

Abstract
That regulatory T cells (Tregs) have a crucial role in controlling allergic diseases such as asthma is now undisputed. The cytokines most commonly implicated in Treg-mediated suppression of allergic asthma are TGF-β and IL-10. In addition to naturally occurring Tregs, adaptive Tregs, induced in response to foreign antigens, have been demonstrated in recent studies. The concept of inducible/adaptive Tregs (iTregs) has considerable significance in preventing asthma if generated early enough in life. This is because cytokines such as IL-4 and IL-6 inhibit Foxp3 induction in naïve CD4+ T cells and therefore de novo generation of Tregs can be expected to be less efficient when it is concomitant with effector cell development in response to an allergen. However, if iTregs can be induced, the process of infectious tolerance would facilitate expansion of the iTreg pool as suggested in the recent literature. It is tempting to speculate that there is a window of opportunity in early life in the context of a relatively immature immune system that is permissive for the generation of iTregs specific to a spectrum of allergens that would regulate asthma lifelong. The focus of this review is the relevance of nTregs and iTregs in controlling asthma from early life into adulthood, the mechanisms underlying Treg function and the prospects for utilizing our current concepts to harness the full potential of Tregs to limit disease development and progression.
Keywords: Tregs, asthma, tolerance, TGF-β, IL-10
Various lines of evidence indicate that T helper cells (CD4+ T cells) and their secreted products play a central role in orchestrating the unique inflammatory response in the airways of asthmatics. Local allergen challenge has been shown to induce an influx of CD4+ T cells and eosinophils into the airways of asthmatics 1. In a mouse model, depletion of CD4+ T-cells with a monoclonal antibody to CD4 antigen reduced airway eosinophilia and eliminated airway hyperresponsiveness 2. T helper 2 type (Th2) cells are believed to augment the inflammatory response observed in asthma by expressing multiple cytokines such as IL-4, IL-5, IL-9 and IL-13, each of which could potentially augment the eosinophilic inflammation in asthma 3–7. In addition to eosinophils, other cell types are also involved in asthma pathogenesis as reviewed recently 8. For example, neutrophilic asthma is also well recognized and is associated with severe airflow obstruction, which is difficult to control by corticosteroids 9. A more recently identified T helper subset, Th17, has been implicated in neutrophil-dominated asthma 10–12 and was shown to be responsible for steroid resistance in a murine model of asthma 13. The effector function of all T helper cells, Th1, Th2 and Th17, is regulated by Tregs. This ensures protection from autoimmune diseases such as diabetes and multiple sclerosis as well as allergic diseases such as eczema and asthma. Here, we discuss recent findings that suggest an important role for Tregs in the induction of tolerance to environmental allergens and in limiting disease when allergic airway inflammation is induced. Also discussed are current concepts about development of Tregs and Treg-expressed mediators that have relevance in controlling asthma and how this knowledge might be exploited to enhance Treg function. Table 1 enlists Treg-associated nomenclature commonly used in the literature to describe different types of Tregs and their regulatory role in controlling inflammation in the lung or the gut.
Table 1.
Treg Nomenclature and Relevance in Immune Regulation in the Lung and the Gut
| Types of Tregs | Phenotype and Original Identification | Mucosal relevance(References) |
|
|---|---|---|---|
| Lung | Gut | ||
| Thymically derived nTregs | CD4+CD25hi Foxp3+ (mice)14 | 16–20 | |
| CD4+CD25hi Foxp3+CD49d− (human)15 | 21–24 | 25 | |
Peripherally induced Adaptive Tregs (iTregs)
|
Peripherally induced from CD4+CD25− cells. The regulatory T cells could be Foxp3+ or Foxp3− | ||
| CD4+CD25hi Foxp3+ (mice)26 | 26–30 | ||
| IL-10-expressing CD4+CD25hi Foxp3−31 | 32–39 | 31,40–42 | |
| TGF-β-expressing CD4+ T cells 43 | Have yet to be associated with immune suppression in the lung or the gut | ||
| ExFoxp3 | CD4+CD25hi Foxp3+ Treg cells that have spontaneously lost Foxp3 expression and secrete pro-inflammatory cytokines 44,45 | 45 | |
From birth to adulthood Tregs make a difference
The phenomenon of tolerance was introduced by Owen in 1945 in his seminal observations of “mosaicism” (red cell chimerism) in adult cattle dizygotic twins 46. A few years later in 1953, acquisition of tolerance to foreign allograft antigens in utero was demonstrated in mice by Billingham, Brent and Medawar 47. Some 50 years later, the concept of acquired tolerance to foreign antigens is highly significant not only in the context of transplantation tolerance but also in the realm of allergic and autoimmune diseases. Enmeshed in the concept of acquired immune tolerance is the fabric of the “hygiene hypothesis” which was postulated to explain the increased prevalence of allergic diseases in developed countries in recent years. The basic tenet of this hypothesis is that early childhood exposures to pathogen-associated products inversely correlates with the incidence of allergic diseases in adulthood. Initially postulated in 198948, the underlying mechanism associated with this hypothesis was Th1/Th2 cross-regulation. However, after epidemiological studies showed that Th2-inducing parasitic infections could protect from atopic diseases which are also engendered by Th2 cells, it became clear that additional mechanisms needed to be invoked to explain the protective effect of microbial exposures 49,50. The need for a more general immunoregulatory mechanism was realized when the same beneficial effect of microbes on Th1-induced diseases such as autoimmune diseases was also evident. With therapid progress of research in the field of regulatory T cells (Tregs) in recent years, it appears that microbial infections are conducive to the development of these cells 51,52 which are then able to exercise their immunosuppressive functions to dampen unwarranted immune responses against foreign or self-antigens.
There is significant interest in understanding the development of the immune system from the fetus into adulthood in order to help define the triggers and brakes in the etiology of allergic diseases. It is now well recognized that microbes can induce the production of the suppressive cytokines TGF-β and IL-10 49,51,53–55. Of note, IL-10 knockout mice develop colitis and a recent study has shown that it is the secretion of this cytokine from myeloid-derived cells stimulated by microbiota that prevents the induction of colitis 40,41. Similarly, mice bearing a dominant negative mutation in the TGF-β receptor on CD4 T cells exhibit profound inflammation in various organs including the lungs, liver and the pancreatic islets suggesting impaired development of regulatory T cells 56.
Recent studies in humans and mice show the importance of Tregs in the promotion of tolerance to foreign antigens transmitted via the placenta to the fetus 57. The mother during pregnancy is exposed to several antigens from the environment and these antigens termed “non-inherited maternal antigens” constantly traverse the placenta and have the capacity to evoke an immune response. How is the mounting of immune response to these foreign antigens stumped in the developing fetus? A study in humans showed that maternal cells traffic in large numbers to fetal lymph nodes to induce CD4+CD25+Foxp3+ cells with suppressive functions that prevent antimaternal immunity lasting until early adulthood. This study also challenges the preconceived notion that fetal lymphocytes are incapable of mounting an immune response 58 (Figure 1). In fact the data from this study show that fetal lymphocytes secrete high levels of cytokines in response to non-inherited maternal antigens(NIMA) when Tregs have been selectively depleted. In a study in mice in which the protocol of aerosolized OVA was used to induce airway tolerance in mothers, transfer of OVA and TGF-β via breast milk into suckling neonates induced suppressive CD4+ T cells whose generation depended on a functional TGF-β receptor 59 (Figure 1). Similarly, when pregnant female mice were tolerized to antigen, the offspring were also tolerized to the same antigen, which was sustained only when the infants were nursed by the tolerized mothers 60. These studies suggest that foreign antigens can be transferred across the placentaor via breast milk to fetuses or infants respectively that can induce Foxp3-expressing Tregs in the presence of TGF-β. The findings not only underscore the importance of Tregs in tempering immune responses in the developing fetus but they also highlight a window of opportunity for the development of novel therapeutics in early life to ward off disease 61,62. Thus, it may not be too fanciful to propose administration of minute doses of a cocktail of allergens to expectant mothers or neonates to develop antigen-specific Tregs that would provide life-long protection from developing allergic diseases such as asthma (Figure 1).
Figure 1.
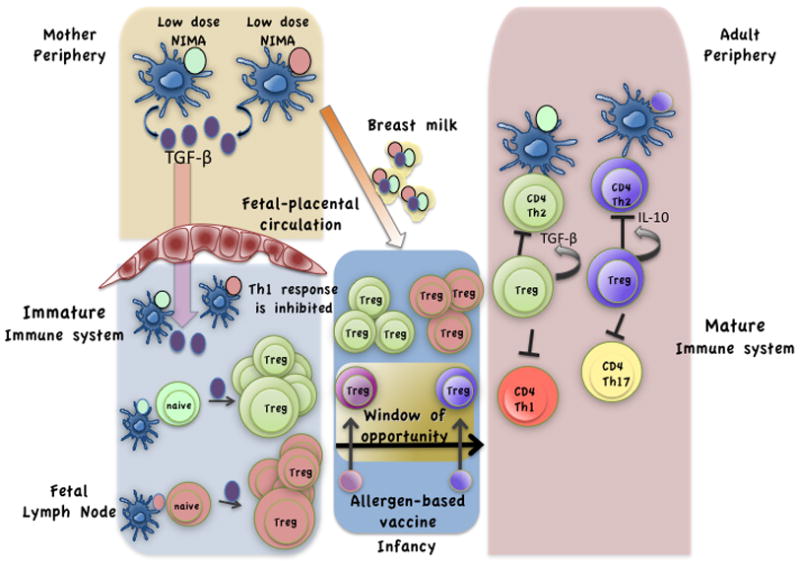
Induced Tregs specific to allergens in early life may be the elixir of asthma-free life. As shown recently 58, the fetus is constantly exposed to non-inherited maternal antigens (NIMA) which traverse the placenta and trigger the development of Tregs in the fetal lymph nodes. Tregs constitute a high percentage of total lymphocytes in the fetal lymph nodes. This pool of Tregs aids in tempering fetal immune responses against maternal antigens. During or after pregnancy, transfer of antigen and TGF-β across the placenta or via breast milk induces antigen-specific Tregs 59,60. The immune system’s ability to induce antigen-specific Tregs in early life suggests a unique window of opportunity to administer allergen based vaccines to infants for the induction of protective allergen-specific Tregs.
Which mechanism is relevant-Foxp3+ Tregs or IL-10-expressing Tr1-like cells?
It is clear that individuals with defective or suboptimal Foxp3 expression due to mutations in the Foxp3 gene (IPEX syndrome) or in genes that promote Foxp3 expression such as STAT5b are susceptible to allergic disease 23. This section will review studies in both humans and rodents that suggest the importance of both Foxp3-expressing Tregs and IL-10-the source of the latter not necessarily being Foxp3-expressing cells, in regulating allergic asthma. CD4+ T cells that do not express Foxp3 but secrete IL-10 and suppress effector functions of T helper cells are known as Tr1 cells 31.
In humans, along with typical disease manifestations such as elevated IgE levels, eczema, and insulin-dependent diabetes mellitus (IDDM), intestinal inflammation, asthma and other recurrent respiratory disorders have also been associated with IPEX 21,24,63. Although eczema is a common feature of IPEX-deficiency, there is less documentation of asthma in IPEX patients. The most likely reason for this is that there is inherently significant uncertainty in the diagnosis of asthma in young children. While wheezing is a phenotype of asthma, wheezing is not a definitive diagnosis of this disease in young children. A vast majority of infants and children less than 6 years of age show wheezing during recurrent respiratory infections, but the children outgrow the phenotype with age 64. Due to the difficulty in diagnosing asthma in early life, asthma is not as commonly associated with IPEX as is eczema, which manifests in infants and is easily diagnosed. For example, in one study of 14 patients, all of whom were diagnosed early with severe disease symptoms including eczema, only one was diagnosed with asthma. This patient underwent spontaneous remission of early diarrhea and all traces of autoimmunity at age 4, but was left with mild eczema, relatively high serum IgE levels and occasional upper respiratory tract infections 65. Whether asthma also persisted is not documented in the report. This patient had two mutations in Foxp3− one a silent mutation at the intron 4/exon 5 boundary that presumably influences RNA splicing and another non-conservative mutation in the FKH domain. It is possible that these mutations allowed some level of expression of functional Foxp3 in later life that was adequate to suppress autoimmunity but not allergic disease. In another study, CD25 deficiency was associated with reduced IL-10 expression and the patient developed symptoms indistinguishable from those observed in IPEX patients with the patient progressing to develop asthma 42. Since it is now well recognized that IL-2 is required to maintain Foxp3+ Tregs in the periphery 66, it is not surprising that this patient showed an IPEX-like phenotype. Most importantly, the development of asthma in the patient suggests an important regulatory role for Foxp3+ Tregs in asthma. In another study, maintenance of tolerance to beestings in beekeepers by seasonal exposure to bee stings was found to be associated with increased bee venom antigen-specific IL-10-producing CD4+ Tr1-like cells 34,37. So, what are the sources of IL-10 and TGF-β and their contribution in suppression of allergic asthma when expressed from a Foxp3-expressing cell or a Foxp3− cell? Interestingly, two types of nTregs that develop in the thymus have been recently described-ICOS− and ICOS+ that use TGF-β and IL-10 respectively for their suppressive functions 16.
A review of animal studies on the role of Tregs in regulating allergic asthma reveals an interesting difference in the involvement of IL-10 versus Foxp3-expressing Tregs depending on whether or not the animals were sensitized through the airways. In multiple studies that used the intraperitoneal (i.p.) mode of antigen sensitization, where non-airway DCs (such as splenic DCs) would be involved in priming naïve T helper cells, a dependence on IL-10 for suppression of effector T cell responses and airway pathology was noted. An exception to this general trend is the results of one study that used intranasal delivery of adjuvant-free OVA where an involvement of IL-10 in suppressing allergic airway inflammation was noted32 that involved the ICOS-ICOS ligand pathway 33. There does not appear to be an agreement in the different studies with respect to the source of IL-10. In one study, the source was found to be not the Foxp3-expressing cells but actually bystander CD4+ T cells 67. In contrast, a different study showed that deletion of IL-10 in Foxp3-expressing CD4+ T cells promoted allergic airways disease 30. In a recent study, where peptides derived from the cat allergen Fel d 1 were delivered intradermally to promote immunotherapy in mice previously immunized by the i.p. route, IL-10-producing cells but not TGF-β-expressing Foxp3+ Tregs were found to mediate immune suppression 36. In our own studies of tolerance induction via repeated exposure to a low dose of aerosolized antigen, a protocol initially established by Holt and colleagues 68, an involvement of Foxp3+ Tregs expressing membrane-bound TGF-β in mediating resistance to development of antigen-induced airway inflammation was identified 27,69. In another study of oral tolerance, Foxp3+Tregs were shown to inhibit allergic airway inflammation that was dependent on TGF-β and not IL-10 26. Similarly, when animals were immunized by intratracheal delivery of the allergen house dust mite, depletion of CD25hi population of cells induced airway hyperresponsiveness in a normally resistant mouse strain C3H 17. In this study, no involvement of IL-10 was apparent 17. Thus, it is clear that no single mechanism of Treg-mediated suppression of allergic airways disease has emerged from multiple studies of experimental asthma reported to date.
Since in models where the antigen was delivered through the respiratory route, a dependence on IL-10 for suppression was not evident 17,26,69, which was the opposite when a non-airway mode was adopted 18,30,36,37,67, it appears that the cell types that are engaged as suppressive cells in the two settings of antigen exposure are distinct. When animals were repeatedly exposed to inhaled antigen without any adjuvant such that sufficient maturation of airway dendritic cells (DCs) would not occur, Foxp3+ Tregs expressing membrane-bound TGF-β were found to mediate inhibition of allergic inflammation in the airways 69. Conversely, conversion of a normally resistant mouse strain susceptible to development of an asthma phenotype by depletion of CD25hi cells was a direct effect of loss of attenuation of lung DC costimulatory functions by Tregs promoting unopposed inflammatory effects of T helper cells 17. If on the other hand, effector cells and their regulators were primarily induced at a distant site, such as the skin 36 or the spleen 18,67, IL-10 appeared to be an important negative regulator of pulmonary disease development as was also suggested in the study of bee keepers 34,37. The primary reason for this difference maybe that when Tregs are activated locally in the lung-draining lymph nodes, particularly in the presence of a low dose of antigen, IL-10 gene expression is not favored either from the Tregs or from bystander cells in the tissue. If, however, IL-10 gene expression is induced when priming occurs elsewhere, the source of the cytokine maybe the Foxp3-expressing cell 30 or a bystander cell 67. It seems likely that IL-10 production is determined by the dose and TCR affinity of the antigen, the adjuvant used or other properties for a complex allergen such as ligands for pattern recognition receptors. A topic for future exploration is differential distribution of Foxp3+Tregs that use TGF-β versus IL-10 in the periphery.
Another possible explanation for the involvement of Foxp3+Tregs expressing membrane-bound TGF-β rather than IL-10 in mediating tolerance in the airways is the relative enrichment of Foxp3+Tregs in mucosal tissues and in lymph nodes draining the lung and the gut. Although a proportion of these Tregs may be induced de novo, polyclonally expanded locally residing ICOS− nTregs may be also involved in disease suppression (discussed in the following section). However, when Tregs are induced post-immunization with antigen and adjuvant delivered via the skin or spleen, IL-10 and not TGF-β produced by Foxp3+Tregs (probably ICOS−)16 or Foxp3− Tr1-like cells (probably ICOS+)16 limit the severity of inflammation. It is clear, however, that Foxp3 deficiency promotes allergic disease 21,24. Therefore, regardless of whether or not IL-10 or TGF-β is the mediator of suppression of asthma or other allergic diseases such as eczema, Foxp3-expressing Tregs are important negative regulators of allergic disease. It will be useful to determine in future studies what conditions induce IL-10 production from a Foxp3-expressing cell even when it is not innately programmed to express IL-10 16. If such conditions could be established, in situations where Foxp3 is mutated or its expression is suboptimal, as observed in IPEX patients, IL-10 could potentially substitute for Foxp3-dependent suppressive mechanisms. Since eczema/atopic dermatitis develops early in life in most IPEX patients, such strategies may need to be directed to the skin, which may also protect the lung (peptide immunotherapy).
Role of Thymus-derived Tregs (nTregs) versus peripherally induced Tregs (iTregs) in regulating allergic asthma
The generation of Foxp3+ Tregs in vitro by activation of CD4+ T cells in the presence of TGF-β showed that acquisition of Foxp3 is possible in a non-thymic environment 70. While the identification of CD4+CD25+Foxp3+ T cells after adoptive transfer of naive CD4+ T cells into recipient TCR transgenic mice devoid of nTregs also suggested similar de novo generation in the periphery, the conversion of CD25−Foxp3+ cells into CD25+Foxp3+ cells could not be ruled out in these experiments since the naïve cells were not depleted of Foxp3+ cells prior to adoptive transfer 71. Close on the heels of these findings, it was more definitively shown using genetic approaches that naïve CD4+ T cells in the periphery can indeed be induced to express Foxp3 under appropriate conditions and that the cells have suppressive function similar to nTregs 26,72–74. Using OVA-specific TCR transgenic mice crossed to scurfy mice that harbor a mutated Foxp3 gene, it was shown that a protocol of oral tolerance induced by supplying OVA in the drinking water of mice induced Tregs in the periphery that were important to control allergic sensitization 28. The findings in this study suggest that the induction of airway tolerance by delivery of aerosolized antigen68,69 most likely also involves iTregs, although the involvement of nTregs activated in a bystander fashion cannot be ruled out.
Induction of tolerance by delivery of antigen via the airways 27,69 or the oral route 26,28 almost completely thwarts the development of effector T cells if animals are subsequently immunized with antigen and adjuvant. Using TCR transgenic mice, it was shown that iTregs are also induced along with effector T cells post-sensitization and antigen challenge and these Tregs limit the severity of allergic airway inflammation and also minimize its dissemination to extrapulmonary locations 28. The question that arises from the results of this study is whether the iTregs were induced during the challenge phase and not during priming in the inflammation model. This is because the presence of IL-4 and IL-6 during priming in the spleen should have inhibited Foxp3 induction. However, during challenge by inhaled antigen without adjuvant, low cytokine levels would have been conducive to iTreg generation in the lung-draining lymph nodes. In humans and in specific animal models, where sensitization and challenge both occur by inhalation of allergen, iTreg generation may be compromised by the inhibitory effects of IL-4 and IL-6. However, these same cytokines would permit polyclonal proliferation of nTregs as discussed below.
It has been suggested that TCR specificity guides peripheral Treg development that is also influenced by the location 75. Thus, the contribution of iTregs induced by allergens versus nTregs in the regulation of allergic asthma would be dictated both by the allergen and the local microenvironment. In this regard, a possibility that can be entertained is that iTregs that regulate allergic airway inflammation are peripherally induced at sites other than the lung to be subsequently recruited by antigen-induced inflammation to the lung. A likely mucosal site for this is the gastrointestinal tract where Tregs are required to maintain intestinal homeostasis by upholding tolerance to food antigens and gut microflora, the absence of which could lead to experimental colitis 76–79. This speculation is based on knowledge derived from various studies that show intestine and associated lymphoid tissue as a site for proliferation79 and induction of Foxp3+Tregs from naïve precursors 53,80 and the observations that changes in gut microflora81 and probiotic treatments influence airway hyperresponsiveness and allergies82–84. When viewed in combination, it seems likely that the gut serves as a “hub” or “depot” for inducing Tregs, which then home to various sites of infection/inflammation to exert suppressive activity. The repertoire of these induced Tregs would be mainly governed by the nature of the intestinal microflora, subtle changes in which could lead to susceptibility or resistance to a particular antigen locally or more distally in the lung. Needless to say, interplay between genetics and the immune mechanisms would drive the net outcome in every individual. So, how do iTregs or nTregs elicit immune suppression?
The process of infectious tolerance was recently shown to involve Foxp3+Tregs expressing membrane-bound TGF-β coupled to latency-associated peptide 85. These membrane-bound TGF-β-LAP expressing Tregs induced Foxp3 expression in naïve Foxp3− CD4+ T cells when stimulated by plate-bound anti CD3 which in turn acquired suppressive function 85. Given that Tregs have been shown to inhibit the immunogenic potential of pulmonary DCs by downregulating expression of MHC class II and costimulatory molecules 17,18, an expanded population of Tregs expressing TGF-β may also serve to downregulate DC function both in draining lymph nodes and in the tissue. This raises the question of the involvement of antigen-specific Tregs in regulating allergic asthma. It is difficult to imagine that the large pool of Tregs that infiltrate the airways during allergic inflammation comprise only antigen-specific iTregs. Even if the process of infectious tolerance is operative in the induction and activation of many of these Tregs, a substantial fraction the Tregs likely consists of polyclonal nTregs. Thus, in the first study that generated Tregs in vitro from naïve CD4+ T cells in the presence of TGF-β, the adoptive transfer of a polyclonal population of Tregs into mice was highly efficient in suppressing house dust mite-induced allergic airway inflammation 70. Another recent study of experimental asthma also suggests that suppression of allergic inflammation in the airways involves locally activated polyclonal Tregs 19. Activation of Tregs is important for efficient suppression since freshly isolated Tregs from naïve mice have weak suppressive function unless activated to boost their potency 86. Taken together, it seems reasonable to conclude that the most efficient Treg-mediated suppression of allergic asthma can be achieved by establishment of mucosal tolerance to a range of allergens in early life. This would involve induction of antigen-specific Tregs enhanced by the process of infectious tolerance that would last lifelong to limit generation of allergen-specific effector T cells. However, the potential to induce such Tregs would be less post antigen-sensitization due to cytokine inhibition.
Any discussion of efficiency of iTreg generation should also consider context. For example, in one study, when CD4+ Foxp3− T cells were adoptively transferred into transgenic mice with TCR specificity for myelin basic protein (MBP) and the mice were subsequently immunized with MOG peptide to induce autoimmune encephalomyelitis (EAE), de novo conversion of Foxp3− to Foxp3+ cells was minimal in the draining lymph nodes 87. Since this model is a driver of intense Th17 differentiation, it illustrates the point that specific pro-inflammatory cytokines produced in the lymph nodes exercise a strong influence on iTreg development. Somewhat conterintuitively, these same cytokines that block iTreg development promote the proliferation of nTregs and help maintain their Foxp3 expression. This would explain why in our study as well as in the EAE and multiple other studies in humans and animals, Tregs can be found in large numbers at the site of inflammation. As can be concluded from the study of Lafaille and colleagues 28, the Tregs that accumulate at the site of inflammation help to temper the inflammation as well as to contain it. Lastly, an important question that needs to be addressed in future studies is whether the efficiency of iTreg development is dissimilar in different lymph nodes.
As discussed previously, there maybe a window of opportunity in early life to develop lifelong tolerance to harmful antigens such as allergens via development of antigen-specific Tregs before sufficient maturation of the immune system and generation of effector T cells has taken place (Figure 1)61,62. Interestingly, a recent study of colorectal cancer patients has shown that Tregs specific to only a limited set of tumor-associated antigens control antitumor effector responses to the same set of antigens 25. Thus, knowledge of antigen specificity of Tregs in disease would undoubtedly be immensely helpful to promote anti-tumor responses or to suppress effector responses in allergic and autoimmune diseases.
Blockade of Treg development by Pro-inflammatory Cytokines and ExFoxp3 Tregs
It is clear that both Th2 and Tregs coexist at the site of allergic inflammation 28,88. Therefore, a logical question that arises is what effects the pro-inflammatory cytokines secreted by the Th2 cells exert on Tregs. Here we focus on 2 cytokines IL-6 and IL-4, both of which promote Th2 but block Treg development. IL-6 also promotes Th17 development in combination with TGF-β 89–92. The IL-6 receptor exists in both membrane-bound and soluble forms (sIL-6R) and the complex of IL-6 with its soluble receptor upon interaction with gp130 expressed on cells induces downstream signaling in a process called IL-6 trans-signaling 93. sIL-6R is found in large quantities in chronic inflammatory diseases. The IL-6/sIL-6R complex has been associated with inhibition of development of CD4+CD25+Foxp3+ cells and also impairment of their suppressive function in models of asthma and inflammatory bowel disease 94,95. In T cells, loss of STAT3, which is activated by IL-6, upregulated Foxp3 expression 96. Thus, targeting IL-6 signaling should improve Treg-mediated suppression in asthma. IL-4-induced signaling in T cells promotes Th2 differentiation but inhibits Treg development. STAT6 is activated by IL-4 in naïve CD4+ T cells and upregulates GATA-3 expression, which is the master regulator of Th2 cell differentiation 97,98. Both STAT6 99 and GATA-3 100 can bind to the Foxp3 promoter to inhibit Foxp3 gene transcription. Together, the cytokines IL-4 and IL-6 would not only drive T helper cell differentiation but would also block iTreg development.
As discussed above, it can be assumed that a large fraction of Tregs that accumulate in the airways during allergic inflammation are nTregs that are induced to proliferate and express CCR4 in the lung-draining lymph nodes resulting in their recruitment to the site of inflammation 20. An additional level of complexity recently identified by us is that chronic STAT6 signaling in the effector cells imparts resistance to suppression by Tregs 88. Thus, although IL-4 promotes nTreg proliferation and maintains Foxp3 expression, Tregs are unable to completely block Th2 effector function in situ. The opposing actions of IL-4 and IL-6 on T helper versus Treg development illustrate an important role of cytokines in regulating the dynamics of inflammation. At the same time a cytokine would allow T helper cell differentiation and induce resistance to Treg-mediated suppression, it would inhibit iTreg development and infectious tolerance. However, under these conditions, nTregs would actually be allowed to proliferate and traffic to the tissue since IL-4 is a proliferative factor for already developed Tregs 88,101. This would ensure that when the effector response eventually contracts, the Tregs are poised to take over and restore homeostasis. In a similar fashion, in sarcoidosis, a granulomatous disease of the lung driven by Th1 cells, spontaneous IL-2 secretion by lung effector T cells has been implicated in Treg expansion around the granulomas102 which was also noted in the study of EAE 87. The competing effects of IL-4 and IL-2 on Tregs versus T effector function likely determine the magnitude of the induced inflammation. Accumulation of a large pool of Tregs in the inflamed lung may also be undesirable in light of the recent unexpected finding that Tregs are susceptible to loss of Foxp3 at inflamed sites 44,45 and that along with Foxp3 they coexpress transcription factors induced in effector T cells 103–105. Whether these factors promote the stability of Tregs will be interesting to determine in the future.
Recently, some unexpected features of Tregs have been identified. Tregs in inflamed tissues were shown to lose Foxp3 (resulting cells termed exFoxp3) and secrete pro-inflammatory cytokines 44,45. Second, Tregs were shown to express transcriptipon factors characteristic of T helper cells such as T-bet and IRF4 104,105. IRF4 promotes IL-4 gene expression in Th2 cells and T-bet is a Th1-specific transcription factor. It has been proposed that expression of specific effector T cell transcription factors in Tregs helps limit the function of the effector T cell in which the same factor is expressed 103–105. The mechanism by which this is achieved is unclear. If this dual expressing Treg then loses Foxp3 but retains the other transcription factor such as IRF4, it may cause expression of pro-inflammatory cytokines from that cell as was observed for exFoxp3 cells recovered from different tissues of diabetic animals 44. It is, however, unclear whether such a cell would indeed be capable of producing all of the Th2 cytokines since IRF4 alone is not enough for the expression of Th2 cytokine genes. Given that both STAT6 and GATA-3, which are expressed specifically in Th2 cells, inhibit Foxp3 expression 99,100, it is unlikely that a Foxp3+ Treg would be replete with the full spectrum of Th2 factors. Tregs can coexpress T-bet and Foxp3 whereas RORγt and Foxp3 have a reciprocal relationship 106–108. Therefore, loss of Foxp3 in Tregs in a model of diabetes might promote secretion of IFN-γ or IL-17A. This, however, does not necessarily imply that Foxp3-deficient Tregs would behave as Th2 cells. Whether these would instead develop into Th17 cells in the context of allergic inflammation is an interesting possibility given that IRF4 is also important for the Th17 phenotype and loss of Foxp3 may promote RORγt expression. This is of significant concern since Tregs express CCR4, which is also expressed by Th2 cells 109, and thus the Tregs with lost Foxp3 would still coexist with Th2 cells at the site of inflammation. These exFoxp3 cells may secrete IL-17A to promote neutrophilic asthma, which is a characteristic of severe asthma. In fact, it may not be too far fetched to consider the possibility that retention of the exFoxp3 cell by virtue of CCR4 expression in the lung, and stimulation by self antigen-derived peptides resulting in IL-17A secretion, is one of the recipes for the development of non-atopic/intrinsic asthma.
Mechanisms by which Tregs inhibit effector T cell function in asthma
As discussed above, the two molecules that have received the most attention with respect to Treg-mediated suppression of allergic airway inflammation are TGF-β and IL-10. Our investigations showed that membrane-bound TGF-β expressed by Tregs in mice tolerized by inhaled antigen69 activates Notch inducing expression of its downstream target gene Hes1 (hairy and enhancer of split 1) in naïve CD4+ T cells 27. Hes1 is a potent repressor of gene expression 110,111. Simultaneous engagement of TCR, CD28 and Notch was shown to inhibit T cell activation, which was associated with upregulation of Hes1 expression 112. Given that membrane-bound TGF-β, originally identified on activated Tregs 113, has now been implicated in infectious tolerance 85, Hes1 may have an important role in the induction and/or stabilization of Foxp3 expression in the CD4+ T cell destined to become an iTreg. Indeed, Notch activation in Tregs was recently shown to promote Foxp3 expression and stabilize Tregs in vivo 114. Figure 2 merges all of these concepts in the context of mucosal tolerance. iTreg development is at a disadvantage during allergen-induced T helper cell differentiation which promotes nTreg proliferation (Figure 2).
Figure 2.
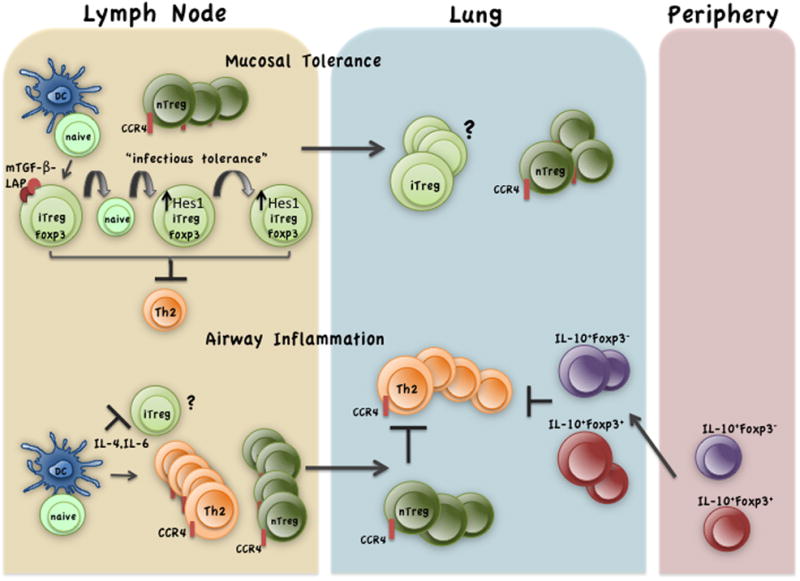
Mucosal tolerance favors iTreg development in lung-draining lymph nodes that have free reign in tolerance but compete with nTregs during inflammation. LAP associated membrane-bound (mTGF-β) on iTregs acts via infectious tolerance to increase its pool size 85. mTGF-β induces Notch activation and upregulation of the downstream repressor Hes1 in naïve CD4+ T cells 27. Hes1 has been shown to stabilize Foxp3 expression in Tregs 114. The iTregs efficiently suppress effector cell development (shown is Th2) in antigen-tolerized animals 26–28,69. In allergen-sensitized animals, the cytokines IL-4 and IL-6 released during priming events suppress Treg induction 94,99,100,224. However, the same cytokines promote nTreg proliferation while inducing Th2 differentiation88,94,224 (Th17 cells are also induced by allergens). The Th2 cells and nTregs traffic to the tissue in recall response to inhaled allergen where they downmodulate costimulatory molecules on DCs 17,18(not shown). If priming occurs at a distant site (skin, spleen), IL-10-expressing Tregs are favored18,30,36,67 which are recruited to the lung in response to allergen challenge to suppress effector T cell functions.
Although Notch activation has been also associated with T helper cell differentiation 115, it is likely that the strength of Notch ligation and duration of expression of its target genes such as Hes1 differs when T cells are activated versus when they are suppressed. IL-10 induces T cell anergy and it is believed to be due to inhibition of costimulation of T cells 116,117. IL-10 also has effects on Ig isotypes promoting IgG4 and inhibiting IgE switching 118. With regard to other mechanisms employed by Tregs in curbing asthma, a recent study showed that Treg-expressed OX40 upon ligation of OX40L on mast cells inhibited their degranulation 119. While indoleamine 2, 3 dioxygenase (IDO) has been associated with Treg-mediated suppression 120, our studies and that of others suggest an involvement of IDO in promotion of Th2 responses using the model allergen, OVA 121,122. In mice infected with Aspergillus fumigatus, a fungus associated with development of allergic bronchopulmonary aspergillosis (ABPA), a role for IDO in control of fungal burden, allergic response and Treg function late in infection was noted 123. More studies are needed in different animal models and in humans to elucidate the role of IDO in regulating effector responses in the airways.
With the demonstration of a specific role for Treg-expressed CTLA-4 in suppressive function via downregulation of CD80/CD86 on DCs 124, CTLA-4 may be a key player in inhibition of expression of costimulatory molecules by lung DCs in allergic airway inflammation 17,18. Another cytokine, IL-35, secreted by Tregs has been implicated in the suppressive effects of these cells 125. In a recent study, a complex of IL-2:anti-IL-2 monoclonal antibody was administered into mice sensitized via the i.p. route with Schistosome antigen or OVA/alum and subsequently challenged with antigen 39. The regimen of cytokine-antibody complex diminished allergic airways disease and required IL-10-producing Tregs 39. Interestingly, an increase in IL-35 production was observed in IL-10-competent but not deficient animals in this study 39. This observation was in line with previous findings that observed IL-10-promoting effects of IL-35. Table 2 is a summary of the literature that associated either Foxp3+ or Foxp3− Tregs with either induction of effector T cells or their function.
Table 2.
Treg-Mediated Suppression of Effector CD4+ T Cell Induction or Function in Allergic Disease
| Treg Type | Regulation of Effector T Cell | |
|---|---|---|
| Induction | Function | |
| Foxp3+ | 26–28,59,69 | 17–19,39,94,105,126 |
| Foxp3− | 32,33,37 | 30,36,67 |
microRNA control of Tregs
The role of microRNAs (miRNA) in Treg function in the periphery is not sufficiently understood at the present time. Treg-specific deletion of Dicer, the processing enzyme for precursors of miRNAs, had only minimal effect on Treg development, seeding, proliferation and survival in the periphery. However, the mice developed splenomegaly and lymphadenopathy in >5 week-old mice and died at 6–8 weeks of age 127,128. The role of a specific miRNA, miR-155, in Treg development and function was addressed by using miR-155−/− mice since it is expressed at higher levels in nTregs as compared to that in double positive thymocytes or CD4+ T cells. Although the frequency and number of Tregs in miR-155−/− mice were reduced by 2 to 3 fold in the thymus and spleen, their suppressive function was not impaired 129,130. miR-155-deficient mice show a bias towards Th2 development and produce more IL-10-producing CD4+ T cells 131,132. Thus, generation of IL-10-producing Tregs may involve downregulation of miR-155 expression.
Use of various treatments to expand Tregs in the context of asthma Instability of in vitro generated Tregs
All of the compelling evidence in the literature suggests that Tregs are ideal candidates for developing effective therapies to treat diseases such as asthma. We have already discussed about the possibility of developing allergen-specific Tregs in neonates for lifelong protection from atopic asthma (Figure 1). What about treating asthmatics who already have allergen-specific effector cells? One possible strategy that has been most studied is Treg cell transfer mediated immunotherapy where antigen specific Treg cells are expanded/induced in vitro and transferred back into the host. The major hurdles in expanding the existing antigen-specific cells from the repertoire were a) low frequency of antigen-specific Treg cells, especially in tissues, b) poor proliferation in vitro 133,134, and c) expansion of contaminating effector cells in the presence of IL-2134,135. Given that polyclonal Tregs have been also shown to exercise suppressive effects 70, a possibility exists in using in vitro expanded polyclonal Tregs. However, it was shown that in vitro-generated human Foxp3+ CD4+ T cells lack suppressive function and produce effector cytokines 136. Additionally, in the absence of exogenous TGF-β, the majority of in vitro-induced Tregs lose their Foxp3 expression 137,138. As stable Foxp3 expression is quintessential for the maintenance, sustenance and suppressive functions of Treg cells 139, the efficacy of TGF b-induced Foxp3+ CD4+ T cells in comparison to nTregs is doubtful. The following section discusses the role of various agents that have been shown to promote Treg generation and/or function and the relevance of these findings in suppressing allergic responses in the lung.
Retinoic acid
In vitro, TGF-β-induced conversion to Foxp3+ Tregs has been shown to be enhanced and stabilized by the vitamin A metabolite, retinoic acid 80,99,140–146. Retinoic acid was shown to induce de novo conversion to Treg cells even at high levels of co-stimulation, suggesting that by some unknown mechanism, retinoic acid overrides the inhibitory effect of co-stimulation on the induction of FoxP3 expression 146. Recently, it was shown that promotion of Foxp3+ iTregs by retinoic acid is not due to its direct effect on the responder cells but due to inhibition of cytokine production by CD44hi effector memory T cells 143.
Similar synergistic relationship between TGF-β and retinoic acid in inducing, expanding and maintaining Tregs has been observed in vivo 143,147. To understand the function of retinoic acid in the generation of Tregs in vivo, we need to understand its metabolism. Retinoic acid production from retinol is a sequential process that ultimately involves retinal to retinoic acid conversion by retinal dehydrogenases (RALDH). Among the various biologically active retinoids, all-trans retinoic acid (ATRA) is the most potent known metabolite 148. Extensive research has characterized ATRA as the physiological ligand for the retinoic acid receptor (RAR) family of nuclear hormone receptors 149,150. Multiple studies have recently shown production of retinoic acid or expression of retinal dehydrogenases(RALDH1 and/or RALDH2) in different cell types in the gut promoting Treg development. Even though both vitamin A intake and its metabolism to retinoic acid have been demonstrated to be essential in lung development and maintaining pulmonary homeostasis, the cellular participants responsible for generating retinoic acid in the lungs are still unclear. In one study151, isolated lung lipid interstitial cells (LIC) were found to be capable of converting all-trans retinol to an acidic retinoid with properties that are similar and possibly identical to those of ATRA. Most of the retinoic acid produced by the LIC was secreted in the lung alveoli where it could be taken up by other lung cells to exert immunodulatory effects. Recently, airway epithelial cell-expressed MMP-7 was shown to regulate RALDH1 which promoted the development of immunosuppressive Tregs causing attenuation of allergic responses 29. This finding was in contrast to reports that demonstrated exacerbated allergic responses following in vivo administration of ATRA 152,153. It should be noted that in the steady state, retinoic acid is bound to albumin and circulates in plasma in nanomolar concentrations with a typical half-life, in rodents, of less than an hour 154. Most studies that have demonstrated the biological activities and importance of retinoic acid in regulating immune responses have used supraphysiological concentrations added either to cultured cells or, less frequently, administered to animals in vivo. The dose of ATRA used in the in vivo studies has been variable, depending on the mouse model used. Although such experiments have demonstrated the potential scope of retinoic acid-mediated induction of Tregs and suppression of inflammatory responses in the gut, in the lung the suppressive effect of retinoic acid is clearly not uniformly observed. The reasons for the divergent results need to be determined in future studies especially because there are studies showing Th2-inducing potential of retinoic acid via RA-RAR mediated signaling 152,153,155–157. Iwata et al suggest that this effect of RA on Th2 differentiation is stage-dependent and retinoic acid enhances Th2 responses only if it is added in initial stages of T helper cell differentiation 157.
Vitamin D
Independently, several investigations have shown a direct relationship between vitamin D and asthma 158–161. 1,25-dihydroxyvitamin D3 [1,25(OH)2D3)], the active metabolite of vitamin D3, is a lypophilic molecule that exerts its actions through a nuclear receptor, the VitD3 receptor (VDR) 162–164. A number of recent studies have consistently demonstrated the ability of vitamin D3 and VDR agonists to act as strong immunosuppressors 35,38. Vitamin D3 has also been shown to directly enhance suppressive function of Tregs 165. As an immunosuppressant, 1,25(OH)2D3 has been implicated in inhibition of Th1 differentiation, as studied in Th1 dominated animal models 35,166,167. Interestingly, Vitamin D3-VDR signaling and VDR agonists have been shown to either promote or inhibit proliferation and differentiation of Th2 cells35,161,168–170. Such opposite effects of vitamin D3 in regulating Th2 cell responses were also observed in models of experimental allergic asthma where 1,25(OH)2D3 treatment was either beneficial 160,161,171, without effect 172 or deleterious160,170,172. As with retinoic acid, the discrepancies in the findings could be due to variations in dose, route of administration, duration of exposure and the choice of experimental model.
Allergen-specific immunotherapy
Another strategy to expand antigen specific Treg cell repertoire in vivo involves exposure to low doses of antigen without adjuvants. Such protocols induce antigen-specific immunosuppression and are designated allergen specific immunotherapy (SIT) pioneered by Noon in 1911 173,174. SIT involves the administration (usually subcutaneous) of increasing doses of allergen in order to achieve hyporesponsiveness to it. Various effects of SIT have been documented including induction of Tregs secreting IL-10 and TGF-β 36,175–178. In the clinical setting, SIT is the only treatment that induces specific Treg cells in human subjects 179. Although administration of specific allergens has been shown to be effective in rhinitis180 and insect venom allergy 181,182, the role of this intervention in allergic asthma remains controversial 183–185. SIT has proven efficacious in treating mild asthma 186, as well as for preventing the progression to asthma in patients suffering from rhinitis187,188. However, it is not yet recommended for treatment of moderate to severe asthma 189. Furthermore, the major drawbacks of this treatment are the risks for inducing rare but life-threatening systemic reactions mediated by IgE in patients requiring long term administration of SIT 184,190.
Strategies to improve the safety of allergen immunotherapy were initially focused on reducing the allergenicity of the preparation by using modified allergens (allergoids), novel adjuvants and use of alternate routes of administration 191–193. Along these lines, peptide immunotherapy, involving use of peptide fragments from allergens corresponding to T-cell epitopes to induce immunologic tolerance has met with success in experimental models of allergic disease 194. Peptide immunotherapy is an advantage over SIT as it does not result in adverse IgE responses due to their relatively small size and therefore inability to cross-link allergen-specific IgE 195,196 Peptide treatment suppressed allergen-specific T cell proliferation and production of cytokines IL-4, IL-13 and IFNγ but induced IL-10 production 197–199 and IL-10-dependent functional Tregs200. Because of the successful induction of tolerance and suppression of inflammatory responses, peptide immunotherapy has emerged as an ideal alternative to SIT. Currently, clinical data is available for 2 allergens that have been targeted with this approach which are the cat allergen Fel d 1197,198,201 and the bee venom allergen Apim1 (phospholipase A2) 181,202,203.
Despite accumulation of considerable evidence demonstrating a beneficial role of allergen-derived peptide immunotherapy, there have been instances where late asthmatic reactions followed by bronchial hyperresponsvieness to the peptides were reported in patients 197. These adverse effects could be related to peptide dose and immunogenicity. Further attempts are being made to fine-tune the existing protocols that can provide maximum benefit at lower doses of peptides. Recently, linked epitope suppression, a key immunologic mechanism of peptide immunotherapy was demonstrated where treatment with selected epitopes from a single allergen, Fel d 1, resulted in suppression of immune responses to other “linked” epitopes within the same molecule 36. Additional evaluation of appropriate immunogenic peptides from different allergens would increase the applicability of peptide immunotherapy to severe asthmatics.
Corticosteroids and rapamycin
Corticosteroids or glucocorticoids are by far the most routinely used and most effective anti-inflammatory therapy for both acute and chronic allergic diseases and asthma. Among the various mechanisms identified for glucocorticoid-mediated suppression, promotion of Foxp3 expression and suppressive function in CD4+ T cells was also noted 204. The increase in Foxp3 mRNA subsequent to glucocorticoid treatment, though transient, was tightly correlated with increase in IL-10 mRNA, though the expression of TGF-β remained unchanged. Similar increases in IL-10 synthesis and reduction in proinflammatory cytokine production subsequent to inhalation of corticosteroids have been reported in asthma patients 205,206. A more conclusive study recently demonstrated that an inhaled corticosteroid could reverse the observed poor numbers of CD4+CD25hi cells in the BAL fluid of asthmatic children 22. However, treatment with corticosteroids (dexamethasone) has been also shown to inhibit induction of IL-10 and development of Tregs207. The authors have cautioned against long-term use of corticosteroids, especially in treating chronic conditions. They suggest that the inhibitory effect of corticosteroids on Treg development may cause excessive pro-inflammatory responses when the individual is re-exposed to the allergen. In contrast, Barrat et al have demonstrated that dexamethasone, in synergism with 1α, 25-dihydroxyvitamin D3 (calcitriol), the active form of vitamin D, directly induces IL-10 secreting regulatory T cells (Tr1 cells) 35, 38.
Rapamycin, a small molecule drug, chemically defined as a macrocyclic lactone, routinely used as an immunosuppressive drug in organ transplantation, has been found to promote in vitro expansion of both murine 208,209 and human Tregs210–213, which could be highly significant in generating clinically relevant quantities of Tregs 214. The mechanisms underlying the selective and preferential expansion of Tregs and maintenance of suppressive activity in the presence of rapamycin are unclear. Some studies attribute it to selective survival advantage 213, whereas others demonstrate induction of Treg like phenotype in T effector cells in the presence of rapamycin 215–217. Recently, a study demonstrated that the molecule Pim2, which is selectively upregulated in Foxp3 expressing cells, is responsible for providing the survival signal218. This preferential expansion or survival of Treg cells has also been demonstrated following in vivo administration of rapamycin219. In independent studies, rapamycin has been shown to inhibit asthma and airway remodeling in experimental models 220, 221, 222,223, thereby highlighting another drug that can be used to expand antigen/allergen-specific Tregs.
Concluding Remarks
Both human and animal studies show that Tregs are regulators of allergic airways disease. There does not appear to be a unique mechanism that underlies their regulatory function in this disease as has been realized in other diseases as well. It is clear that thymus-derived nTregs are not the only Tregs that are involved in suppression of allergic disease. iTregs have been identified in early life and the challenge in future years is to capitalize on this realization to induce allergen-specific Tregs. However, multiple issues and controversies need to be resolved before Treg cell-based immunotherapy becomes routine practice. More knowledge about the molecular mechanisms driving Treg cell proliferation, activation, and survival is required. Investigations pertaining to stability of Foxp3 in ex vivo-manipulated and later adoptively transferred induced/expanded Treg population is critical, especially in light of the concept of exFoxp3 cells44,45. Given the intense interest in these cells in the field of immunology as a whole, it is hoped that the basic knowledge gained about these cells would translate into dependable Treg-mediated patient care in the not too distant future.
Acknowledgments
The authors thank Stephanie L. Poe for excellent assistance with the artwork. This work was supported by U.S. National Institutes of Health grants HL 077430 and AI 048927 (to A.R), HL060207 (to P.R.) and HL 084932 (to A.R. and P.R.). The authors regret inadvertent omission of additional relevant publications.
Footnotes
Disclosures: No conflict
References
- 1.Metzger WJ, et al. Local allergen challenge and bronchoalveolar lavage of allergic asthmatic lungs: description of the model and local airway inflammation. Am Rev Respir Dis. 1987;135:433–440. doi: 10.1164/arrd.1987.135.2.433. [DOI] [PubMed] [Google Scholar]
- 2.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 3.Corrigan CJ, Hartnell A, Kay AB. T lymphocyte activation in acute severe asthma. Lancet. 1988;1:1129–1132. doi: 10.1016/s0140-6736(88)91951-4. [DOI] [PubMed] [Google Scholar]
- 4.Wierenga EA, et al. Evidence for compartmentalization of functional subsets of CD2+ T lymphocytes in atopic subjects. J Immunol. 1990;144:4651–4656. [PubMed] [Google Scholar]
- 5.Hamid Q, et al. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J Clin Invest. 1991;87:1541–1546. doi: 10.1172/JCI115166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson DS, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 7.Kay AB, et al. Messenger RNA expression of the cytokine gene cluster IL-3, IL-4, IL-5 and GM-CSF in allergen-induced late-phase cutaneous reactions in atopic subjects. J Exp Med. 1991;173:775–778. doi: 10.1084/jem.173.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett NA, Austen KF. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity. 2009;31:425–437. doi: 10.1016/j.immuni.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J Allergy Clin Immunol. 2007;119:1055–1062. doi: 10.1016/j.jaci.2007.01.015. quiz 1063–1054. [DOI] [PubMed] [Google Scholar]
- 10.Bullens DM, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellings PW, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 12.Molet S, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 13.McKinley L, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–4097. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 15.Kleinewietfeld M, et al. CD49d provides access to “untouched” human Foxp3+ Treg free of contaminating effector cells. Blood. 2009;113:827–836. doi: 10.1182/blood-2008-04-150524. [DOI] [PubMed] [Google Scholar]
- 16.Ito T, et al. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity. 2008;28:870–880. doi: 10.1016/j.immuni.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewkowich IP, et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J Exp Med. 2005;202:1549–1561. doi: 10.1084/jem.20051506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strickland DH, et al. Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. J Exp Med. 2006;203:2649–2660. doi: 10.1084/jem.20060155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joetham A, et al. Naturally occurring lung CD4(+)CD25(+) T cell regulation of airway allergic responses depends on IL-10 induction of TGF-beta. J Immunol. 2007;178:1433–1442. doi: 10.4049/jimmunol.178.3.1433. [DOI] [PubMed] [Google Scholar]
- 20.Saito K, et al. Differential regulatory function of resting and preactivated allergen-specific CD4+ CD25+ regulatory T cells in Th2-type airway inflammation. J Immunol. 2008;181:6889–6897. doi: 10.4049/jimmunol.181.10.6889. [DOI] [PubMed] [Google Scholar]
- 21.Bennett CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 22.Hartl D, et al. Quantitative and functional impairment of pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. J Allergy Clin Immunol. 2007;119:1258–1266. doi: 10.1016/j.jaci.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 23.Torgerson TR, Ochs HD. Regulatory T cells in primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol. 2007;7:515–521. doi: 10.1097/ACI.0b013e3282f1a27a. [DOI] [PubMed] [Google Scholar]
- 24.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol. 2007;120:744–750. doi: 10.1016/j.jaci.2007.08.044. quiz 751–742. [DOI] [PubMed] [Google Scholar]
- 25.Bonertz A, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest. 2009;119:3311–3321. doi: 10.1172/JCI39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mucida D, et al. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostroukhova M, et al. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta. J Clin Invest. 2006;116:996–1004. doi: 10.1172/JCI26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curotto de Lafaille MA, et al. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–126. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Goswami S, et al. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10:496–503. doi: 10.1038/ni.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 31.Groux H, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 32.Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001;2:725–731. doi: 10.1038/90667. [DOI] [PubMed] [Google Scholar]
- 33.Akbari O, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med. 2002;8:1024–1032. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 34.Akdis M, et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med. 2004;199:1567–1575. doi: 10.1084/jem.20032058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrat FJ, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)-and Th2-inducing cytokines. J Exp Med. 2002;195:603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell JD, et al. Peptide immunotherapy in allergic asthma generates IL-10-dependent immunological tolerance associated with linked epitope suppression. J Exp Med. 2009;206:1535–1547. doi: 10.1084/jem.20082901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meiler F, et al. In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J Exp Med. 2008;205:2887–2898. doi: 10.1084/jem.20080193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Garra A, Barrat FJ. In vitro generation of IL-10-producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by Th1-and Th2-inducing cytokines. Immunol Lett. 2003;85:135–139. doi: 10.1016/s0165-2478(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 39.Wilson MS, et al. Suppression of murine allergic airway disease by IL-2:anti-IL-2 monoclonal antibody-induced regulatory T cells. J Immunol. 2008;181:6942–6954. doi: 10.4049/jimmunol.181.10.6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berg DJ, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 42.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119:482–487. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 44.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oldenhove G, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owen RD. Immunogenetic Consequences of Vascular Anastomoses between Bovine Twins. Science. 1945;102:400–401. doi: 10.1126/science.102.2651.400. [DOI] [PubMed] [Google Scholar]
- 47.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 48.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Biggelaar AH, et al. Decreased atopy in children infected with Schistosoma haematobium: a role for parasite-induced interleukin-10. Lancet. 2000;356:1723–1727. doi: 10.1016/S0140-6736(00)03206-2. [DOI] [PubMed] [Google Scholar]
- 50.Yazdanbakhsh M, Kremsner PG, van Ree R. Allergy, parasites, and the hygiene hypothesis. Science. 2002;296:490–494. doi: 10.1126/science.296.5567.490. [DOI] [PubMed] [Google Scholar]
- 51.Wilson MS, et al. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. 2005;202:1199–1212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGuirk P, Mills KH. Pathogen-specific regulatory T cells provoke a shift in the Th1/Th2 paradigm in immunity to infectious diseases. Trends Immunol. 2002;23:450–455. doi: 10.1016/s1471-4906(02)02288-3. [DOI] [PubMed] [Google Scholar]
- 53.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jankovic D, et al. Conventional T-bet(+)Foxp3(−) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kursar M, et al. Cutting Edge: Regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol. 2007;178:2661–2665. doi: 10.4049/jimmunol.178.5.2661. [DOI] [PubMed] [Google Scholar]
- 56.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 57.Dutta P, Burlingham WJ. Tolerance to noninherited maternal antigens in mice and humans. Curr Opin Organ Transplant. 2009;14:439–447. doi: 10.1097/MOT.0b013e32832d6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mold JE, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008;322:1562–1565. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verhasselt V, et al. Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat Med. 2008;14:170–175. doi: 10.1038/nm1718. [DOI] [PubMed] [Google Scholar]
- 60.Polte T, Hansen G. Maternal tolerance achieved during pregnancy is transferred to the offspring via breast milk and persistently protects the offspring from allergic asthma. Clin Exp Allergy. 2008;38:1950–1958. doi: 10.1111/j.1365-2222.2008.03096.x. [DOI] [PubMed] [Google Scholar]
- 61.Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. 2008;8:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- 62.Sly PD, et al. Early identification of atopy in the prediction of persistent asthma in children. Lancet. 2008;372:1100–1106. doi: 10.1016/S0140-6736(08)61451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–435. doi: 10.1097/00002281-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 64.Zuidgeest MG, et al. Prescription of respiratory medication without an asthma diagnosis in children: a population based study. BMC Health Serv Res. 2008;8:16. doi: 10.1186/1472-6963-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gambineri E, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. 2008;122:1105–1112. e1101. doi: 10.1016/j.jaci.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 66.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 67.Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med. 2005;202:1539–1547. doi: 10.1084/jem.20051166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McMenamin C, Pimm C, McKersey M, Holt PG. Regulation of IgE responses to inhaled antigen in mice by antigen-specific gamma delta T cells. Science. 1994;265:1869–1871. doi: 10.1126/science.7916481. [DOI] [PubMed] [Google Scholar]
- 69.Ostroukhova M, et al. Tolerance induced by inhaled antigen involves CD4(+) T cells expressing membrane-bound TGF-beta and FOXP3. J Clin Invest. 2004;114:28–38. doi: 10.1172/JCI20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25− T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. 2004;173:7259–7268. doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- 72.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cobbold SP, et al. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol. 2004;172:6003–6010. doi: 10.4049/jimmunol.172.10.6003. [DOI] [PubMed] [Google Scholar]
- 74.Kretschmer K, et al. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 75.Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med. 2008;205:3105–3117. doi: 10.1084/jem.20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aranda R, et al. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J Immunol. 1997;158:3464–3473. [PubMed] [Google Scholar]
- 77.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammationin C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 78.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–271. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 79.Uhlig HH, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect Immun. 2005;73:30–38. doi: 10.1128/IAI.73.1.30-38.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hisbergues M, et al. In vivo and in vitro immunomodulation of Der p 1 allergen-specific response by Lactobacillus plantarum bacteria. Clin Exp Allergy. 2007;37:1286–1295. doi: 10.1111/j.1365-2222.2007.02792.x. [DOI] [PubMed] [Google Scholar]
- 83.Feleszko W, et al. Probiotic-induced suppression of allergic sensitization and airway inflammation is associated with an increase of T regulatory-dependent mechanisms in a murine model of asthma. Clin Exp Allergy. 2007;37:498–505. doi: 10.1111/j.1365-2222.2006.02629.x. [DOI] [PubMed] [Google Scholar]
- 84.Karimi K, Inman MD, Bienenstock J, Forsythe P. Lactobacillus reuteri-induced regulatory T cells protect against an allergic airway response in mice. Am J Respir Crit Care Med. 2009;179:186–193. doi: 10.1164/rccm.200806-951OC. [DOI] [PubMed] [Google Scholar]
- 85.Andersson J, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korn T, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pillemer BB, et al. STAT6 activation confers upon T helper cells resistance to suppression by regulatory T cells. J Immunol. 2009;183:155–163. doi: 10.4049/jimmunol.0803733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 90.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 91.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 92.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 93.Rose-John S, Neurath MF. IL-6 trans-signaling: the heat is on. Immunity. 2004;20:2–4. doi: 10.1016/s1074-7613(04)00003-2. [DOI] [PubMed] [Google Scholar]
- 94.Doganci A, et al. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–325. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dominitzki S, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179:2041–2045. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 96.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 97.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 98.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 99.Takaki H, et al. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008;283:14955–14962. doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mantel PY, et al. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007;5:e329. doi: 10.1371/journal.pbio.0050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pace L, Rizzo S, Palombi C, Brombacher F, Doria G. Cutting edge: IL-4-induced protection of CD4+CD25− Th cells from CD4+CD25+ regulatory T cell-mediated suppression. J Immunol. 2006;176:3900–3904. doi: 10.4049/jimmunol.176.7.3900. [DOI] [PubMed] [Google Scholar]
- 102.Miyara M, et al. The immune paradox of sarcoidosis and regulatory T cells. J Exp Med. 2006;203:359–370. doi: 10.1084/jem.20050648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chaudhry A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koch MA, et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zheng Y, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou L, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 107.Zhou L, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sasai Y, Kageyama R, Tagawa Y, Shigemoto R, Nakanishi S. Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 1992;6:2620–2634. doi: 10.1101/gad.6.12b.2620. [DOI] [PubMed] [Google Scholar]
- 111.Kageyama R, Ohtsuka T, Tomita K. The bHLH gene Hes1 regulates differentiation of multiple cell types. Mol Cells. 2000;10:1–7. doi: 10.1007/s10059-000-0001-0. [DOI] [PubMed] [Google Scholar]
- 112.Eagar TN, et al. Notch 1 signaling regulates peripheral T cell activation. Immunity. 2004;20:407–415. doi: 10.1016/s1074-7613(04)00081-0. [DOI] [PubMed] [Google Scholar]
- 113.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor β. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Samon JB, et al. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. 2008;112:1813–1821. doi: 10.1182/blood-2008-03-144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Amsen D, Antov A, Flavell RA. The different faces of Notch in T-helper-cell differentiation. Nat Rev Immunol. 2009;9:116–124. doi: 10.1038/nri2488. [DOI] [PubMed] [Google Scholar]
- 116.Becker JC, Czerny C, Brocker EB. Maintenance of clonal anergy by endogenously produced IL-10. Int Immunol. 1994;6:1605–1612. doi: 10.1093/intimm/6.10.1605. [DOI] [PubMed] [Google Scholar]
- 117.Enk AH, Saloga J, Becker D, Mohamadzadeh M, Knop J. Induction of hapten-specific tolerance by interleukin 10 in vivo. J Exp Med. 1994;179:1397–1402. doi: 10.1084/jem.179.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Akdis CA, et al. Epitope-specific T cell tolerance to phospholipase A2 in bee venom immunotherapy and recovery by IL-2 and IL-15 in vitro. J Clin Invest. 1996;98:1676–1683. doi: 10.1172/JCI118963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gri G, et al. CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity. 2008;29:771–781. doi: 10.1016/j.immuni.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Odemuyiwa SO, et al. Cutting edge: human eosinophils regulate T cell subset selection through indoleamine 2,3-dioxygenase. J Immunol. 2004;173:5909–5913. doi: 10.4049/jimmunol.173.10.5909. [DOI] [PubMed] [Google Scholar]
- 122.Xu H, et al. Indoleamine 2,3-Dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation but is not required for tolerance. Proc Natl Acad Sci USA. 2008;105:6690–6695. doi: 10.1073/pnas.0708809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Montagnoli C, et al. Immunity and tolerance to Aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J Immunol. 2006;176:1712–1723. doi: 10.4049/jimmunol.176.3.1712. [DOI] [PubMed] [Google Scholar]
- 124.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 125.Collison LW, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 126.Pillemer BL, et al. STAT6 Activation Confers Upon T Helper Cells Resistance To Suppression By Regulatory T Cells. J Immunol. 2009;183:155–163. doi: 10.4049/jimmunol.0803733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhou X, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205:1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kohlhaas S, et al. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- 130.Lu LF, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rodriguez A, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 133.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 134.Fisson S, et al. Therapeutic potential of self-antigen-specific CD4+ CD25+ regulatory T cells selected in vitro from a polyclonal repertoire. Eur J Immunol. 2006;36:817–827. doi: 10.1002/eji.200535445. [DOI] [PubMed] [Google Scholar]
- 135.Masteller EL, Tang Q, Bluestone JA. Antigen-specific regulatory T cells--ex vivo expansion and therapeutic potential. Semin Immunol. 2006;18:103–110. doi: 10.1016/j.smim.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 136.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Selvaraj RK, Geiger TL. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J Immunol. 2007;179:11. p following 1390. [PubMed] [Google Scholar]
- 138.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 140.Mucida D, et al. Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity. 2009;30:471–472. doi: 10.1016/j.immuni.2009.03.008. author reply 472–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang J, Huizinga TW, Toes RE. De novo generation and enhanced suppression of human CD4+CD25+ regulatory T cells by retinoic acid. J Immunol. 2009;183:4119–4126. doi: 10.4049/jimmunol.0901065. [DOI] [PubMed] [Google Scholar]
- 142.Xiao S, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–2284. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hill JA, et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity. 2008;29:758–770. doi: 10.1016/j.immuni.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Moore C, et al. Transforming growth factor-beta and all-trans retinoic acid generate ex vivo transgenic regulatory T cells with intestinal homing receptors. Transplant Proc. 2009;41:2670–2672. doi: 10.1016/j.transproceed.2009.06.130. [DOI] [PubMed] [Google Scholar]
- 146.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 148.Ross AC. On the sources of retinoic acid in the lung: understanding the local conversion of retinol to retinoic acid. Am J Physiol Lung Cell Mol Physiol. 2004;286:L247–248. doi: 10.1152/ajplung.00234.2003. [DOI] [PubMed] [Google Scholar]
- 149.Mucida D, Park Y, Cheroutre H. From the diet to the nucleus: vitamin A and TGF-beta join efforts at the mucosal interface of the intestine. Semin Immunol. 2009;21:14–21. doi: 10.1016/j.smim.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dirami G, et al. Lung retinol storing cells synthesize and secrete retinoic acid, an inducer of alveolus formation. Am J Physiol Lung Cell Mol Physiol. 2004;286:L249–256. doi: 10.1152/ajplung.00140.2003. [DOI] [PubMed] [Google Scholar]
- 152.Maret M, et al. Liposomal retinoic acids modulate asthma manifestations in mice. J Nutr. 2007;137:2730–2736. doi: 10.1093/jn/137.12.2730. [DOI] [PubMed] [Google Scholar]
- 153.Dawson H, et al. Localized Th1-, Th2-, T regulatory cell-, and inflammation-associated hepatic and pulmonary immune responses in Ascaris suum-infected swine are increased by retinoic acid. Infect Immun. 2009;77:2576–2587. doi: 10.1128/IAI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.el Mansouri S, et al. Time-and dose-dependent kinetics of all-trans-retinoic acid in rats after oral or intravenous administration(s) Drug Metab Dispos. 1995;23:227–231. [PubMed] [Google Scholar]
- 155.Ueki S, et al. Retinoic acids are potent inhibitors of spontaneous human eosinophil apoptosis. J Immunol. 2008;181:7689–7698. doi: 10.4049/jimmunol.181.11.7689. [DOI] [PubMed] [Google Scholar]
- 156.Dawson HD, et al. Direct and indirect effects of retinoic acid on human Th2 cytokine and chemokine expression by human T lymphocytes. BMC Immunol. 2006;7:27. doi: 10.1186/1471-2172-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Iwata M, Eshima Y, Kagechika H. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol. 2003;15:1017–1025. doi: 10.1093/intimm/dxg101. [DOI] [PubMed] [Google Scholar]
- 158.Hypponen E, et al. Infant vitamin d supplementation and allergic conditions in adulthood: northern Finland birth cohort 1966. Ann N Y Acad Sci. 2004;1037:84–95. doi: 10.1196/annals.1337.013. [DOI] [PubMed] [Google Scholar]
- 159.Wjst M, Dold S. Genes, factor X, and allergens: what causes allergic diseases? Allergy. 1999;54:757–759. doi: 10.1034/j.1398-9995.1999.00193.x. [DOI] [PubMed] [Google Scholar]
- 160.Matheu V, Back O, Mondoc E, Issazadeh-Navikas S. Dual effects of vitamin D-induced alteration of TH1/TH2 cytokine expression: enhancing IgE production and decreasing airway eosinophilia in murine allergic airway disease. J Allergy Clin Immunol. 2003;112:585–592. doi: 10.1016/s0091-6749(03)01855-4. [DOI] [PubMed] [Google Scholar]
- 161.Topilski I, et al. The anti-inflammatory effects of 1,25-dihydroxyvitamin D3 on Th2 cells in vivo are due in part to the control of integrin-mediated T lymphocyte homing. Eur J Immunol. 2004;34:1068–1076. doi: 10.1002/eji.200324532. [DOI] [PubMed] [Google Scholar]
- 162.Sigmundsdottir H, Butcher EC. Environmental cues, dendritic cells and the programming of tissue-selective lymphocyte trafficking. Nat Immunol. 2008;9:981–987. doi: 10.1038/ni.f.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wjst M. The vitamin D slant on allergy. Pediatr Allergy Immunol. 2006;17:477–483. doi: 10.1111/j.1399-3038.2006.00456.x. [DOI] [PubMed] [Google Scholar]
- 164.Szatmari I, Nagy L. Nuclear receptor signalling in dendritic cells connects lipids, the genome and immune function. EMBO J. 2008;27:2353–2362. doi: 10.1038/emboj.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gorman S, et al. Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J Immunol. 2007;179:6273–6283. doi: 10.4049/jimmunol.179.9.6273. [DOI] [PubMed] [Google Scholar]
- 166.Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J Nutr. 1998;128:68–72. doi: 10.1093/jn/128.1.68. [DOI] [PubMed] [Google Scholar]
- 167.Cantorna MT, Woodward WD, Hayes CE, DeLuca HF. 1,25-dihydroxyvitamin D3 is a positive regulator for the two anti-encephalitogenic cytokines TGF-beta 1 and IL-4. J Immunol. 1998;160:5314–5319. [PubMed] [Google Scholar]
- 168.Staeva-Vieira TP, Freedman LP. 1,25-dihydroxyvitamin D3 inhibits IFN-gamma and IL-4 levels during in vitro polarization of primary murine CD4+ T cells. J Immunol. 2002;168:1181–1189. doi: 10.4049/jimmunol.168.3.1181. [DOI] [PubMed] [Google Scholar]
- 169.O’Kelly J, et al. Normal myelopoiesis but abnormal T lymphocyte responses in vitamin D receptor knockout mice. J Clin Invest. 2002;109:1091–1099. doi: 10.1172/JCI12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wittke A, et al. Vitamin D receptor expression by the lung micro-environment is required for maximal induction of lung inflammation. Arch Biochem Biophys. 2007;460:306–313. doi: 10.1016/j.abb.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Taher YA, van Esch BC, Hofman GA, Henricks PA, van Oosterhout AJ. 1alpha, 25-dihydroxyvitamin D3 potentiates the beneficial effects of allergen immunotherapy in a mouse model of allergic asthma: role for IL-10 and TGF-beta. J Immunol. 2008;180:5211–5221. doi: 10.4049/jimmunol.180.8.5211. [DOI] [PubMed] [Google Scholar]
- 172.Wittke A, Weaver V, Mahon BD, August A, Cantorna MT. Vitamin D receptor-deficient mice fail to develop experimental allergic asthma. J Immunol. 2004;173:3432–3436. doi: 10.4049/jimmunol.173.5.3432. [DOI] [PubMed] [Google Scholar]
- 173.Noon L. Prophylactic inoculation of hay fever. Lancet. 1911;1:1572–1573. [Google Scholar]
- 174.Noon L. Prophylactic inoculation against hay fever (historical document) Ann Allergy. 1955;13:713–716. passim. [PubMed] [Google Scholar]
- 175.Francis JN, Till SJ, Durham SR. Induction of IL-10+CD4+CD25+ T cells by grass pollen immunotherapy. J Allergy Clin Immunol. 2003;111:1255–1261. doi: 10.1067/mai.2003.1570. [DOI] [PubMed] [Google Scholar]
- 176.Akdis CA, Blesken T, Akdis M, Wuthrich B, Blaser K. Role of interleukin 10 in specific immunotherapy. J Clin Invest. 1998;102:98–106. doi: 10.1172/JCI2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jutel M, et al. IL-10 and TGF-beta cooperate in the regulatory T cell response to mucosal allergens in normal immunity and specific immunotherapy. Eur J Immunol. 2003;33:1205–1214. doi: 10.1002/eji.200322919. [DOI] [PubMed] [Google Scholar]
- 178.O’Hehir RE, et al. House dust mite sublingual immunotherapy: the role for transforming growth factor-beta and functional regulatory T cells. Am J Respir Crit Care Med. 2009;180:936–947. doi: 10.1164/rccm.200905-0686OC. [DOI] [PubMed] [Google Scholar]
- 179.Akdis CA, Akdis M. Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J Allergy Clin Immunol. 2009;123:735–746. doi: 10.1016/j.jaci.2009.02.030. quiz 747–738. [DOI] [PubMed] [Google Scholar]
- 180.Wilson DR, Torres LI, Durham SR. Sublingual immunotherapy for allergic rhinitis. Cochrane Database Syst Rev. 2003:CD002893. doi: 10.1002/14651858.CD002893. [DOI] [PubMed] [Google Scholar]
- 181.Muller U, et al. Successful immunotherapy with T-cell epitope peptides of bee venom phospholipase A2 induces specific T-cell anergy in patients allergic to bee venom. J Allergy Clin Immunol. 1998;101:747–754. doi: 10.1016/S0091-6749(98)70402-6. [DOI] [PubMed] [Google Scholar]
- 182.Pereira-Santos MC, et al. Expansion of circulating Foxp3+)D25 bright CD4+ T cells during specific venom immunotherapy. Clin Exp Allergy. 2008;38:291–297. doi: 10.1111/j.1365-2222.2007.02887.x. [DOI] [PubMed] [Google Scholar]
- 183.Abramson MJ, Puy RM, Weiner JM. Is allergen immunotherapy effective in asthma? A meta-analysis of randomized controlled trials. Am J Respir Crit Care Med. 1995;151:969–974. doi: 10.1164/ajrccm.151.4.7697274. [DOI] [PubMed] [Google Scholar]
- 184.Bousquet J, Michel FB. Specific immunotherapy in asthma: is it effective? J Allergy Clin Immunol. 1994;94:1–11. doi: 10.1016/0091-6749(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 185.Bousquet J, Michel FB, Malling HJ. Is allergen immunotherapy effective in asthma? A meta-analysis of randomized clinical trials. Am J Respir Crit Care Med. 1995;152:1737–1738. doi: 10.1164/ajrccm.152.5.7582321. [DOI] [PubMed] [Google Scholar]
- 186.Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev. 2003:CD001186. doi: 10.1002/14651858.CD001186. [DOI] [PubMed] [Google Scholar]
- 187.Moller C, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT-study) J Allergy Clin Immunol. 2002;109:251–256. doi: 10.1067/mai.2002.121317. [DOI] [PubMed] [Google Scholar]
- 188.Jacobsen L, et al. Specific immunotherapy has long-term preventive effect of seasonal and perennial asthma: 10-year follow-up on the PAT study. Allergy. 2007;62:943–948. doi: 10.1111/j.1398-9995.2007.01451.x. [DOI] [PubMed] [Google Scholar]
- 189.Rolland JM, Gardner LM, O’Hehir RE. Allergen-related approaches to immunotherapy. Pharmacol Ther. 2009;121:273–284. doi: 10.1016/j.pharmthera.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 190.Bousquet J, Michel FB, Vignola AM. Allergen immunotherapy: therapeutic vaccines for asthma. Clin Allergy Immunol. 2004;18:511–528. [PubMed] [Google Scholar]
- 191.Larche M, Akdis CA, Valenta R. Immunological mechanisms of allergen-specific immunotherapy. Nat Rev Immunol. 2006;6:761–771. doi: 10.1038/nri1934. [DOI] [PubMed] [Google Scholar]
- 192.Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol. 2007;119:780–791. doi: 10.1016/j.jaci.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 193.Broide DH. Immunomodulation of allergic disease. Annu Rev Med. 2009;60:279–291. doi: 10.1146/annurev.med.60.041807.123524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Larche M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11:S69–76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 195.Larche M. Immunotherapy with peptides. Eur Ann Allergy Clin Immunol. 2006;38:240–243. [PubMed] [Google Scholar]
- 196.Larche M. Peptide immunotherapy for allergic diseases. Allergy. 2007;62:325–331. doi: 10.1111/j.1398-9995.2006.01309.x. [DOI] [PubMed] [Google Scholar]
- 197.Oldfield WL, Kay AB, Larche M. Allergen-derived T cell peptide-induced late asthmatic reactions precede the induction of antigen-specific hyporesponsiveness in atopic allergic asthmatic subjects. J Immunol. 2001;167:1734–1739. doi: 10.4049/jimmunol.167.3.1734. [DOI] [PubMed] [Google Scholar]
- 198.Oldfield WL, Larche M, Kay AB. Effect of T-cell peptides derived from Fel d 1 on allergic reactions and cytokine production in patients sensitive to cats: a randomised controlled trial. Lancet. 2002;360:47–53. doi: 10.1016/s0140-6736(02)09332-7. [DOI] [PubMed] [Google Scholar]
- 199.Van Overtvelt L, et al. IL-10-inducing adjuvants enhance sublingual immunotherapy efficacy in a murine asthma model. Int Arch Allergy Immunol. 2008;145:152–162. doi: 10.1159/000108140. [DOI] [PubMed] [Google Scholar]
- 200.Verhoef A, Alexander C, Kay AB, Larche M. T cell epitope immunotherapy induces a CD4+ T cell population with regulatory activity. PLoS Med. 2005;2:e78. doi: 10.1371/journal.pmed.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Alexander C, Ying S, ABK, Larche M. Fel d 1-derived T cell peptide therapy induces recruitment of CD4+ CD25+; CD4+ interferon-gamma+ T helper type 1 cells to sites of allergen-induced late-phase skin reactions in cat-allergic subjects. Clin Exp Allergy. 2005;35:52–58. doi: 10.1111/j.1365-2222.2005.02143.x. [DOI] [PubMed] [Google Scholar]
- 202.Fellrath JM, et al. Allergen-specific T-cell tolerance induction with allergen-derived long synthetic peptides: results of a phase I trial. J Allergy Clin Immunol. 2003;111:854–861. doi: 10.1067/mai.2003.1337. [DOI] [PubMed] [Google Scholar]
- 203.Tarzi M, et al. Induction of interleukin-10 and suppressor of cytokine signalling-3 gene expression following peptide immunotherapy. Clin Exp Allergy. 2006;36:465–474. doi: 10.1111/j.1365-2222.2006.02469.x. [DOI] [PubMed] [Google Scholar]
- 204.Karagiannidis C, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol. 2004;114:1425–1433. doi: 10.1016/j.jaci.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 205.John M, et al. Inhaled corticosteroids increase interleukin-10 but reduce macrophage inflammatory protein-1alpha, granulocyte-macrophage colony-stimulating factor, and interferon-gamma release from alveolar macrophages in asthma. Am J Respir Crit Care Med. 1998;157:256–262. doi: 10.1164/ajrccm.157.1.9703079. [DOI] [PubMed] [Google Scholar]
- 206.Stelmach I, Jerzynska J, Kuna P. A randomized, double-blind trial of the effect of glucocorticoid, antileukotriene and beta-agonist treatment on IL-10 serum levels in children with asthma. Clin Exp Allergy. 2002;32:264–269. doi: 10.1046/j.1365-2222.2002.01286.x. [DOI] [PubMed] [Google Scholar]
- 207.Stock P, Akbari O, DeKruyff RH, Umetsu DT. Respiratory tolerance is inhibited by the administration of corticosteroids. J Immunol. 2005;175:7380–7387. doi: 10.4049/jimmunol.175.11.7380. [DOI] [PubMed] [Google Scholar]
- 208.Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol. 2007;7:1819–1824. doi: 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Coenen JJ, et al. Rapamycin, not cyclosporine, permits thymic generation and peripheral preservation of CD4+ CD25+ FoxP3+ T cells. Bone Marrow Transplant. 2007;39:537–545. doi: 10.1038/sj.bmt.1705628. [DOI] [PubMed] [Google Scholar]
- 210.Battaglia M, et al. Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes. 2006;55:40–49. [PubMed] [Google Scholar]
- 211.Battaglia M, et al. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 212.Keever-Taylor CA, et al. Rapamycin enriches for CD4(+) CD25(+) CD27(+) Foxp3(+) regulatory T cells in ex vivo-expanded CD25-enriched products from healthy donors and patients with multiple sclerosis. Cytotherapy. 2007;9:144–157. doi: 10.1080/14653240601145223. [DOI] [PubMed] [Google Scholar]
- 213.Strauss L, et al. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 214.Miyara M, Wing K, Sakaguchi S. Therapeutic approaches to allergy and autoimmunity based on FoxP3+ regulatory T-cell activation and expansion. J Allergy Clin Immunol. 2009;123:749–755. doi: 10.1016/j.jaci.2009.03.001. quiz 756–747. [DOI] [PubMed] [Google Scholar]
- 215.Long SA, Buckner JH. Combination of rapamycin and IL-2 increases de novo induction of human CD4(+)CD25(+)FOXP3(+) T cells. J Autoimmun. 2008;30:293–302. doi: 10.1016/j.jaut.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. 2006;107:1018–1023. doi: 10.1182/blood-2005-07-3032. [DOI] [PubMed] [Google Scholar]
- 217.Bocian K, et al. Rapamycin, unlike cyclosporine A, enhances suppressive functions of in vitro-induced CD4+CD25+ Tregs. Nephrol Dial Transplant. 2009 doi: 10.1093/ndt/gfp586. [DOI] [PubMed] [Google Scholar]
- 218.Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: Foxp3− mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol. 2008;180:5794–5798. doi: 10.4049/jimmunol.180.9.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Qu Y, et al. The effect of immunosuppressive drug rapamycin on regulatory CD4+CD25+Foxp3+T cells in mice. Transpl Immunol. 2007;17:153–161. doi: 10.1016/j.trim.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 220.Eynott PR, et al. Effects of cyclosporin A and a rapamycin derivative (SAR943) on chronic allergic inflammation in sensitized rats. Immunology. 2003;109:461–467. doi: 10.1046/j.1365-2567.2003.01672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Corrigan CJ. Asthma refractory to glucocorticoids: the role of newer immunosuppressants. Am J RespirMed. 2002;1:47–54. doi: 10.1007/BF03257162. [DOI] [PubMed] [Google Scholar]
- 222.Fujitani Y, Trifilieff A. In vivo and in vitro effects of SAR 943, a rapamycin analogue, on airway inflammation and remodeling. Am J Respir Crit Care Med. 2003;167:193–198. doi: 10.1164/rccm.200205-455OC. [DOI] [PubMed] [Google Scholar]
- 223.Krymskaya VP. Targeting the phosphatidylinositol 3-kinase pathway in airway smooth muscle: rationale and promise. BioDrugs. 2007;21:85–95. doi: 10.2165/00063030-200721020-00003. [DOI] [PubMed] [Google Scholar]
- 224.Pace L, Pioli C, Doria G. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. J Immunol. 2005;174:7645–7653. doi: 10.4049/jimmunol.174.12.7645. [DOI] [PubMed] [Google Scholar]