by the tumor microenvironment as well as anti-neoplastic defense mechanisms conferred by the host immune system 51, 52.
A good example for possible shortcomings of cell culture based in vitro models as compared to animal models was given in a recent report by Curran and co-workers. While spontaneously developing medulloblastomas in Ptch+/−; p53−/− mice were dependent on Hedgehog-signaling and thus showed dramatic response to pharmacological Hedgehog blockade in vivo, no signs of Hedgehog pathway-activity or therapeutic response to Hedgehog-inhibition was observed by the same group in medulloblastoma cells after establishment as cell lines in vitro 53, 54. This was almost certainly as a result of the absence of tumor-stroma interactions in tissue culture models. Some of the key advantages and disadvantages of different in vitro and in vivo model systems of pancreatic cancer with respect to their application in drug discovery are summarized in Table 1.
Table 1.
Key advantages and disadvantages of different in vitro and in vivo model systems of pancreatic cancer in drug discovery
| Model system | Advantages | Disadvantages |
|---|---|---|
| Patient-derived cell lines | Accurately mirror entire spectrum of genetic alterations of human pancreatic cancers Cell-line based studies are comparably cheap and less time- and labor-consuming than studies on animal models Easily accessible Virtually unlimited amounts of cancer cells can be generated with relatively little required technical equipment |
Functional studies generally highly prone to artificial results Purely cell-line based treatment studies aimed at determining efficacy of novel drug candidates are commonly extremely poor in predicting actual therapeutic response or adverse effects in humans Do not allow to study important in vivo factors such as pharmacokinetics and toxicity profiles Do not reflect effects conferred by tumor-stroma interactions May genetically change over time |
| Co-cultures | Can to some degree mimic effects of secreted growth-modulating stimuli and direct physical contact with the ECM conferred by stromal cells | Difficult to co-culture more than two different cell compartments in one experiment Difficult to accurately control for the physiological ratio of neoplastic to stromal cells Most of the other disadvantages listed above for “patient-derived cell lines” apply here as well |
| Patient-derived xenografts | Accurately mirror entire spectrum of genetic alterations of human pancreatic cancers Can be used to enrich for neoplastic cell compartment and eliminate contaminating human stromal cells Testing of candidate drugs or other novel experimental therapeutic strategies by using xenografts usually faster and cheaper as compared to transgenic models Orthotopic xenograft models allow to study effects of locally invasive growth and systemic metastasis |
Xenogenic chimera between human and murine cells Do not accurately mirror effects conferred by host immune system Not suitable to accurately mimic tumor-stroma interactions in humans Different anatomies in mice versus humans May genetically change over time Require experimental exploitation of and potentially inflicting harm on mammals |
| Genetically engineered mouse models | Overall reliable, usually highly reproducible results Allow for distinct studies of virtually any desired combination of genetic alterations in an in vivo setting More recently described models enable to mirror most of the key features of the human disease surprisingly realistically, including local intrapancreatic growth, invasion into surrounding structures, systemic metastasis, development via mPanIN precursor lesions, histological microarchitecture resembling aspects of human pancreatic cancers Readily available for pre-clinical evaluation of novel experimental therapeutic approaches |
Do not reflect the entire spectrum of genetic alterations found in human cancer tissues Xenogenic model systems Different anatomies in mice versus humans Require experimental exploitation of and potentially inflicting harm on mammals Usually highly cost-, labor- and time-intensive studies Though usually providing valuable data on efficacy, pharmacokinetics and toxicity profiles of novel experimental therapeutic interventions, results from these studies nevertheless often fail to accurately predict response in humans, i.e. can not substitute subsequent clinical evaluation |
4.1 In vitro models of tumor-stroma interactions
In humans, pancreatic cancer is characterized by a pronounced deposition of extracellular matrix components and proliferation of stromal cells, namely surrounding fibroblasts, commonly referred to as “desmoplasmic reaction” 55. An increasing body of evidence suggests that this process is centrally involved in regulating neoplastic cell growth, invasiveness and metastatic spread and is thus of pivotal interest both for a deeper understanding of pathophysiological mechanisms governing pancreatic cancer progression as well as in order to identify processes that might be exploited as molecular targets for novel therapeutic approaches (see 56–58 for more comprehensive recent reviews on the topic of tumor-stromal cell interactions in pancreatic cancer). A striking example for the importance of the tumor microenvironment in mediating cancer progression was recently given by studies reporting somatic mutations and copy number alterations within cancer-associated fibroblasts which might be involved in promoting enhanced cancer cell growth 59–66. An ever growing number of secreted growth factors, cytokines and chemokines that are produced by stromal cells have been suggested to be pathophysiologically involved in pancreatic cancer progression, among them transforming growth factor-beta (TGF-beta), connective tissue growth factor (CTGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), insulin-like growth factor (IGF), nerve growth factor (NGF), leukemia inhibitory factor (LIF), oncostatin M, interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-8 (IL-8), chemokine (C-C motif) ligand 2 (CCL2), C-X-C chemokines CXCL1, CXCL2, CXCL8, CXCL12 and winglesstype MMTV integration site family members 1 and 3 (WNT1 and WNT3) 56, 67–69. Moreover, a growing body of evidence suggests that extracellular matrix components expressed by stromal cells can stimulate membrane-bound receptors on pancreatic cancer cells and mediate cell survival and motility 56.
A more recent finding is the observation that pancreatic stellate cells (PSC) are activated in pancreatic cancers and are potentially involved in modulating the malignant phenotype and inducing desmoplasmic reaction 70–74. Cell culture models aimed at studying potential synergistic effects on in vitro growth and motility of pancreatic cancer cells – by either using co-culture techniques or conditioned media - have been tested, but their value and applicability for functional studies in translational research is only beginning to be well understood at present 68, 75–77.
4.2 Functional in vitro assays
Finally, and especially in the case of pancreatic cancer, metastatic dissemination is a major predictor of therapeutic success and overall survival in solid tumors. The complex cascade of metastatic spread involves several steps which a neoplastic cell has to overcome in order to form a metastatic tumor, including proliferation of neoplastic cells at the primary tumor site, active migration and invasion into surrounding tissue, intravasation into lymph or blood vessels, anchorage independent survival in the blood stream, accumulation and extravasation and autonomous growth at a distant organ site, and induction of neo-angiogenesis at the metastatic tumor site (Figure 2). To date, most of these steps are far from being sufficiently well understood on a pathophysiological level and are therefore difficult to simulate in an in vitro setting in a way that would allow to accurately mirror each and every of the biological aspects involved in regulating these events in situ 78, 79.
Figure 2.

Pancreatic cancer does not occur ‘de novo’ but via development of clearly defined tangible precursor lesions that correlate with step-wise accumulation of genetic alterations. In order to develop systemic metastases, an invasive clone must overcome several additional selection barriers, including invasion into surrounding tissue, anchorage-independent survival in the blood stream, active migration and invasion at a distant organ site and establishment of a metastatic tumor lesion. While a variety of in vitro methods exist trying to mimic single aspects of this complex process, it is nevertheless impossible to faithfully mimic in its entirety by means of cell line-based in vitro models alone as of to date.
Of note, an ever growing number of in vitro assays has been developed, attempting to mimic some of the aspects of neoplastic tumor growth and dissemination named above. For example, net tumor cell growth can readily be assessed in vitro by direct cell counting or by colorimetric 3-(4,5-dimethylthiazolyl- 2)-2,5-diphenyltetrazolium bromide (MTT) assays, proliferation can clearly be quantified by assays such as BrdU or tritium incorporation, immunolabeling for Ki67 and other surrogate markers of proliferation, CFSE assays etc., while a significant number of assays exists to accurately quantify different stages of apoptosis in vitro (e.g. DNA fragmentation, TUNEL, annexin binding, caspase activation assays, etc.) 80–83. Also, a number of in vitro assays exist which simulate and quantify some of the more complex steps pointed out above. Cell motility can readily be assessed in vitro by time-lapse observations under the microscope, wound healing or Boyden chamber assays; introduction of a layer of matrigel or other coating material can be used to mimic tumor cell invasion and breach through the epithelial basement membrane 84. Anchorage independent growth can be simulated in vitro by means of soft agar, methylcellulose or other three-dimensional culturing techniques 85, and re-plating of highly diluted cell suspensions and subsequent ability to form colonies has been used as a means to determine clonogenecity in vitro. Finally, various in vitro models of tube formation and angiogenesis are commonly used 86.
All of the above-mentioned assays have been utilized for assessing in vitro-effects of novel candidate drugs on these distinct central cell functions, and in many cases, a reasonable correlation between these in vitro assays and the in vivo scenario has been documented. Moreover, these in vitro assays enable scientists to specifically examine functional effects of manipulating single proteins by means of genetic techniques. Gene function can be specifically abrogated through gene knockout, RNAi-mediated knockdown of gene expression or introduction of dominant negative protein. Enhanced gene function on the other hand is readily achieved by forced overexpression or introduction of constitutively active mutant gene products. This approach can be applied to link specific genes and signaling pathways to distinct cellular functions involved in pancreatic carcinogenesis and metastatic spread, with the final goal being the identification of potential targets for the development of novel therapeutics 87. One promising in vitro screening method to emerge in recent times has been pharmacological synthetic lethal screening, wherein isogenic cell lines differing in a single gene product are screened against large scale chemical libraries to identify small molecules that are lethal only to that line in the isogenic pair which has a defective gene 88. The use of synthetic lethal screen was first employed in yeast to identify genetic pathways that are lethal to survival when mutated in concert, and its extrapolation to cancer cells as a tool for drug discovery was first proposed by Hartwell and colleagues in 1997 89. Recently, using such synthetic lethal screening of isogenic pancreatic cancer cell lines differing only in Dpc4 function, von Hoff and colleagues were able to isolate compounds that are specifically toxic to Dpc4- null cells, a finding with immediate translational significance given that 55% of pancreatic cancer lack Dpc4 function 90.
4.3 In vitro models for genetic studies
While these techniques are undoubtedly of immense value as screening tests to identify potential new drug targets, there are some obvious limitations of these cell culture-based in vitro model systems and the in vivo relevance of obtained in vitro results is often unclear 91. Firstly, as mentioned above, it is almost impossible to accurately mimic the complex network of tumor-host interactions in its entirety. This crosstalk includes direct effects conferred by the physical interaction of neoplastic cells with the extracellular matrix of surrounding stromal tissue, humoral factors, e.g. secretion of cytokines and growth factors by host cells, and effects mediated by the host immune system. Secondly, and possibly more importantly, pancreatic cancer cells might progressively alter genetically over time with increasing duration of in vitro culture.
There are various reports demonstrating that pancreatic cancer cells cultured ex vivo as xenografts or as cell lines in vitro accurately mirror the genetic setup and dependence on known oncogenic signaling pathways of their parent tumors and remain fairly stable over time 36, 92. However, there are also several lines of evidence suggesting that additional genetic alterations might in fact occur during long-term in vitro propagation of pancreatic cancer cells.
Firstly, from a theoretical point of view it seems likely that pancreatic cancer cell lines might not have the ability to maintain their genetic fidelity over longer periods of time. Genomic instability is a key feature observed in almost all cases of pancreatic cancer and is among the earliest aberrations found during pancreatic carcinogenesis 93–96. Therefore, under cell culture conditions optimized for maximum survival of neoplastic cells and in the absence of other selection mechanisms usually found in the in vivo situation (i.e. lack of immune-surveillance, absence of anatomical barriers etc.) there might likely be a greater tendency towards acquiring additional, heterogeneous genetic alterations.
Secondly, there are also various lines of empirical evidence suggesting that accumulation of additional genetic changes might in fact occur in pancreatic cancer cell lines with increasing passage numbers. In a recent global sequence analysis study on 24 cases of pancreatic cancer covering the majority of genes represented in the RefSeq database, Jones et. al found an average of 48 somatic mutations per pancreatic cancer sample (ranging from less than 30 to over 140 mutations in individual samples) 49. While this number is significantly lower than the average number of mutations found in breast (on average 101 per tumor) and colorectal cancers (77 per tumor) using a similar approach 97, it is still by far higher than what might have been expected based on in vitro and in vivo transformation experiments (Table 2).
Table 2.
Studies estimating numbers of genetic alterations required in pancreatic carcinogenesis
| Reference | Summary | Species | Genetic model used | Number of genetic alterations | Comments |
|---|---|---|---|---|---|
| 49 | direct sequencing of 24 human pancreatic cancer samples; DNA isolated from cell lines or murine xenografts | human | Genuine human pancreatic ductal adenocarcinoma cells | average of 48 per tumor sample | first and so far only global sequencing study of human pancreatic cancer samples |
| 91 | pancreas-specific expression of an oncogenic KrasG12D, which is achieved by combination to a lox-stop-lox (LsL) cassette and expression of Cre-recombinase under control of Pdx1 or P48 promoters, led to development of mPanIN precursor lesions, which eventually progressed to fully invasive cancers at low frequency | mouse | LsL-KrasG12D; Pdx1-Cre LSL-KrasG12D; P48+/Cre | 1 | |
| 7 | targeting of oncogenic KrasG12D expression to the adult pancreas by means of an inducible Ela-CreERT2 construct leads to formation of mPanIN lesions but not fully malignant pancreatic cancer | mouse | LsL-KrasG12D; Ela-CreERT2 | 1 | |
| 93 | pancreas-specific targeting of combined KrasG12D and Trp53R172H expression led to development of metastatic pancreatic carcinomas with median survival of 5 months | mouse | LsL-KrasG12D; LsL-Trp53R172H; Pdxl-Cre | 2 | no acquired mutations found in Cdkn2/lnk4a, Cdk4 or Smad4 |
| 94 | development of pancreatic adenocarcinomas with average latency of 6.2 weeks in mice expressing KrasG12D in the absence of p53 und control of Pdx1-Cre construct | mouse | LsL-KrasG12D; p53lox/lox; Pdx1-Cre | 2 | |
| 95 | rapid development of poorly differentiated metastasizing cancers in murine pancreata expressing oncogenic KrasG12D in combination with abrogation of Ink4a/Arf expression | mouse | LsL-KrasG12D; Ink4a/Arflox/lox; Pdx1-Cre | 3 | |
| 97 | development of well-differentiated pancreatic ductal adenocarcinomas with high penetrance and short median survival of only 59 days upon concomitant oncogenic Kras-signaling and Tgfbr2-knockout | mouse | LsL-KrasG12D;Tgfbr2flox/flox; Ptf1acre/+ | 2 | |
| 100 | Immortalization and in vitro transformation of cultured non-neoplastic human pancreatic ductal cells (hTERT-HPNE) | human | hTERT-HPNE; E6/E7; KrasG12D;SV40st | 5 | |
| 19 | Expression of oncogenic Kras in cultured immortalized non-neoplastic human pancreatic ductal cells (HPDE) leads to tumorigenecity in SCID mice in 50% of cases | human | HPDE; E6/E7; Kras4BG12V | 3 | absence of in vitro clonogenecity |
For example, in a recently described genetically engineered mouse model of pancreatic cancer it was found, that introduction of one single genetic alteration, i.e. pancreas-specific overexpression of an oncogenic KrasG12D allele, was sufficient to induce formation of murine pancreatic intraepithelial neoplasia (mPanIN) lesions. In two of 29 mice followed longitudinally (i.e. less than 10%) these precursor lesions progressed into frank malignancy after 6 and 8 months, respectively 98. In this particular model, the long latency and low frequency of actual cancers suggest, that additional mutations must occur in order to result in a fully malignant phenotype, whereas the majority of KrasG12D expressing cells might undergo ras-induced senescence and be eliminated, thereby failing to accumulate additional genetic alterations required for complete malignant transformation 99. These observations are in line with data from our own group showing that expression of KrasG12D is sufficient to generate mPanIN lesions in adult mouse pancreata when targeted under the control of tamoxifen-inducible Ela-CreERT2 or Mist1-CreERT2 constructs 7. However, progression to fully malignant pancreatic cancer was not observed in this model during 12 months of follow-up.
A later report by Hingorani et al. showed that additional expression of a dominant negative form of the tumor suppressor gene p53 (Trp53R172H) under the control of Pdx1-Cre led to development of moderately to well-differentiated metastatic pancreatic cancers with a median survival of 5 months and 100% lethality after 12 months of follow-up 100. As observed in pancreata overexpressing KrasG12D alone, development of pancreatic cancer in this latter model was preceded by the appearance occurrence of mPanIN lesions. Of note, LOH of the p53 gene locus was reproducibly observed in cell lines derived from murine pancreatic cancers in this model system, but expression analysis and direct sequencing did not show any evidence for alterations in the Cdkn2/Ink4a, Cdk4 or Smad4 signaling pathways 100.
These data are in line with another report of specific expression of KrasG12D and concomitant abrogation of p53 expression by Cre-mediated excision under the control of Pdx1-Cre 101. In this model, pancreatic adenocarcinomas occurred with an average latency of 6.2 weeks. In yet another example, DePinho and collegues reported that pancreas-specific targeting of oncogenic KrasG12D expression in combination with abrogated Ink4a/Arf (murine p16 and p19) function by crossing on a Pdx1-Cre genetic background led to the rapid formation of poorly differentiated pancreatic cancers 102. These mice reproducibly die of metastatic disease between 7 to 11 weeks of age 103. Similarly, pancreas-specific KrasG12D expression in combination with targeted silencing of transforming growth factor beta-signaling by specific Tgfbr2- knockout led to the development of well-differentiated ductal adenocarcinomas in the murine pancreata with 100% penetrance and a short median survival of only 59 days 104.
While all of the above mentioned transformation models were done in mice, i.e. in a different species and conclusions drawn from these models can therefore be applied to the human disease only with some caution 105, 106, there are nevertheless also some in vitro transformation models of human cells suggesting that the number of genetic alterations required for malignant transformation might actually be considerably smaller than the average of 48 alterations observed by global direct sequencing 49.
In a recent study, Campbell and collegues found that only 4 genetic alterations are necessary for in vitro transformation of immortalized human pancreatic epithelial cells (HPNE) 20, 107. hTERT-HPNE cells are immortalized through ectopic expression of the catalytic subunit of telomerase (hTERT) 108–110. Upon subsequent stable transfection of the human papillomavirus 16-derived genes E6 and E7 (E6/E7), constitutively active KrasG12D and SV40 small t (st) antigen, HPNE cells readily showed colony formation and anchorage independent growth, as well as spontaneous migration and invasion into a matrigel layer in modified Boyden chamber assays, in line with acquisition of a malignant phenotype.
Using a similar experimental approach, Tsao and collegues found that isolated non-neoplastic human pancreatic ductal epithelial cells, which had previously been immortalized by transfection with the human papillomavirus 16 genes E6 and E7 (E6/E7) 16, 17, showed xenograft tumor formation upon subcutaneous injection of one million cells subcutaneously into SCID mice in 4/7 cases (57%) and in 2/5 cases (40%) upon orthotopic (intrapancreatic) injection of two million cells 19. Curiously, these cells did not shown signs of in vitro transformation, specifically no colony formation was observed in soft agar assays.
Taken together, these observations suggest that the actual number of genetic alterations required for pancreatic carcinogenesis in humans might be considerably smaller than the average of 48 alterations found in the global sequencing approach by Jones et. al 49. Not only is this of immense interest for our understanding of underlying pathophysiological mechanisms leading to the development of pancreatic cancer, but this idea also holds extremely promising implications for the identification of molecular targets for therapeutic intervention. Despite the lack of clinically tangible progress in the development of novel therapeutic strategies for pancreatic cancer over the last decades, it seems likely that in the end pancreatic cancer is caused by only a limited number of defined genetic alterations, many or most of which are already known to date and hence readily accessible for the development of novel therapeutics.
5. Expert opinion
From the discussion above, it is fair to conclude that panels of freshly generated low-passage pancreatic cancer cell lines mirror genetic features and response to therapeutic interventions with better fidelity than cell lines which have been cultured in vitro for longer periods of time 36, 47, 48.
In the case of pancreatic cancer, a significant limitation for the use of in vitro models for translational research and drug discovery is the lack of cell line models for pancreatic cancer precursor lesions. In recent years it has increasingly become commonly accepted that virtually all cases of pancreatic cancer arise via the development of tangible and genetically defined precursor lesions, including pancreatic intraepithelial neoplasia (PanIN), mucinous cystic neoplasms (MCN) and intraductal papillary mucinous neoplasms (IPMN) 111, 112. Better understanding of the genetics of these precursor lesions has direct implications for early detection as well as for secondary prophylaxis. Moreover, studying the genetics of precursor lesions allows us to separate “early” (disease initiating) alterations from “late” (disease promoting) changes. While nearly all or majority of genetic changes in precursors are likely to be “driver” lesions, many of the genetic alterations in frank malignancies are likely to be “passenger” mutations, acquired as a result of progressive genetic instability.
While genetically engineered mouse models mimicking mPanINs 98, MCNs 113, 114 and IPMNs 115 could be successfully developed in recent years, long term in vitro cultures of the human counterparts of these precursor lesions are currently not available. The lack of such cell line models seems to be most likely due to technical difficulties. For example, PanIN lesions are, by definition, microscopic in nature and nearly impossible to localize macroscopically, such that enrichment for these cells through ex vivo passage in mice is not an option with currently available techniques. Moreover, due to their likely limited growth potential, the ability of PanINs to engraft and be serially propagated in immunocompromised mice is also questionable. It remains an interesting point of speculation, whether 17 or not improvements of ex vivo and in vitro culturing techniques will enable generation of such cell line models of pancreatic cancer precursor lesions in the near future.
One can conclude from the mentioned points that although cell line models are undoubtedly of immense value for drug discovery in translational research related to pancreatic cancer, the use of animal models is nevertheless indispensible for a more concise preclinical evaluation of novel drug candidates. This is even more so, since acquisition of toxicity data and estimation of adverse effect profiles can only be done using test animals in a preclinical setting. Estimation of toxicity profiles based on cell line data alone is almost uniformly less useful.
Therefore, the main scope for the use of cell line based models remains in the early phase of drug discovery, wherein rapid, repetitive screening of large numbers of candidate substances, often in an automated or semi-automated manner, is required. As opposed to the use of experimental animal models, cell lines enable to cheaply and rapidly obtain virtually unlimited amounts of cancer cells and are therefore often ideal for such screening setups.
Also, at an early phase of drug discovery, where a direct molecular target or oncogenic signaling pathway has already been identified, genetically manipulated cell line models can often be used successfully for high-throughput efficacy screening in vitro. Candidate small molecules with satisfactory efficacy can then undergo further biological testing, including assessment of toxicity profiles, in vivo pharmacokinetics and pharmacodynamics in animal models.
A recent example for this approach is the search for small-molecule Hedgehog inhibitors as novel cancer drug candidates. Aberrantly re-activated Hedgehog signaling has been described in basal cell carcinomas 116, medulloblastomas 117, 118 as well as in various cancers of the gastrointestinal 119 and respiratory tract 120, 121, including pancreatic cancer 119, 122. Pharmacological blockade of Hedgehog signaling by means of the plant alkaloid cyclopamine 123, 124 has been identified as valid experimental therapeutic approach in preclinical in vivo and in vitro model systems 117, 121. However, cyclopamine itself is not an ideal drug candidate for clinical application due to its poor water solubility and bioavailability as well as a relatively low affinity to Smo. Therefore, cell based high-throughput screens have been used to identify other small molecule Hedgehog inhibitors with more favorable pharmacokinetics and – dynamics that are more suitable as drug candidates 125. The in vitro model system for this screen consisted of C3H.10T1/2 cells stably transfected with a Hh-responsive (Gli8x-luciferase) reporter construct, and a lipid-modified form of Hedgehog-ligand with enhanced potency which had an octyl moiety attached to its N-terminus (ShhOCT) 126 was used for basal induction of Hedgehog pathway activity. This reporter cell line was assayed against a library of ~100,000 small synthetic organic molecules. One small molecule Hedgehog-inhibitor thus identified, designated Hh-Antag691 127, inhibited pathway activity at ~10fold lower concentrations as compared to cyclopamine and was subsequently shown to inhibit medulloblastoma growth in a genetically engineered Ptch1+/− p53+/− mouse model 54. Similar reporter systems used in other studies included stably transfected NIH-3T3 cells and a palmitoyl- and cholesteryl-modified ShhN polypeptide (ShhN) 128, 129. Of note, these examples also unmasked some of the inherent drawbacks of this approach for drug discovery, namely that pharmacokinetics, in vivo efficacy and toxicity profiles in humans are often difficult to estimate based on the initial preclinical data alone. Moreover, potential off-target-effects of a given substance in the in vivo situation can greatly affect its suitability as a drug candidate – either by conferring undesirable adverse effects or by contributing additional but unexpected favorable effects on tumor cell survival. Therefore, in scenarios like the one mentioned here, it is quite common that drug candidates which performed well in the initial discovery screens and early preclinical characterization phases fail at later stages during preclinical and clinical development, so that they need to be modified in chemical composition and reevaluated or completely abandoned from further development towards clinical application. Only a minority of candidate substances identified in discovery screens can be developed all the way towards successful application as novel drugs in the clinical arena. In the case of Hedgehog-Antagonists described here follow-up drug candidate small molecule Hedgehog-inhibitors are currently evaluated in clinical phase I and II trials in various malignant solid tumors including pancreatic cancer (www.clinicaltrials.gov) and others are likely to follow in the near future.
Another example demonstrating the amount of tedious work and time-consuming steps involved in translating an identified molecular target into clinical application as novel therapeutic strategy is given by attempts to target tumor-neoangiogenesis in pancreatic cancer as well as other solid tumors over recent years. While various studies reported striking successes of such strategies in mouse models of various cancers beginning over a decade ago 130–135, drugs resulting from these efforts have only recently been introduced into the clinical arena and have so far shown overall disappointing results in terms of improving overall survival of pancreatic cancer patients, although additional evaluations are still ongoing 136 (www.clinicaltrials.gov).
Additional examples of signaling pathways recently identified to be pathogenetically involved in genesis and maintenance of pancreatic cancer and thus representing potential targets for the development of novel therapeutics, for which similar screening and preclinical evaluation strategies are being followed include the Notch 137–140, Wnt 141, 142, TGF-beta 143, EGF 144–146, Raf/Mek/Erk 147, 148, PI3K/Akt 149 and mTOR 150–153 signaling pathways.
Acknowledgments
Declaration of interests
The authors are supported by the Sol Goldman Pancreatic Cancer Research Center, the Michael Rolfe Foundation, NIH Specialized Programs of Research Excellence in Gastrointestinal Cancer grants P50CA62924 and NIH R01CA113669.
Bibliography
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008 Mar–Apr;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.ACS. Cancer Facts & Figures 2008. Atlanta: American Cancer Society; 2008. [Google Scholar]
- 3.Carpelan-Holmstrom M, Nordling S, Pukkala E, Sankila R, Luttges J, Kloppel G, et al. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data of the Finnish Cancer Registry. Gut. 2005 Mar;54(3):385–7. doi: 10.1136/gut.2004.047191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jamieson JD. Prospectives for cell and organ culture systems in the study of pancreatic carcinoma. J Surg Oncol. 1975;7(2):139–41. doi: 10.1002/jso.2930070209. [DOI] [PubMed] [Google Scholar]
- 5.Longnecker DS, Wiebkin P, Schaeffer BK, Roebuck BD. Experimental carcinogenesis in the pancreas. Int Rev Exp Pathol. 1984;26:177–229. [PubMed] [Google Scholar]
- 6.Hall PA, Lemoine NR. Rapid acinar to ductal transdifferentiation in cultured human exocrine pancreas. J Pathol. 1992 Feb;166(2):97–103. doi: 10.1002/path.1711660203. [DOI] [PubMed] [Google Scholar]
- 7.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008 Dec 2;105(48):18913–8. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park SW, Davison JM, Rhee J, Hruban RH, Maitra A, Leach SD. Oncogenic KRAS induces progenitor cell expansion and malignant transformation in zebrafish exocrine pancreas. Gastroenterology. 2008 Jun;134(7):2080–90. doi: 10.1053/j.gastro.2008.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murtaugh LC, Leach SD. A case of mistaken identity? Nonductal origins of pancreatic “ductal” cancers. Cancer Cell. 2007 Mar;11(3):211–3. doi: 10.1016/j.ccr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 10.Githens S. Pancreatic duct cell cultures. Annu Rev Physiol. 1994;56:419–43. doi: 10.1146/annurev.ph.56.030194.002223. [DOI] [PubMed] [Google Scholar]
- 11.Githens S. The pancreatic duct cell: proliferative capabilities, specific characteristics, metaplasia, isolation, and culture. J Pediatr Gastroenterol Nutr. 1988 Jul–Aug;7(4):486–506. [PubMed] [Google Scholar]
- 12.Bonner-Weir S, Toschi E, Inada A, Reitz P, Fonseca SY, Aye T, et al. The pancreatic ductal epithelium serves as a potential pool of progenitor cells. Pediatric diabetes. 2004;5 Suppl 2:16–22. doi: 10.1111/j.1399-543X.2004.00075.x. [DOI] [PubMed] [Google Scholar]
- 13.Jones RT, Barrett LA, van Haaften C, Harris CC, Trump BF. Carcinogenesis in the pancreas. I. Long-term explant culture of human and bovine pancreatic ducts. J Natl Cancer Inst. 1977 Mar;58(3):557–65. doi: 10.1093/jnci/58.3.557. [DOI] [PubMed] [Google Scholar]
- 14.Oda D, Savard CE, Nguyen TD, Swenson ER, Lee SP. Culture of human main pancreatic duct epithelial cells. In Vitro Cell Dev Biol Anim. 1998 Mar;34(3):211–6. doi: 10.1007/s11626-998-0126-6. [DOI] [PubMed] [Google Scholar]
- 15.Trautmann B, Schlitt HJ, Hahn EG, Lohr M. Isolation, culture, and characterization of human pancreatic duct cells. Pancreas. 1993 Mar;8(2):248–54. doi: 10.1097/00006676-199303000-00017. [DOI] [PubMed] [Google Scholar]
- 16.Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao MS. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol. 1996 Jun;148(6):1763–70. [PMC free article] [PubMed] [Google Scholar]
- 17.Ouyang H, Mou L, Luk C, Liu N, Karaskova J, Squire J, et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000 Nov;157(5):1623–31. doi: 10.1016/S0002-9440(10)64800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KM, Yasuda H, Hollingsworth MA, Ouellette MM. Notch 2-positive progenitors with the intrinsic ability to give rise to pancreatic ductal cells. Lab Invest. 2005 Aug;85(8):1003–12. doi: 10.1038/labinvest.3700298. [DOI] [PubMed] [Google Scholar]
- 19.Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005 Jun 15;65(12):5045–53. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 20.Campbell PM, Lee KM, Ouellette MM, Kim HJ, Groehler AL, Khazak V, et al. Ras-driven transformation of human nestin-positive pancreatic epithelial cells. Methods Enzymol. 2008;439:451–65. doi: 10.1016/S0076-6879(07)00431-4. [DOI] [PubMed] [Google Scholar]
- 21.Bendayan M, Duhr MA, Gingras D. Studies on pancreatic acinar cells in tissue culture: basal lamina (basement membrane matrix promotes three-dimensional reorganization. Eur J Cell Biol. 1986 Oct;42(1):60–7. [PubMed] [Google Scholar]
- 22.Longnecker DS, Lilja HS, French J, Kuhlmann E, Noll W. Transplantation of azaserine-induced carcinomas of pancreas in rats. Cancer Lett. 1979 Aug;7(4):197–202. doi: 10.1016/s0304-3835(79)80080-4. [DOI] [PubMed] [Google Scholar]
- 23.Ulrich AB, Schmied BM, Standop J, Schneider MB, Pour PM. Pancreatic cell lines: a review. Pancreas. 2002 Mar;24(2):111–20. doi: 10.1097/00006676-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Esni F, Miyamoto Y, Leach SD, Ghosh B. Primary explant cultures of adult and embryonic pancreas. Methods Mol Med. 2005;103:259–71. doi: 10.1385/1-59259-780-7:259. [DOI] [PubMed] [Google Scholar]
- 25.Hober C, Benhamou PY, Watt PC, Watanabe Y, Nomura Y, Stein E, et al. A new culture method for human pancreatic islets using a biopore membrane insert. Pancreas. 1997 Mar;14(2):199–204. doi: 10.1097/00006676-199703000-00014. [DOI] [PubMed] [Google Scholar]
- 26.Kenmochi T, Miyamoto M, Une S, Nakagawa Y, Moldovan S, Navarro RA, et al. Improved quality and yield of islets isolated from human pancreata using a two-step digestion method. Pancreas. 2000 Mar;20(2):184–90. doi: 10.1097/00006676-200003000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Lucas-Clerc C, Massart C, Campion JP, Launois B, Nicol M. Long-term culture of human pancreatic islets in an extracellular matrix: morphological and metabolic effects. Molecular and cellular endocrinology. 1993 Jul;94(1):9–20. doi: 10.1016/0303-7207(93)90046-m. [DOI] [PubMed] [Google Scholar]
- 28.Yuan S, Rosenberg L, Paraskevas S, Agapitos D, Duguid WP. Transdifferentiation of human islets to pancreatic ductal cells in collagen matrix culture. Differentiation. 1996 Oct;61(1):67–75. doi: 10.1046/j.1432-0436.1996.6110067.x. [DOI] [PubMed] [Google Scholar]
- 29.Beattie GM, Itkin-Ansari P, Cirulli V, Leibowitz G, Lopez AD, Bossie S, et al. Sustained proliferation of PDX-1+ cells derived from human islets. Diabetes. 1999 May;48(5):1013–9. doi: 10.2337/diabetes.48.5.1013. [DOI] [PubMed] [Google Scholar]
- 30.Lu J, Gu YP, Xu X, Liu ML, Xie P, Song HP. Adult islets cultured in collagen gel transdifferentiate into duct-like cells. World J Gastroenterol. 2005 Jun 14;11(22):3426–30. doi: 10.3748/wjg.v11.i22.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray HE, Paget MB, Bailey CJ, Downing R. Sustained insulin secretory response in human islets co-cultured with pancreatic duct-derived epithelial cells within a rotational cell culture system. Diabetologia. 2009 Jan 8; doi: 10.1007/s00125-008-1247-x. [DOI] [PubMed] [Google Scholar]
- 32.Mueller BM, Reisfeld RA. Potential of the scid mouse as a host for human tumors. Cancer Metastasis Rev. 1991 Oct;10(3):193–200. doi: 10.1007/BF00050791. [DOI] [PubMed] [Google Scholar]
- 33.Pantelouris EM. Absence of thymus in a mouse mutant. Nature. 1968 Jan 27;217(5126):370–1. doi: 10.1038/217370a0. [DOI] [PubMed] [Google Scholar]
- 34.van Weerden WM, Romijn JC. Use of nude mouse xenograft models in prostate cancer research. Prostate. 2000 Jun 1;43(4):263–71. doi: 10.1002/1097-0045(20000601)43:4<263::aid-pros5>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 35.Ikeda Y, Ezaki M, Hayashi I, Yasuda D, Nakayama K, Kono A. Establishment and characterization of human pancreatic cancer cell lines in tissue culture and in nude mice. Jpn J Cancer Res. 1990 Oct;81(10):987–93. doi: 10.1111/j.1349-7006.1990.tb03336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006 Aug 1;12(15):4652–61. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 37.Walter K, Eshleman J, Goggins M. Xenografting and harvesting human ductal pancreatic adenocarcinomas for DNA analysis. Methods Mol Med. 2005;103:103–11. doi: 10.1385/1-59259-780-7:103. [DOI] [PubMed] [Google Scholar]
- 38.Pretlow TG, Delmoro CM, Dilley GG, Spadafora CG, Pretlow TP. Transplantation of human prostatic carcinoma into nude mice in Matrigel. Cancer Res. 1991 Jul 15;51(14):3814–7. [PubMed] [Google Scholar]
- 39.Elsasser HP, Lehr U, Agricola B, Kern HF. Establishment and characterisation of two cell lines with different grade of differentiation derived from one primary human pancreatic adenocarcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61(5):295–306. doi: 10.1007/BF02890431. [DOI] [PubMed] [Google Scholar]
- 40.Kobari M, Hisano H, Matsuno S, Sato T, Kan M, Tachibana T. Establishment of six human pancreatic cancer cell lines and their sensitivities to anti-tumor drugs. Tohoku J Exp Med. 1986 Nov;150(3):231–48. doi: 10.1620/tjem.150.231. [DOI] [PubMed] [Google Scholar]
- 41.Bleday R, Tzanakakis GN, Schwalke MA, Wanebo HJ, Vezeridis MP. Epidermal growth factor stimulation and metastatic rate in human pancreatic carcinoma cell lines. J Surg Res. 1990 Sep;49(3):276–9. doi: 10.1016/0022-4804(90)90133-m. [DOI] [PubMed] [Google Scholar]
- 42.Silberberg JM, Gordon S, Zucker S. Identification of tissue factor in two human pancreatic cancer cell lines. Cancer Res. 1989 Oct 1;49(19):5443–7. [PubMed] [Google Scholar]
- 43.Kyriazis AP, McCombs WB, 3rd, Sandberg AA, Kyriazis AA, Sloane NH, Lepera R. Establishment and characterization of human pancreatic adenocarcinoma cell line SW-1990 in tissue culture and the nude mouse. Cancer Res. 1983 Sep;43(9):4393–401. [PubMed] [Google Scholar]
- 44.Chang BK, Gutman R, Chou TC. Schedule-dependent interaction of alphadifluoromethylornithine and cis-diamminedichloroplatinum(II) against human and hamster pancreatic cancer cell lines. Cancer Res. 1987 May 1;47(9):2247–50. [PubMed] [Google Scholar]
- 45.Elsasser HP, MacDonald R, Dienst M, Kern HF. Characterization of a transglutaminase expressed in human pancreatic adenocarcinoma cells. Eur J Cell Biol. 1993 Aug;61(2):321–8. [PubMed] [Google Scholar]
- 46.Suwa H, Yoshimura T, Yamaguchi N, Kanehira K, Manabe T, Imamura M, et al. K-ras and p53 alterations in genomic DNA and transcripts of human pancreatic adenocarcinoma cell lines. Jpn J Cancer Res. 1994 Oct;85(10):1005–14. doi: 10.1111/j.1349-7006.1994.tb02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int J Cancer. 1975 May 15;15(5):741–7. doi: 10.1002/ijc.2910150505. [DOI] [PubMed] [Google Scholar]
- 48.Jaffee EM, Schutte M, Gossett J, Morsberger LA, Adler AJ, Thomas M, et al. Development and characterization of a cytokine-secreting pancreatic adenocarcinoma vaccine from primary tumors for use in clinical trials. Cancer J Sci Am. 1998 May–Jun;4(3):194–203. [PubMed] [Google Scholar]
- 49.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008 Sep 26;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore PS, Sipos B, Orlandini S, Sorio C, Real FX, Lemoine NR, et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001 Dec;439(6):798–802. doi: 10.1007/s004280100474. [DOI] [PubMed] [Google Scholar]
- 51.Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn. 2008 Mar;10(2):111–22. doi: 10.2353/jmoldx.2008.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grippo PJ, Sandgren EP. Modeling pancreatic cancer in animals to address specific hypotheses. Methods Mol Med. 2005;103:217–43. doi: 10.1385/1-59259-780-7:217. [DOI] [PubMed] [Google Scholar]
- 53.Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, et al. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006 Apr 15;66(8):4215–22. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- 54.Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004 Sep;6(3):229–40. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 55.Maitra A, Hruban RH. Pancreatic cancer. Annual review of pathology. 2008;3:157–88. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007 Apr;6(4):1186–97. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 57.Kleeff J, Beckhove P, Esposito I, Herzig S, Huber PE, Lohr JM, et al. Pancreatic cancer microenvironment. Int J Cancer. 2007 Aug 15;121(4):699–705. doi: 10.1002/ijc.22871. [DOI] [PubMed] [Google Scholar]
- 58.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annual review of pathology. 2006;1:119–50. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 59.Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008 May;40(5):650–5. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002 Nov;32(3):355–7. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 61.Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998 Jan 1;58(1):23–8. [PubMed] [Google Scholar]
- 62.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000 May 1;60(9):2562–6. [PubMed] [Google Scholar]
- 63.Weber F, Shen L, Fukino K, Patocs A, Mutter GL, Caldes T, et al. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am J Hum Genet. 2006 Jun;78(6):961–72. doi: 10.1086/504090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004 Oct 15;64(20):7231–6. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 65.Fukino K, Shen L, Patocs A, Mutter GL, Eng C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. Jama. 2007 May 16;297(19):2103–11. doi: 10.1001/jama.297.19.2103. [DOI] [PubMed] [Google Scholar]
- 66.Tuhkanen H, Anttila M, Kosma VM, Heinonen S, Juhola M, Helisalmi S, et al. Frequent gene dosage alterations in stromal cells of epithelial ovarian carcinomas. Int J Cancer. 2006 Sep 15;119(6):1345–53. doi: 10.1002/ijc.21785. [DOI] [PubMed] [Google Scholar]
- 67.Lohr M, Schmidt C, Ringel J, Kluth M, Muller P, Nizze H, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001 Jan 15;61(2):550–5. [PubMed] [Google Scholar]
- 68.Shek FW, Benyon RC, Walker FM, McCrudden PR, Pender SL, Williams EJ, et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am J Pathol. 2002 May;160(5):1787–98. doi: 10.1016/s0002-9440(10)61125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato N, Maehara N, Goggins M. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res. 2004 Oct 1;64(19):6950–6. doi: 10.1158/0008-5472.CAN-04-0677. [DOI] [PubMed] [Google Scholar]
- 70.Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007 Jan;117(1):50–9. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneiderhan W, Diaz F, Fundel M, Zhou S, Siech M, Hasel C, et al. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J Cell Sci. 2007 Feb 1;120(Pt 3):512–9. doi: 10.1242/jcs.03347. [DOI] [PubMed] [Google Scholar]
- 72.Erkan M, Kleeff J, Gorbachevski A, Reiser C, Mitkus T, Esposito I, et al. Periostin creates a tumorsupportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology. 2007 Apr;132(4):1447–64. doi: 10.1053/j.gastro.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 73.Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Hamada S, et al. Fibrinogen induces cytokine and collagen production in pancreatic stellate cells. Gut. 2008 Dec 3; doi: 10.1136/gut.2008.154401. [DOI] [PubMed] [Google Scholar]
- 74.Vonlaufen A, Phillips PA, Xu Z, Goldstein D, Pirola RC, Wilson JS, et al. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 2008 Oct 1;68(19):7707–10. doi: 10.1158/0008-5472.CAN-08-1132. [DOI] [PubMed] [Google Scholar]
- 75.Manapov F, Muller P, Rychly J. Translocation of p21(Cip1/WAF1) from the nucleus to the cytoplasm correlates with pancreatic myofibroblast to fibroblast cell conversion. Gut. 2005 Jun;54(6):814–22. doi: 10.1136/gut.2003.036491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang HB, Xu M, Wang XP. Pancreatic stellate cells promote proliferation and invasiveness of human pancreatic cancer cells via galectin-3. World J Gastroenterol. 2008 Apr 7;14(13):2023–8. doi: 10.3748/wjg.14.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kanno A, Satoh K, Masamune A, Hirota M, Kimura K, Umino J, et al. Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int J Cancer. 2008 Jun 15;122(12):2707–18. doi: 10.1002/ijc.23332. [DOI] [PubMed] [Google Scholar]
- 78.Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008 Mar;8(3):212–26. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004 Jun;4(6):448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 80.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994 May 2;171(1):131–7. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 81.Nolte T, Kaufmann W, Schorsch F, Soames T, Weber E. Standardized assessment of cell proliferation: the approach of the RITA-CEPA working group. Exp Toxicol Pathol. 2005 Nov;57(2):91–103. doi: 10.1016/j.etp.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 82.Heatwole VM. TUNEL assay for apoptotic cells. Methods Mol Biol. 1999;115:141–8. doi: 10.1385/1-59259-213-9:141. [DOI] [PubMed] [Google Scholar]
- 83.Aitken RJ, De Iuliis GN. Value of DNA integrity assays for fertility evaluation. Soc Reprod Fertil Suppl. 2007;65:81–92. [PubMed] [Google Scholar]
- 84.Albini A, Iwamoto Y, Kleinman HK, Martin GR, Aaronson SA, Kozlowski JM, et al. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 1987 Jun 15;47(12):3239–45. [PubMed] [Google Scholar]
- 85.Lee J, Cuddihy MJ, Kotov NA. Three-dimensional cell culture matrices: state of the art. Tissue Eng Part B Rev. 2008 Mar;14(1):61–86. doi: 10.1089/teb.2007.0150. [DOI] [PubMed] [Google Scholar]
- 86.Staton CA, Stribbling SM, Tazzyman S, Hughes R, Brown NJ, Lewis CE. Current methods for assaying angiogenesis in vitro and in vivo. Int J Exp Pathol. 2004 Oct;85(5):233–48. doi: 10.1111/j.0959-9673.2004.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Caldwell JS. Cancer cell-based genomic and small molecule screens. Adv Cancer Res. 2007;96:145–73. doi: 10.1016/S0065-230X(06)96006-0. [DOI] [PubMed] [Google Scholar]
- 88.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005 Sep;5(9):689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 89.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997 Nov 7;278(5340):1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Han H, Von Hoff DD. Identification of an Agent Selectively Targeting DPC4 (Deleted in Pancreatic Cancer Locus 4)-Deficient Pancreatic Cancer Cells. Cancer Res. 2006 Oct 1;66(19):9722–30. doi: 10.1158/0008-5472.CAN-05-4602. [DOI] [PubMed] [Google Scholar]
- 91.Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005 Jun 15;65(12):4975–8. doi: 10.1158/0008-5472.CAN-05-0481. [DOI] [PubMed] [Google Scholar]
- 92.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008 Mar 18;105(11):4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hansel DE, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Annu Rev Genomics Hum Genet. 2003;4:237–56. doi: 10.1146/annurev.genom.4.070802.110341. [DOI] [PubMed] [Google Scholar]
- 94.Yamano M, Fujii H, Takagaki T, Kadowaki N, Watanabe H, Shirai T. Genetic progression and divergence in pancreatic carcinoma. Am J Pathol. 2000 Jun;156(6):2123–33. doi: 10.1016/S0002-9440(10)65083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002 Nov;161(5):1541–7. doi: 10.1016/S0002-9440(10)64432-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luttges J, Galehdari H, Brocker V, Schwarte-Waldhoff I, Henne-Bruns D, Kloppel G, et al. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol. 2001 May;158(5):1677–83. doi: 10.1016/S0002-9440(10)64123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007 Nov 16;318(5853):1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 98.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003 Dec;4(6):437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 99.Bardeesy N, Sharpless NE. RAS unplugged: negative feedback and oncogene-induced senescence. Cancer Cell. 2006 Dec;10(6):451–3.31. doi: 10.1016/j.ccr.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 100.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005 May;7(5):469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 101.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006 Apr 11;103(15):5947–52. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003 Dec 15;17(24):3112–26. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008 Oct;57(10):1420–30. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006 Nov 15;20(22):3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002 Nov 14;347(20):1593–603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- 106.Renan MJ. How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog. 1993;7(3):139–46. doi: 10.1002/mc.2940070303. [DOI] [PubMed] [Google Scholar]
- 107.Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3- kinase signaling. Cancer Res. 2007 Mar 1;67(5):2098–106. doi: 10.1158/0008-5472.CAN-06-3752. [DOI] [PubMed] [Google Scholar]
- 108.Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun. 2003 Feb 21;301(4):1038–44. doi: 10.1016/s0006-291x(03)00086-x. [DOI] [PubMed] [Google Scholar]
- 109.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999 Jul 29;400(6743):464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 110.Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):14723–8. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg. 2007;14(3):224–32. doi: 10.1007/s00534-006-1166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feldmann G, Maitra A. Molecular pathology of precursor lesions of pancreatic cancer: pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN) In: Neoptolemos J, Abbruzzese J, Buchler M, Urrutia R, editors. Handbook of Pancreatic Cancer. New York: Springer; 2009. (in press) [Google Scholar]
- 113.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006 Oct 15;66(20):10171–8. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007 Mar;11(3):229–43. doi: 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 115.Siveke JT, Einwachter H, Sipos B, Lubeseder-Martellato C, Kloppel G, Schmid RM. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007 Sep;12(3):266–79. doi: 10.1016/j.ccr.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 116.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996 Jun 14;85(6):841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 117.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002 Aug 30;297(5586):1559–61. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 118.Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005 Jun 9;24(25):4026–36. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 119.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003 Oct 23;425(6960):846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 120.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003 Mar 20;422(6929):313–7. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 121.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006 Dec;5(12):1026–33. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 122.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003 Oct 23;425(6960):851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Binns W, James LF, Shupe JL, Everett G. A Congenital Cyclopian-Type Malformation in Lambs Induced by Maternal Ingestion of a Range Plant, Veratrum Californicum. Am J Vet Res. 1963 Nov;24:1164–75. [PubMed] [Google Scholar]
- 124.Keeler RF, Binns W. Teratogenic compounds of Veratrum californicum (Durand). V. Comparison of cyclopian effects of steroidal alkaloids from the plant and structurally related compounds from other sources. Teratology. 1968 Feb;1(1):5–10. doi: 10.1002/tera.1420010103. [DOI] [PubMed] [Google Scholar]
- 125.Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci U S A. 2003 Apr 15;100(8):4616–21. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taylor FR, Wen D, Garber EA, Carmillo AN, Baker DP, Arduini RM, et al. Enhanced potency of human Sonic hedgehog by hydrophobic modification. Biochemistry. 2001 Apr 10;40(14):4359–71. doi: 10.1021/bi002487u. [DOI] [PubMed] [Google Scholar]
- 127.Gabay L, Lowell S, Rubin LL, Anderson DJ. Deregulation of dorsoventral patterning by FGF confers trilineage differentiation capacity on CNS stem cells in vitro. Neuron. 2003 Oct 30;40(3):485–99. doi: 10.1016/s0896-6273(03)00637-8. [DOI] [PubMed] [Google Scholar]
- 128.Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000 Aug 31;406(6799):1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 129.Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998 May 29;273(22):14037–45. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- 130.Hotz HG, Reber HA, Hotz B, Sanghavi PC, Yu T, Foitzik T, et al. Angiogenesis inhibitor TNP-470 reduces human pancreatic cancer growth. J Gastrointest Surg. 2001 Mar–Apr;5(2):131–8. doi: 10.1016/s1091-255x(01)80024-x. [DOI] [PubMed] [Google Scholar]
- 131.Kawarada Y, Ishikura H, Kishimoto T, Saito K, Takahashi T, Kato H, et al. Inhibitory effects of the antiangiogenic agent TNP-470 on establishment and growth of hematogenous metastasis of human pancreatic carcinoma in SCID beige mice in vivo. Pancreas. 1997 Oct;15(3):251–7. doi: 10.1097/00006676-199710000-00006. [DOI] [PubMed] [Google Scholar]
- 132.Kato H, Ishikura H, Kawarada Y, Furuya M, Kondo S, Yoshiki T. Anti-angiogenic treatment for peritoneal dissemination of pancreas adenocarcinoma: a study using TNP-470. Jpn J Cancer Res. 2001 Jan;92(1):67–73. doi: 10.1111/j.1349-7006.2001.tb01049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Solorzano CC, Baker CH, Bruns CJ, Killion JJ, Ellis LM, Wood J, et al. Inhibition of growth and metastasis of human pancreatic cancer growing in nude mice by PTK 787/ZK222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases. Cancer Biother Radiopharm. 2001 Oct;16(5):359–70. doi: 10.1089/108497801753354267. [DOI] [PubMed] [Google Scholar]
- 134.Bruns CJ, Shrader M, Harbison MT, Portera C, Solorzano CC, Jauch KW, et al. Effect of the vascular endothelial growth factor receptor-2 antibody DC101 plus gemcitabine on growth, metastasis and angiogenesis of human pancreatic cancer growing orthotopically in nude mice. Int J Cancer. 2002 Nov 10;102(2):101–8. doi: 10.1002/ijc.10681. [DOI] [PubMed] [Google Scholar]
- 135.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971 Nov 18;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 136.Burris H, 3rd, Rocha-Lima C. New therapeutic directions for advanced pancreatic cancer: targeting the epidermal growth factor and vascular endothelial growth factor pathways. The oncologist. 2008 Mar;13(3):289–98. doi: 10.1634/theoncologist.2007-0134. [DOI] [PubMed] [Google Scholar]
- 137.van Es JH, Clevers H. Notch and Wnt inhibitors as potential new drugs for intestinal neoplastic disease. Trends Mol Med. 2005 Nov;11(11):496–502. doi: 10.1016/j.molmed.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 138.Kopan R, Goate A. A common enzyme connects notch signaling and Alzheimer’s disease. Genes Dev. 2000 Nov 15;14(22):2799–806. doi: 10.1101/gad.836900. [DOI] [PubMed] [Google Scholar]
- 139.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gammasecretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005 Jun 16;435(7044):959–63. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 140.Mullendore M, Koorstra JB, Li Y, Offerhaus GJ, Fan X, Henderson C, et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res. 2009 doi: 10.1158/1078-0432.CCR-08-2004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dihlmann S, von Knebel Doeberitz M. Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int J Cancer. 2005 Feb 10;113(4):515–24. doi: 10.1002/ijc.20609. [DOI] [PubMed] [Google Scholar]
- 142.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, et al. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004 Jan;5(1):91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 143.Lahn M, Kloeker S, Berry BS. TGF-beta inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2005 Jun;14(6):629–43. doi: 10.1517/13543784.14.6.629. [DOI] [PubMed] [Google Scholar]
- 144.Jimeno A, Tan AC, Coffa J, Rajeshkumar NV, Kulesza P, Rubio-Viqueira B, et al. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 2008 Apr 15;68(8):2841–9. doi: 10.1158/0008-5472.CAN-07-5200. [DOI] [PubMed] [Google Scholar]
- 145.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007 May 20;25(15):1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 146.Laurent-Puig P, Taieb J. Lessons from Tarceva in pancreatic cancer: where are we now, and how should future trials be designed in pancreatic cancer? Curr Opin Oncol. 2008 Jul;20(4):454–8. doi: 10.1097/CCO.0b013e32830218d6. [DOI] [PubMed] [Google Scholar]
- 147.Ng R, Chen EX. Sorafenib (BAY 43–9006): review of clinical development. Curr Clin Pharmacol. 2006 Sep;1(3):223–8. doi: 10.2174/157488406778249325. [DOI] [PubMed] [Google Scholar]
- 148.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008 Oct;7(10):3129–40. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cheng GZ, Park S, Shu S, He L, Kong W, Zhang W, et al. Advances of AKT pathway in human oncogenesis and as a target for anti-cancer drug discovery. Curr Cancer Drug Targets. 2008 Feb;8(1):2–6. [PubMed] [Google Scholar]
- 150.Mita M, Sankhala K, Abdel-Karim I, Mita A, Giles F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Investig Drugs. 2008 Dec;17(12):1947–54. doi: 10.1517/13543780802556485. [DOI] [PubMed] [Google Scholar]
- 151.Rini BI. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin Cancer Res. 2008 Mar 1;14(5):1286–90. doi: 10.1158/1078-0432.CCR-07-4719. [DOI] [PubMed] [Google Scholar]
- 152.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The rapamycin analog CCI-779 is a potent inhibitor of pancreatic cancer cell proliferation. Biochem Biophys Res Commun. 2005 May 27;331(1):295–302. doi: 10.1016/j.bbrc.2005.03.166. [DOI] [PubMed] [Google Scholar]
- 153.Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR Inhibitor Everolimus in Patients With Gemcitabine-Refractory Metastatic Pancreatic Cancer. J Clin Oncol. 2009 Jan 10;27(2):193–8. doi: 10.1200/JCO.2008.18.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]