

Abstract
In higher vertebrates, sulfatases belong to a conserved family of enzymes that are involved in the regulation of cell metabolism and in developmental cell signaling. They cleave the sulfate from sulfate esters contained in hormones, proteins, and complex macromolecules. A highly conserved cysteine in their active site is post-translationally converted into formylglycine by the formylglycine-generating enzyme encoded by SUMF1 (sulfatase modifying factor 1). This post-translational modification activates all sulfatases. Sulfatases are extensively glycosylated proteins and some of them follow trafficking pathways through cells, being secreted and taken up by distant cells. Many proteoglycans, glycoproteins, and glycolipids contain sulfated carbohydrates, which are sulfatase substrates. Indeed, sulfatases operate as decoding factors for a large amount of biological information contained in the structures of the sulfated sugar chains that are covalently linked to proteins and lipids. Modifications to these sulfate groups have pivotal roles in modulating specific signaling pathways and cell metabolism in mammals.
Keywords: Sulfatases, Sulfatase modifying factor 1, Multiple sulfatase deficiency, Lysosomal storage disease, Cell signaling
Introduction
Sulfatases are hydrolytic enzymes that can cut sulfate groups from sulfate esters (CO–S) and sulfamates (CN–S) according to the reaction depicted below [1].
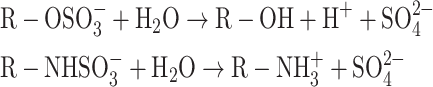 |
These mechanisms are common to all sulfatases, although each has its own substrate specificity (see Table 1). To date, bioinformatic analyses have revealed the existence of 17 distinct genes that can code for sulfatases in humans, and 14 in rodents. [2].
Table 1.
The complete set of human sulfatases: their subcellular localization and secretion, their physiological and synthetic substrates, and the N-glycosylation prediction is reported
| Gene symbol | Localization | Secretion | Substrate | N-glycosylation predictiona | Glycosylation sites X-ray solved |
|---|---|---|---|---|---|
| Arylsulfatase A (ARSA) | Lysosomal | Yes |
4MUSb pNCSb ascorbic acid-2S cerebroside-3S seminolipid-3S psychosine-3S tyrosine-S |
Positions 158,184,350 | Positions 158,184,350 [8] |
| Arylsulfatase B (ARSB) | Lysosomal | Yes |
4MUSb GalN4S-(CS/DS) tyrosine-S |
Positions 188, 279, 366, 426, 458 | Positions 279,426,366 [102] |
| Arylsulfatase C, isozyme S (steroid sulfatase) (STS) (ARSC) | Microsomal | No |
4MUSb pNPSb estrone-S pregnolone-S cholesterol-S DHEA-S testosterone-S vitamin D3 S |
Positions 47, 259, 333, 459 | Positions 47, 259, Possible sites 333,459 [101] |
| Arylsulfatase D (ARSD) | ER | N.D. | 4MUSb | Positions 58, 277 | N.D. |
| Arylsulfatase E (ARSE) | Golgi | N.D. | 4MUSb | Positions 58, 125, 258, 344 | N.D. |
| Arylsulfatase F (ARSF) | ER | N.D. | 4MUSb | Positions 50, 117, 336 | N.D. |
| Arylsulfatase G (ARSG) | ER | Yes | 4MUSb | Positions 47, 145, 286, 427 | N.D. |
| Arylsulfatase H (ARSH) | N.D. | N.D. | 4MUSb | Positions 27, 95, 314 | N.D. |
| Arylsulfatase I (ARSI) | ER | N.D. | 4MUSb | Positions 206, 218, 396, 426 | N.D. |
| Arylsulfatase J (ARSJ) | ER | N.D. | 4MUSb | Positions 157, 254, 306, 318, 431,497, 527 | N.D. |
| Arylsulfatase K (ARSK) | N.D. | N.D. | 4MUSb | Positions108, 166, 193, 262,375, 413,498 | N.D. |
| Galactosamine (N-acetyl)-6-sulfate sulfatase (GALNS) | Lysosomal | Yes |
4MUSb GalNAc6S-(CS) Gal6S-(KS) |
Positions 204, 423 | N.D. |
| Glucosamine (N-acetyl)-6-sulfatase (G6S) (GNS) | Lysosomal | Yes |
4MUSb GlcNAc6S-(HS) GlcNAc6S-(KS) GlcNAc6S |
Positions 111, 117, 183, 198, 210, 279, 317, 362, 387, 405, 422, 449, 480 | N.D. |
| Iduronate 2-sulfatase (IDS) | Lysosomal | Yes |
4MUSb IdoA2S-(HS) |
Positions 31, 115, 144, 246, 280, 325, 513, 537 | N.D. |
| N-sulfoglucosamine sulfohydrolase (SGSH) | Lysosomal | Yes |
4MUSb GalNS-(HS) GalNS-IdoA2S GalNS |
Positions 41, 142, 151, 264, 413 | N.D. |
| Sulfatase 1 (SULF1) | Cell Surface | Yes |
4MUSb GlcN6S (endo) |
Positions 41, 61, 78, 100, 127, 170, 553, 703, 713 | N.D. |
| Sulfatase 2 (SULF2) | Cell Surface | Yes |
4MUSb GlcN6S (endo) |
Positions 65, 112, 132, 149, 171, 198, 241,561, 608, 717, 754, 764 | N.D. |
N.D. No data
aN-glycosylation prediction by looking at the N(S/T) N-glycosylation consensus sequence using the Protein Pattern Find tool (http://www.ualberta.ca/~stothard/javascript/protein_pattern.html)
bSynthetic substrate
Human sulfatases can be classified, on the basis of their subcellular localization and their pH-dependent activities, as: (1) lysosomal and acidic pH-dependent sulfatases, including arylsulfatases A and B (ARSA, B), galactosamine-N-acetyl-6-sulfate-sulfatase (GALNS), N-acetylglucosamine-6-sulfatase (GNS), heparan N-sulfatase (SGSH) and iduronate-2-sulfatase (IDS); (2) non-lysosomal [endoplasmic reticulum (ER) and Golgi localized] and neutral pH-dependent sulfatases, including arylsulfatases C-H, J and K (ARSC-H, J, K); and (3) extracellular and neutral–basic pH-dependent sulfatases, including Sulfatase 1 (Sulf1) and Sulfatase 2 (Sulf2).
Sulfatases share remarkable sequence homology over their entire protein length (between 20 and 60%) across eukaryotic and prokaryotic species. The protein structures are characterized by four domains: A, B and C, highly conserved domains, participate in the formation of N-terminal region with the B domain including the active site (Fig. 1a, b), while the D domain includes the less conserved C-terminal region. In the B domain, the aminoacids reach ≥90% of similarity across all sulfatases, revealing evolutionarily constrained regions (ECRs) that could be essential for the enzyme function (Fig. 1c). The ECRs contain aminoacids with known and potential function in substrate hydrolysis involved in the formation of the active site pocket [2]. Interestingly, the three-dimensional structure of the N-terminal domain also shares high homology with the alkaline phosphatase protein family, suggesting a common enzymatic function and evolution from common ancestral genes [2].
Fig. 1.

a Cristal structure of human Arylsulfatase A (ARSA). The drawing shows the N-terminal part that contains the A and C, and the C-terminal region that includes the D domain. b Detailed representation of N-terminal B domain of hARSA that contains the FGly generation consensus sequence CXP(X/S)R in the substrate binding pocket. c ClustalW multiple protein sequence alignments of all the 17 human sulfatases. The FGly generation consensus sequence CXP(X/S)R is conserved in the active site of all sulfatases. Amino acids with the same gray shadingintensities are conserved in all of the analyzed sequences, or share similar chemical characteristics
The conserved N-terminal region includes a consensus motif that contains a unique active-site aldehyde residue, α-formylglycine (FGly); this is essential for sulfatase catalytic activity and it is generated post-translationally from a highly conserved cysteine (Cys) [3, 4]. Mutagenesis experiments have identified the minimal consensus motif that allows this Cys conversion into FGly. This is formed by 11 amino acids and includes the essential CXP(X/S)R aminoacidic stretch (Fig. 1c). Mutations in this active site or in the flanking regions result in decreased substrate affinities or sulfatase stabilities [3, 5, 6].
The crystal structures of hARSA, hARSB, and hARSC sulfatases have been solved by X-ray diffraction: they appear as globular monomers that are divided in two domains [7]. The N-terminal part contains α-helices that are surrounded by ten large β-sheets. The C-terminal part is constituted by a small anti-parallel β-sheet, with a tightly associated α-helix (Fig. 1a). The sulfatase active sites contain a divalent metal ion located in the substrate-binding pocket [7]. Of note, the secondary structure and tertiary fold is quite identical with the exception of the length of some loops, due to the insertion or deletion of aminoacid sequences. The most conserved region found in ARSA and present in all sulfatases, correspond to 19–146 and 219–363 residues [8]. This means that the major part of the β-sheets and α-helices are conserved in all the family. The ARSA portion, corresponding to 6–9 β-sheets, lacks of similarity in the primary and secondary structure among the sulfatases [8]. Interestingly, in this region of ARSC, an hydrophobic insertion is present that was demonstrated to anchor the enzyme to the microsomes membrane and that allows the contemporary exposition of N- and C-terminal parts at the lumen side [9]. This part was moreover found in ARSD and ARSE sulfatases.
Sulfatases are glycoproteins that contain asparagines that link complex oligosaccharides. N-linked glycan can strongly influence protein structures by regulating the correct folding and stability [10]. Whereas most N-linked oligosaccharides may be structurally dispensable in the mature protein, they are often crucial during glycoprotein folding and oligomerization. [11, 12]. The correct folding of sulfatases might also be dependent on the sugar chains. Human lymphoblastoid cells isolated from Hunter patients, who carry loss of function mutations in the IDS gene, were transfected with the wild-type IDS cDNA and cultured in the presence of tunicamycin. These cells synthesized a 60-kDa unglycosylated and inactive polypeptide which was not further processed, indicating that N-glycosylation was essential for folding, catalytic activity, and processing of IDS [13]. In many cases, unglycosylated precursors of lysosomal enzymes, such as that of sulfatases, are misfolded and degraded in the endoplasmic reticulum [14]. Thus, we have predicted the N-glycosylation pattern of all human sulfatases using a bio-informatic approach (Table 1). This data can be useful for further studies on the function of the glycosylation content of human sulfatases.
In addition to controlling correct protein folding, the biological function of glycosylation is also to regulate sulfatase subcellular targeting. A key step in the cellular targeting of newly synthesized sulfatases is the formation of phosphomannosyl residues on the high mannose oligosaccharide chains. The transfer of GlcNac-phosphate to mannose occurs in the intermediate compartment of the secretory pathway, while a second mannose phosphorylation step occurs in the cis-Golgi, after the removal of the terminal mannose residue [15]. The sorting of lysosomal enzymes, such as that of sulfatases, occurs via the mannose-6-phosphate receptor (M6PR), which recycles from the trans-Golgi network (TGN) to lysosomes and the plasma membrane [16, 17]. Part of the newly synthesized sulfatases escapes the saturable M6PR intracellular targeting and is secreted.
Upon secretion, lysosomal sulfatases, such as ARSA, ARSB, GALNS, GNS, IDS, and SGSH, can be re-internalized following different retrograde transport pathways via two different M6PRs. These are M6PR 300 or cation independent (CI-MPR) and M6PR 46 or cation dependent (CD-MPR), which are 300 and 46 kDa, respectively; they are localized in the cis or TGN, endosomal and plasma membranes [18, 19] and are post-translationally modified through glycosylation, phosphorylation, and disulfide bond formation [20].
Immunodepletion of M6PR 46 does not increase lysosomal enzyme secretion, suggesting a compensatory role for M6PR 300 [21]. In contrast, M6PR-300-deficient cells showed a 30–40% increase in lysosomal enzyme retention [22–24]. This suggests that M6PR 300 can efficiently localize and re-internalize lysosomal proteins, while even if M6PR 46 recycles from the intracellular membranes to the plasma membrane, it has a minor role controlling endocytosis of lysosomal enzymes probably because of the slightly acidic pH required for its efficient ligand binding [24, 25].
We still do not know why sulfatases can be secreted and re-internalized. This trafficking might control or tune modifications of extracellular compounds; it might contribute to lysosome function and homeostasis; it might control sulfatase recruitment to specific tissues or organs that over-express M6PRs on the cell surface membrane upon different stimuli; or it might even rescue loss of function somatic mutations of specific sulfatases.
The extracellular sulfatases Sulf1 and Sulf2 control development and cell signaling by remodeling the heparan sulfate proteoglycans
Sulfated oligosaccharides are synthesized by sulfotransferases, glycosyltransferases, and related enzymes. The sulfate group is transferred from 3′-phosphoadenosine 5′-phosphosulfate to precursor oligosaccharides in the lumen of the Golgi apparatus by Golgi-associated sulfotransferases [26]. The sulfate group represents a unique structural feature of carbohydrates bound to proteins and lipids, and it can confer cell-, tissue- and organ-specific functions. Important advances in different biological fields have demonstrated that concerted sulfotransferase/sulfatase activities are not only responsible for modulation of well-tuned catabolic pathways, but are also relevant in different cellular processes, such as neuromodulation, immunomodulation/autoimmunity, and development [27].
Indeed, sulfotransferases produce extensively sulfated glycosaminoglycans (GAGs) by creating a cell-, tissue- and organ-specific code that allows modulation of multiple processes. Glycosaminoglycans are carbohydrate polymers, consisting of repeating disaccharide units, with an amino sugar, either N-acetyl glucosamine (GlcNAc) or N-acetyl galactosamine (GalNAc), and a uronic acid (Glucuronic acid or Iduronic acid) [27]. At least one glycosaminoglycan side chain is covalently attached to the protein core thereby forming complex macromolecules called proteoglycans. Different sulfation patterns produce different proteoglycan types, such as chondroitin sulfate, dermatan sulfate, heparan sulfate (HS), and keratan sulfate [28].
In particular, the heparan sulfate proteoglycans (HSPGs) have a key role in the extracellular matrix, which is a reservoir of growth factors and cytokines such as Wnts, fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), Noggin, Sonic hedgehog (Shh) and vascular endothelial growth factors (VEGFs). All these morphogens are stored at the interface between the plasma membrane and the extracellualr matrix and are associated to the HS chains of the HSPGs [29, 30]. Upon specific signals that produce low binding affinities with the HSPGs, the morphogens can interact with their coupled receptors [28].
Biosynthetic modifications within the HSs of HSPGs are therefore implicated in the strength and outcome of HS–ligand interactions. The identification of mutations in enzymes involved in HS chain biosynthesis in Drosophila and mice have shown the critical roles of HSPGs in developmental processes and specific signaling pathways. Mutations in the homologue of UDP-D-glucose dehydrogenase [31, 32] or in N-deacetylase/N-sulphotransferase [33] in Drosophila, result in the severe impairment of Wnt, FGFs and Shh signaling pathways. Drosophila mutants in the pipe gene, which encodes a putative 2-O-sulphotransferase, show defects in the establishment of dorsal–ventral (D–V) polarity in the embryo [34]. The abrogation of the murine homologous of the pipe gene, named Hs2st, produce developmental failure of embryos after mid-gestation [35].
Sulfatases 1 and 2 (Sulf1, 2) are “special” sulfatases, in that by cleaving the sulfate from the HSPG chains, they translate the sulfated information into a biological event [36–39]. They are associated exclusively to the external portion of the plasma membrane where they remodel 6-O sulfation of HS chains to promote signaling. Both Sulf1 and Sulf2 sulfatases exert the same substrate specificity toward HSPGs S-domain formed by UA-GlcNS(6S) and UA(2S)-GlcNS(6S) units [40, 41].
6-O-glucosamine sulfate removal by Sulf1 and Sulf2 produces low-affinity binding between growth factors (such as Wnts, FGFs, BMPs, Noggin) and HS, allowing growth factor interactions with their cognate receptors and modulation of signaling pathways [42–44]. The latter include activation of the canonical Wnt/β-catenin pathway [36], destabilization of the trimeric complex 2HSPGs:2FGFs:2FGF receptor, with the consequent reduction in FGF signaling [38] and modulation of the BMP/Noggin pathway [39]. Sulf1 and Sulf2 activities modulate developmental and homeostatic processes, although to date, no genetic disorders have been correlated to Sulf1 or Sulf2 gene mutations.
Sulf1 and Sulf2 are involved in a variety of processes, such as neuroanatomical development, synaptic plasticity, satellite-cell differentiation, muscle regeneration, and tumor growth [45, 46]. Single and double Sulf knockout mutant mice were generated. Sulf1/Sulf2 double knockout mice showed significant developmental flaws and high neonatal lethality associated with reduced body weight and multiple developmental defects, including skeletal and renal abnormalities [47, 48]. In addition, Sulf1/Sulf2 double knockout mice display comparable developmental defects with the Hs2st null mice [35].
Metabolism and cell homeostasis is controlled by lysosomal sulfatases
The lysosomal sulfatases, including ARSA, ARSB, GALNS, GNS, SGSH, and IDS sulfatases, are ubiquitous housekeeping enzymes which are involved in the catabolic and sequential degradation steps of several substrates. These include cerebroside-3-sulfate, chondroitin/keratan/dermatan, and heparan sulfate compounds [1, 49]. Lysosomal sulfatases can recognize specific sugar–sulfate epitopes, and their catalytic action allows the accessibility, by other lysosomal degrading enzymes such as glycosidases, of specific regions within complex lipids and proteoglycan macromolecules.
ARSA hydrolyzes a number of sulfated substrates. The major physiological substrates are sulfatides, and specifically sphingolipids with galactose-3S (Gal-3S) head groups [50, 51]. Furthermore, it has been suggested that ascorbic acid-2S is another natural substrate [52]. Mutations in the ARSA gene have been associated with the inherited genetic disorder metachromatic leukodystrophy, which is characterized by extensive neuron demyelination in the nervous system. This disorder results from improper catabolism of the sulfatide cerebroside-3S, which is one of the major structural components of the myelin sheath [53–55]. Additional studies indicate that ARSA may also have a role in non-lysosomal environments. It has been detected on sperm surfaces, where it is thought to adhere to sulfated egg glycoproteins and modulate steps in the fertilization process [56].
The other lysosomal sulfatases have different substrate specificities. ARSB specifically cleaves sulfate esters at the 4S position of GalNAc, which are residues in dermatan sulfate and chondroitin sulfate [57–59]; SGSH catalyzes the hydrolysis of N-linked sulfamates of glucosamine residues in heparin and heparan sulfate [60]; GALNS is responsible for the hydrolysis of galactose-N6S (GalN6S) residues of keratan sulfate [61, 62]; GNS removes the sulfates from glucose-N6S residues in the heparan and keratan sulfate chains [63, 64]; and IDS removes the sulfate group from the 2O position of l-iduronic acid in the heparan and dermatan sulfate GAGs [62, 65]. In addition, all sulfatases can cleave a variety of synthetic substrates (Table 1). Mutations in the genes encoding for these lysosomal sulfatases results in mucopolysaccharidosis (MPS), a group of lysosomal storage disorders that arise from deficiencies in GAG catabolism [7] (Table 2).
Table 2.
Lysosomal sulfatase genes and their chromosomal localization. Mutations in each one of these genes causes the reported types of mucopolysaccharidosis (MPS) in humans
| Sulfatase | Chromosomal localization | Associated genetic disorder |
|---|---|---|
| Arylsulfatase B (ARSB) | 5q11-q13 | Maroteaux-Lamy Syndrome (MPSVI) |
| Galactosamine6-Sulfatase (GALNS) | 16q24.3 | Morquio A Syndrome (MPSIVA) |
| Glucosamine6-Sulfatase (GNS) | 12q14 | Sanfilippo D Syndrome (MPSIIID) |
| HeparanN-Sulfatase (SGSH) | 17q25.3 | Sanfilippo A Syndrome (MPSIIIA) |
| Iduronidate2-Sulfatase (IDS) | Xq28 | Hunter Syndrome (MPSII) |
Clinically, MPS disorders are associated with progressive disease phenotypes involving multiple organs and tissues, including severe inflammation and neurodegeneration. The accumulation of non-catabolized substrates severely impairs lysosomal function, resulting in defects in protein, glycoconjugate, lipid, nucleic acid, and phosphate metabolism, with the consequent perturbation of cell homeostasis [66].
A block of autophagy, which results in defective fusion between lysosomes and autophagosomes, occurs in many cases of mucopolysaccharidosis [67]. The mechanisms by which autophagosome–lysosome fusion is impaired in lysosomal storage disease remain to be clarified, although they could be based upon microtubule-based transport deficiencies or changes in the lipid composition of lysosomal membranes, resulting in an impairment of vescicular trafficking [68]. In addition, alterations in the lysosome lipid composition that can occur in these diseases could produce lysosomal membrane permeabilization and cathepsin release. Cathepsins have been shown to induce the activation or mitochondrial release of pro-apoptotic factors that mediate cell death programming [69, 70]. Sulf1 and Sulf2 can connect lysosomal function and cell signaling. Indeed, the unique ability of Sulf1 and Sulf2 to down-regulate FGF signaling could in fact decrease p38 phosphorylation and pro-apoptotic factors activation [71]; to this purpose, it would be interesting to generate a mucopolysaccharidosis (MPS) cell line over-expressing Sulf1 and Sulf2, to demonstrate block of apoptosis. Interestingly, not only the lysosomal sulfatases were associated with devasting genetic disorders, but also ARSC and ARSE, which are localized in the microsomes and Golgi, respectively.
ARSC, or steroid sulfatase (STS), is a ubiquitous enzyme detectable in different tissues and with a distribution varying across the different mammals. It hydrolyses aryl and alkyl [dehydroepiandrosterone (DHEA) sulfate (DHEAS)] steroid sulfates (estrone sulfate) [72]. STS mRNA level and enzyme activity were found to be increased in 74% of breast carcinoma cells [73]. STS-dependent estrogen desulfation activity produces, in fact, high levels of biologically active compounds that are able to bind the estrogen receptor increasing the incidence of malignant transformation [74]. In addition, STS deficiency has been associated with the X-linked Ichthyosis, a human genetic skin disorder that affects males [75]. Lipids have an important role in the formation of the skin corneum stratum structure, and alterations in cholesterol sulfate processing or quantity due to unefficient ARSC activity, produce the scaling of the skin, particularly on the neck, trunk, and lower extremities [72, 76].
Another X-linked human genetic disorder is the chondrodysplasia punctata (CDPX), which is due to mutations in ARSE. Arylsulfatase E is a Golgi localized enzyme with unknown physiologic substrates. CDPX is a rare genetic disorder characterized by skin and hair alterations, and causes short stature with skeletal abnormalities, cataracts, and deafness. Skeletal dysplasias is characterized by abnormal calcium deposition in regions of enchondral bone formation [77].
SUMF1: structure, subcellular localization and function
All sulfatases need to be activated by SUMF1/FGE. The SUMF1 gene was discovered by two different and complementary approaches. Biochemical purification of the formylglycine-generating enzyme (FGE) from the soluble fraction (reticuloplasm) of bovine testis microsomes was carried out through its ability to bind to and modify the Cys within a 23-mer peptide from ARSA. The isolated protein was subjected to sequence analysis and the cDNA cloned [78]. We used a genetic approach that was coupled to complementation functional assays. By microcell-mediated chromosome transfer and irradiation microcell-mediated chromosome transfer, we first identified chromosome 3 as the complementing chromosome, then a 2.6-Mb section of chromosome 3 as the minimal complementing region. By direct sequencing of all of the genes within this minimal region using genomic DNA from a collection of multiple sulfatase deficiency (MSD) patient cell lines, we identified an unknown open-reading frame that we called SUMF1 [79]. The gene is located at 3p26 and encodes the FGE. This protein is highly conserved during evolution, from bacteria to mammalians. Co-expression of SUMF1 with a single sulfatase cDNA strongly increased the sulfatase activity, demonstrating that SUMF1 is an essential and limiting factor for the activity of sulfatases [79].
Based on crystal structure studies, Dierks and co-workers demonstrated that FGE functions as an oxygenase, using oxygen to generate FGly via the cysteine sulfenic acid intermediate. This catalytic mechanism was elucidated from six high resolution structures under different redox conditions [80].
SUMF1 is a glycoprotein that is present into the ER and in the secretory pathway (Golgi, lysosomes, endosomes), and it can be secreted into the extracellular space [81, 82]. It contains a single N-linked glysosylation site at asparagine 141, and the intracellular SUMF1 forms contain high mannose carbohydrates, comprising 4–9 hexoses attached to two N-acetylhexosamine residues. The secreted forms contain a family of complex type oligosaccharides, with a core carbohydrate consisting of 2× N-acetylhexosamine, 3× hexose (mannose), and 1× deoxyesose (fucose) residues that carry up to 3× lactosamine, 2× sialic acid and 1× deoxyesose residues [81]. The heterogeneity of these intracellular and extracellular SUMF1 forms are probably related to different and yet unknown biological activities.
The SUMF1 N141A mutant and Xenopus SUMF1 encode for unglycosylated forms of the SUMF1 protein. Upon over-expression, they correctly localize in the ER, but they have low enhancing activities for co-expressed sulfatases. The absence of glycosylation could produce aberrant protein folding or a reduction in the binding affinity for SUMF1 to sulfatases. Nevertheless, the mutant proteins can be secreted into the culture media [82].
The balance between ER retention and secretion of SUMF1 is a finely tuned process that is regulated by SUMF1 protein interactors [83]. Protein disulfide isomerase (PDI) is a redox-dependent chaperone that mediates the formation, isomerization, and reduction of numerous substrates [84], and can interact with SUMF1 [83]. By changing the levels of PDI in the cells, it is possible to modulate the ER retention of SUMF1 versus its secretion. SUMF1 also interacts with ER-Golgi intermediate compartment protein ERGIC-53 [85, 86] via its carbohydrate-recognition domain (CRD). This interaction promotes the trafficking of SUMF1 through the secretory pathway [83]. Finally, SUMF1 can bind to the endoplasmic reticulum protein (ERp)44 [87–90] that mediates ER-retrieval transport of SUMF1 [83, 91]. Overall, a fine-tuned multistep mechanism controls SUMF1 retention and secretion [83].
After secretion, SUMF1 can be taken up in distant cells and tissues. It is retrogradely re-internalized via a receptor-mediated mechanism as an active enzyme into the ER. Interestingly, unglycosylated SUMF1 protein, SUMF1 N141A mutant, and the Xenopus SUMF1 retain the ability to be secreted into the medium, but they cannot be taken up from the extracellular space, suggesting that the sugar content is involved in SUMF1 re-internalization [82]. ER targeting from the extracellular space has been otherwise shown only for a few toxins, such as Shiga toxin and ricin. Further studies will be necessary to dissect out the exact route that allows SUMF1 retrograde transport into the ER.
Sumf1-/- mice have shown that mammals have a single sulfatase modification system. The Sumf1 null mice displayed a drastic reduction of their life span, growth retardation, skeletal abnormalities, and neurological defects due to the lysosomal storage, but also developmental failure, strictly associated to the Sulfs activity abrogation. The overall phenotype contribute to the trigger of a generalized inflammatory process [92].
Interestingly, Sumf1-/- mice display a severe failure of hematopoietic differentiation, that is due to the increase of FGF signaling, to the activation of the Wnt/β-catenin and Notch pathways. This phenotype is specifically due to the loss of function of Sulf1 and Sulf2 in Sumf1-/- mice (Buono et al., unpublished results). Likewise, alterations in the proteoglycan desulfation process, due to the inactivity of Sulf1 and Sulf2 sulfatases in Sumf1-/- mice affect several aspects of chondrocyte biology, such as proliferation and differentiation during skeletal development, through modulation of growth factor signaling and also by influencing lysosomal function. Of note, the Sumf1-/- mice lacking one copy of Fgf18 show a restoration of chondrocyte viability and function demonstrating that these defects were due to the constitutive activation of FGF signaling pathway controlled by Sulf1/Sulf2 sulfatases [93].
Clinical perspectives and therapies
Mutations in sulfatase genes produce metabolic and developmental syndromes that involve multiple tissues and organs. A large amount of data has been produced over the last few years, which has led to clarification of the actions of each individual sulfatase and of their activator SUMF1, including their localization, structure, and function. However, ARSH, ARSI, ARSG, and ARSK are novel sulfatases that were identified through bioinformatics analysis, and they remain to be characterized. The cloning and expression of these genes will allow a full understanding of this large protein family.
Not all the sulfatase substrates have been identified. The isolation of an enzyme–substrate complex is not an easy task, due the rapid kinetics of the conversion mechanism. Site-specific mutagenesis of the required Cys residue, so it cannot be converted into FGly, might still preserve the sulfatase ability to bind its substrate, allowing the formation of a stable enzyme–substrate complex. The latter could thus be immunoprecipitated and analyzed by mass spectrometry.
Very little is known about the physiological roles of the sulfatase glycosylation, secretion, and re-internalization routes (in particular for lysosomal sulfatases). However, the discovery of the retrieval route for lysosomal sulfatases and for SUMF1, and the observation that these enzymes retain catalytic activity upon re-internalization, form the basis for enzyme replacement therapies. These make use of glycosylated recombinant sulfatases that are injected directly into the blood to restore the disease defects. Other approaches utilize adeno-associated virus (AVV)- and lentiviral (LV)-mediated gene delivery to obtain systemic expression, or an “organ factory”, that over-secretes the protein into the blood, allowing it to reach all the non-transduced organs. Systemic delivery of AVV2/8-TBG-hIDS vectors leads to the engineering of the liver of a Hunter mouse model resulting in the production and secretion of IDS into the blood. From the blood stream, IDS can be taken up into all non-transduced tissues, therefore resulting in the complete rescue of the pathological phenotype in all the visceral organs of the mice [94]. In addition, we recently showed that a single administration of the AAV2/5CMV-hIDS vector in MPSII mouse pups resulted in high plasmatic levels of hIDS that were sufficient to rescue central nervous system (CNS) and visceral phenotype up to 18 months after therapy. Interestingly, in the treated MPSII animals, this CNS correction arises from the crossing of the blood–brain barrier by the IDS enzyme itself, and not from the brain transduction [95].
Delivery of AVV2/8 TBG ARSB was tested for the treatment of the Maroteaux–Lamy Syndrome (MPSVI) in rats. After systemic or intramuscular administration of AAV, therapeutic levels of circulating ARSB are achieved, resulting in skeletal improvements and significant decrease in GAG storage, inflammation, and apoptosis [96].
Autologous transplantation of hematopoietic stem and progenitor cells transduced with LV-Pgk-ARSA produced over-physiological plasma levels of ARSA that is able to reach multiple organs, providing correction of the CNS symptoms in the metachromatic leucodystrophy mouse model [97].
Since SUMF1 can activate the sulfatases, the co-delivery of SUMF1 with the exogenous sulfatase might also produce a more efficient rescue of the phenotype [98]. The intracerebral delivery of sulfamidase and SUMF1 genes (via the use of the AAV2/5-CMV-SGSH-IRES-SUMF1 vectors) has also provided functional correction of CNS lesions in an MPS-IIIA mouse model [99].
Interestingly, small medical compounds (steroidal and nonsteroidal sulfamates) were recently used to inhibit ARSC. The treatment with 667COUMATE, an irreversible ARSC inhibitor, in postmenopausal women with breast cancer demonstrated that low doses of the drug can efficiently inhibit the enzyme (more than 90%) in peripheral blood. The use of this inhibitor and of drug derivatives with higher in vivo-stability or enhanced efficacy could improve the treatment of malignant transformation [72]. Finally, an in vivo-model to develop gene therapy for X-linked ichthyosis has also been developed. X-linked ichthyosis primary keratinocytes from patients were transduced with retroviruses carrying the ARSC gene and then were grafted onto immunodeficient mice. Transduced keratinocytes, likewise normal keratinocytes grafted as control, were able to generate human epidermis with normal ARSC expression and with the normalization of the barrier function parameters [100].
Studies of the glycosylation content of the sulfatases could also lead to the design of sulfatases carrying modified sugars that might enhance their activity and re-internalization. The engineered sulfatases might reach tissues and organs showing poor uptake capacity, for example, a more efficient crossing of the blood–brain barrier for the treatment of CNS symptoms might be achieved. In conclusion, the study of the cellular trafficking of the sulfatases could be important to develop therapies for a large family of devastating diseases, as the lysosomal storage disorders.
Acknowledgments
The authors thank G. Diez-Roux and D. Di Bernardo for critical reading of the manuscript.
References
- 1.Diez-Roux G, Ballabio A. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- 2.Sardiello M, Annunziata I, Roma G, Ballabio A. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum Mol Genet. 2005;14:3203–3217. doi: 10.1093/hmg/ddi351. [DOI] [PubMed] [Google Scholar]
- 3.Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999;18:2084–2091. doi: 10.1093/emboj/18.8.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landgrebe J, Dierks T, Schmidt B, von Figura K. The human SUMF1 gene, required for posttranslational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes. Gene. 2003;316:47–56. doi: 10.1016/S0378-1119(03)00746-7. [DOI] [PubMed] [Google Scholar]
- 5.Recksiek M, Selmer T, Dierks T, Schmidt B, von Figura K. Sulfatases, trapping of the sulfated enzyme intermediate by substituting the active site formylglycine. J Biol Chem. 1998;273:6096–6103. doi: 10.1074/jbc.273.11.6096. [DOI] [PubMed] [Google Scholar]
- 6.Waldow A, Schmidt B, Dierks T, von Bulow R, von Figura K. Amino acid residues forming the active site of arylsulfatase A. Role in catalytic activity and substrate binding. J Biol Chem. 1999;274:12284–12288. doi: 10.1074/jbc.274.18.12284. [DOI] [PubMed] [Google Scholar]
- 7.Hanson SR, Best MD, Wong CH. Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Int Ed Engl. 2004;43:5736–5763. doi: 10.1002/anie.200300632. [DOI] [PubMed] [Google Scholar]
- 8.Lukatela G, Krauss N, Theis K, Selmer T, Gieselmann V, von Figura K, Saenger W. Crystal structure of human arylsulfatase A: the aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry. 1998;37:3654–3664. doi: 10.1021/bi9714924. [DOI] [PubMed] [Google Scholar]
- 9.Stein C, Hille A, Seidel J, Rijnbout S, Waheed A, Schmidt B, Geuze H, von Figura K. Cloning and expression of human steroid-sulfatase. J Biol Chem. 1989;264:13865–13872. [PubMed] [Google Scholar]
- 10.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 11.Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell. 1994;5:253–265. doi: 10.1091/mbc.5.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kornfeld S. Diseases of abnormal protein glycosylation: an emerging area. J Clin Invest. 1998;101:1293–1295. doi: 10.1172/JCI3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millat G, Froissart R, Maire I, Bozon D. IDS transfer from overexpressing cells to IDS-deficient cells. Exp Cell Res. 1997;230:362–367. doi: 10.1006/excr.1996.3435. [DOI] [PubMed] [Google Scholar]
- 14.Tikkanen R, Enomaa N, Riikonen A, Ikonen E, Peltonen L. Intracellular sorting of aspartylglucosaminidase: the role of N-linked oligosaccharides and evidence of Man-6-P-independent lysosomal targeting. DNA Cell Biol. 1995;14:305–312. doi: 10.1089/dna.1995.14.305. [DOI] [PubMed] [Google Scholar]
- 15.Spiro RG. Role of N-linked polymannose oligosaccharides in targeting glycoproteins for endoplasmic reticulum-associated degradation. Cell Mol Life Sci. 2004;61:1025–1041. doi: 10.1007/s00018-004-4037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y, Doray B, Poussu A, Lehto VP, Kornfeld S. Binding of GGA2 to the lysosomal enzyme sorting motif of the mannose 6-phosphate receptor. Science. 2001;292:1716–1718. doi: 10.1126/science.1060896. [DOI] [PubMed] [Google Scholar]
- 17.Doray B, Ghosh P, Griffith J, Geuze HJ, Kornfeld S. Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science. 2002;297:1700–1703. doi: 10.1126/science.1075327. [DOI] [PubMed] [Google Scholar]
- 18.Brown WJ, Farquhar MG. The mannose-6-phosphate receptor for lysosomal enzymes is concentrated in cis Golgi cisternae. Cell. 1984;36:295–307. doi: 10.1016/0092-8674(84)90223-X. [DOI] [PubMed] [Google Scholar]
- 19.Brown WJ, Farquhar MG. The distribution of 215-kilodalton mannose 6-phosphate receptors within cis (heavy) and trans (light) Golgi subfractions varies in different cell types. Proc Natl Acad Sci USA. 1987;84:9001–9005. doi: 10.1073/pnas.84.24.9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahagian GG, Neufeld EF. Biosynthesis and turnover of the mannose 6-phosphate receptor in cultured Chinese hamster ovary cells. J Biol Chem. 1983;258:7121–7128. [PubMed] [Google Scholar]
- 21.Koster A, Saftig P, Matzner U, von Figura K, Peters C, Pohlmann R. Targeted disruption of the M(r) 46,000 mannose 6-phosphate receptor gene in mice results in misrouting of lysosomal proteins. EMBO J. 1993;12:5219–5223. doi: 10.1002/j.1460-2075.1993.tb06217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabel CA, Goldberg DE, Kornfeld S. Identification and characterization of cells deficient in the mannose 6-phosphate receptor: evidence for an alternate pathway for lysosomal enzyme targeting. Proc Natl Acad Sci USA. 1983;80:775–779. doi: 10.1073/pnas.80.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nolan CM, Creek KE, Grubb JH, Sly WS. Antibody to the phosphomannosyl receptor inhibits recycling of receptor in fibroblasts. J Cell Biochem. 1987;35:137–151. doi: 10.1002/jcb.240350207. [DOI] [PubMed] [Google Scholar]
- 24.Stein M, Zijderhand-Bleekemolen JE, Geuze H, Hasilik A, von Figura K. Mr 46,000 mannose 6-phosphate specific receptor: its role in targeting of lysosomal enzymes. EMBO J. 1987;6:2677–2681. doi: 10.1002/j.1460-2075.1987.tb02559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahms NM, Lobel P, Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem. 1989;264:12115–12118. [PubMed] [Google Scholar]
- 26.Abeijon C, Mandon EC, Hirschberg CB. Transporters of nucleotide sugars, nucleotide sulfate and ATP in the Golgi apparatus. Trends Biochem Sci. 1997;22:203–207. doi: 10.1016/S0968-0004(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 27.Honke K, Taniguchi N. Sulfotransferases and sulfated oligosaccharides. Med Res Rev. 2002;22:637–654. doi: 10.1002/med.10020. [DOI] [PubMed] [Google Scholar]
- 28.Habuchi H, Habuchi O, Kimata K. Sulfation pattern in glycosaminoglycan: does it have a code? Glycoconj J. 2004;21:47–52. doi: 10.1023/B:GLYC.0000043747.87325.5e. [DOI] [PubMed] [Google Scholar]
- 29.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 31.Binari RC, Staveley BE, Johnson WA, Godavarti R, Sasisekharan R, Manoukian AS. Genetic evidence that heparin-like glycosaminoglycans are involved in wingless signaling. Development. 1997;124:2623–2632. doi: 10.1242/dev.124.13.2623. [DOI] [PubMed] [Google Scholar]
- 32.Haerry TE, Heslip TR, Marsh JL, O’Connor MB. Defects in glucuronate biosynthesis disrupt Wingless signaling in Drosophila . Development. 1997;124:3055–3064. doi: 10.1242/dev.124.16.3055. [DOI] [PubMed] [Google Scholar]
- 33.Lin X, Perrimon N. Dally cooperates with Drosophila Frizzled 2 to transduce Wingless signalling. Nature. 1999;400:281–284. doi: 10.1038/22343. [DOI] [PubMed] [Google Scholar]
- 34.Sen J, Goltz JS, Stevens L, Stein D. Spatially restricted expression of pipe in the Drosophila egg chamber defines embryonic dorsal–ventral polarity. Cell. 1998;95:471–481. doi: 10.1016/S0092-8674(00)81615-3. [DOI] [PubMed] [Google Scholar]
- 35.Merry CL, Bullock SL, Swan DC, Backen AC, Lyon M, Beddington RS, Wilson VA, Gallagher JT. The molecular phenotype of heparan sulfate in the Hs2st-/- mutant mouse. J Biol Chem. 2001;276:35429–35434. doi: 10.1074/jbc.M100379200. [DOI] [PubMed] [Google Scholar]
- 36.Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U, Emerson CP., Jr QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP., Jr Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001;293:1663–1666. doi: 10.1126/science.293.5535.1663. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Ai X, Freeman SD, Pownall ME, Lu Q, Kessler DS, Emerson CP., Jr QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci USA. 2004;101:4833–4838. doi: 10.1073/pnas.0401028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Viviano BL, Paine-Saunders S, Gasiunas N, Gallagher J, Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J Biol Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- 40.Ai X, Do AT, Kusche-Gullberg M, Lindahl U, Lu K, Emerson CP., Jr Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J Biol Chem. 2006;281:4969–4976. doi: 10.1074/jbc.M511902200. [DOI] [PubMed] [Google Scholar]
- 41.Lamanna WC, Frese MA, Balleininger M, Dierks T. Sulf loss influences N-, 2-O-, and 6-O-sulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J Biol Chem. 2008;283:27724–27735. doi: 10.1074/jbc.M802130200. [DOI] [PubMed] [Google Scholar]
- 42.Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatase in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamanna WC, Baldwin RJ, Padva M, Kalus I, Ten Dam G, van Kuppevelt TH, Gallagher JT, von Figura K, Dierks T, Merry CL. Heparan sulfate 6-O-endosulfatases: discrete in vivo activities and functional co-operativity. Biochem J. 2006;400:63–73. doi: 10.1042/BJ20060848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. The heparanome—the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol. 2007;129:290–307. doi: 10.1016/j.jbiotec.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 45.Langsdorf A, Do AT, Kusche-Gullberg M, Emerson CP, Jr, Ai X. Sulfs are regulators of growth factor signaling for satellite cell differentiation and muscle regeneration. Dev Biol. 2007;311:464–477. doi: 10.1016/j.ydbio.2007.08.053. [DOI] [PubMed] [Google Scholar]
- 46.Dai Y, Yang Y, MacLeod V, Yue X, Rapraeger AC, Shriver Z, Venkataraman G, Sasisekharan R, Sanderson RD. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem. 2005;280:40066–40073. doi: 10.1074/jbc.M508136200. [DOI] [PubMed] [Google Scholar]
- 47.Holst CR, Bou-Reslan H, Gore BB, Wong K, Grant D, Chalasani S, Carano RA, Frantz GD, Tessier-Lavigne M, Bolon B, French DM, Ashkenazi A. Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. PLoS ONE. 2007;2:e575. doi: 10.1371/journal.pone.0000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ratzka A, Kalus I, Moser M, Dierks T, Mundlos S, Vortkamp A. Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Dev Dyn. 2008;237:339–353. doi: 10.1002/dvdy.21423. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh D. Human sulfatases: a structural perspective to catalysis. Cell Mol Life Sci. 2007;64:2013–2022. doi: 10.1007/s00018-007-7175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jatzkewitz H, Mehl E. Cerebroside-sulphatase and arylsulphatase A deficiency in metachromatic leukodystrophy (ML) J Neurochem. 1969;16:19–28. doi: 10.1111/j.1471-4159.1969.tb10339.x. [DOI] [PubMed] [Google Scholar]
- 51.Mehl E, Jatzkewitz H. Cerebroside 3-sulfate as a physiological substrate of arylsulfatase A. Biochim Biophys Acta. 1968;151:619–627. doi: 10.1016/0005-2744(68)90008-9. [DOI] [PubMed] [Google Scholar]
- 52.Roy AB. l-ascorbic acid 2-sulphate. A substrate for mammalian arylsulphatases. Biochim Biophys Acta. 1975;377:356–363. doi: 10.1016/0005-2744(75)90316-2. [DOI] [PubMed] [Google Scholar]
- 53.Farooqui AA, Mandel P. Recent developments in the biochemistry of globoid and metachromatic leucodystrophies. Biomedicine. 1977;26:232–236. [PubMed] [Google Scholar]
- 54.Fluharty AL, Stevens RL, Goldstein EB, Kihara H. The activity of arylsulfatase A and B on tyrosine O-sulfates. Biochim Biophys Acta. 1979;566:321–326. doi: 10.1016/0005-2744(79)90035-4. [DOI] [PubMed] [Google Scholar]
- 55.Louis AI, Fluharty AL. Activator-dependent hydrolysis of myelin cerebroside sulfate by arylsulfatase A. Dev Neurosci. 1991;13:41–46. doi: 10.1159/000112139. [DOI] [PubMed] [Google Scholar]
- 56.Tantibhedhyangkul J, Weerachatyanukul W, Carmona E, Xu H, Anupriwan A, Michaud D, Tanphaichitr N. Role of sperm surface arylsulfatase A in mouse sperm–zona pellucida binding. Biol Reprod. 2002;67:212–219. doi: 10.1095/biolreprod67.1.212. [DOI] [PubMed] [Google Scholar]
- 57.Matalon R, Arbogast B, Dorfman A. Deficiency of chondroitin sulfate N-acetylgalactosamine 4-sulfate sulfatase in Maroteaux-Lamy syndrome. Biochem Biophys Res Commun. 1974;61:1450–1457. doi: 10.1016/S0006-291X(74)80446-8. [DOI] [PubMed] [Google Scholar]
- 58.Matalon R, Arbogast B, Justice P, Brandt IK, Dorfman A. Morquio’s syndrome: deficiency of a chondroitin sulfate N-acetylhexosamine sulfate sulfatase. Biochem Biophys Res Commun. 1974;61:759–765. doi: 10.1016/0006-291X(74)91022-5. [DOI] [PubMed] [Google Scholar]
- 59.O’Brien JF, Cantz M, Spranger J. Maroteaux-Lamy disease (mucopolysaccharidosis VI), subtype A: deficiency of a N-acetylgalactosamine-4-sulfatase. Biochem Biophys Res Commun. 1974;60:1170–1177. doi: 10.1016/0006-291X(74)90435-5. [DOI] [PubMed] [Google Scholar]
- 60.Anson DS, Bielicki J. Sulphamidase. Int J Biochem Cell Biol. 1999;31:363–367. doi: 10.1016/S1357-2725(98)00148-4. [DOI] [PubMed] [Google Scholar]
- 61.Bielicki J, Hopwood JJ. Human liver N-acetylgalactosamine 6-sulphatase. Purification and characterization. Biochem J. 1991;279:515–520. doi: 10.1042/bj2790515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bielicki J, Fuller M, Guo XH, Morris CP, Hopewood JJ, Anson DS. Expression, purification and characterization of recombinant human N-acetylgalactosamine-6-sulphatase. Biochem J. 1995;311:333–339. doi: 10.1042/bj3110333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Litjens T, Bielicki J, Anson DS, Friderici K, Jones MZ, Hopwood JJ. Expression, purification and characterization of recombinant caprine N-acetylglucosamine-6-sulphatase. Biochem J. 1997;327:89–94. doi: 10.1042/bj3270089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gibson GJ, Saccone GT, Brooks DA, Clements PR, Hopwood JJ. Human N-acetylgalactosamine-4-sulphate sulphatase. Purification, monoclonal antibody production and native and subunit Mr values. Biochem J. 1987;248:755–764. doi: 10.1042/bj2480755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ginsberg LC, Di Ferrante DT, Di Ferrante N. A substrate for direct measurement of l-iduronic acid 2-sulfate sulfatase. Carbohydr Res. 1978;64:225–235. doi: 10.1016/S0008-6215(00)83703-9. [DOI] [PubMed] [Google Scholar]
- 66.Neufeld EFM. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1999. [Google Scholar]
- 67.Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 68.Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 69.Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 70.Erdal H, Berndtsson M, Castro J, Brunk U, Shoshan MC, Linder S. Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis. Proc Natl Acad Sci USA. 2005;102:192–197. doi: 10.1073/pnas.0408592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanaka K, Abe M, Sato Y. Roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the signal transduction of basic fibroblast growth factor in endothelial cells during angiogenesis. Jpn J Cancer Res. 1999;90:647–654. doi: 10.1111/j.1349-7006.1999.tb00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reed MJ, Purohit A, Woo LW, Newman SP, Potter BV. Steroid sulfatase: molecular biology, regulation, and inhibition. Endocr Rev. 2005;26:171–202. doi: 10.1210/er.2004-0003. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki T, Nakata T, Miki Y, Kaneko C, Moriya T, Ishida T, Akinaga S, Hirakawa H, Kimura M, Sasano H. Estrogen sulfotransferase and steroid sulfatase in human breast carcinoma. Cancer Res. 2003;63:2762–2770. [PubMed] [Google Scholar]
- 74.Pasqualini JR, Gelly C, Nguyen BL, Vella C. Importance of estrogen sulfates in breast cancer. J Steroid Biochem. 1989;34:155–163. doi: 10.1016/0022-4731(89)90077-0. [DOI] [PubMed] [Google Scholar]
- 75.Ballabio A, Parenti G, Tippett P, Mondello C, Di Maio S, Tenore A, Andria G. X-linked ichthyosis due to steroid sulphatase deficiency associated with Kallmann syndrome (hypogonadotropic hypogonadism and anosmia): linkage relationships with Xg and cloned DNA sequences from the distal short arm of the X chromosome. Hum Genet. 1986;72:237–240. doi: 10.1007/BF00291885. [DOI] [PubMed] [Google Scholar]
- 76.Richard G. Molecular genetics of the ichthyoses. Am J Med Genet C Semin Med Genet. 2004;131C:32–44. doi: 10.1002/ajmg.c.30032. [DOI] [PubMed] [Google Scholar]
- 77.Franco B, Meroni G, Parenti G, Levilliers J, Bernard L, Gebbia M, Cox L, Maroteaux P, Sheffield L, Rappold GA, Andria G, Petit C, Ballabio A. A cluster of sulfatase genes on Xp22.3: mutations in chondrodysplasia punctata (CDPX) and implications for warfarin embryopathy. Cell. 1995;81:15–25. doi: 10.1016/0092-8674(95)90367-4. [DOI] [PubMed] [Google Scholar]
- 78.Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell. 2003;113:435–444. doi: 10.1016/S0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 79.Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. doi: 10.1016/S0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 80.Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 81.Preusser-Kunze A, Mariappan M, Schmidt B, Gande SL, Mutenda K, Wenzel D, von Figura K, Dierks T. Molecular characterization of the human Calpha-formylglycine-generating enzyme. J Biol Chem. 2005;280:14900–14910. doi: 10.1074/jbc.M413383200. [DOI] [PubMed] [Google Scholar]
- 82.Zito E, Buono M, Pepe S, Settembre C, Annunziata I, Surace EM, Dierks T, Monti M, Cozzolino M, Pucci P, Ballabio A, Cosma MP. Sulfatase modifying factor 1 trafficking through the cells: from endoplasmic reticulum to the endoplasmic reticulum. EMBO J. 2007;26:2443–2453. doi: 10.1038/sj.emboj.7601695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fraldi A, Zito E, Annunziata F, Lombardi A, Cozzolino M, Monti M, Spampanato C, Ballabio A, Pucci P, Sitia R, Cosma MP. Multistep, sequential control of the trafficking and function of the multiple sulfatase deficiency gene product, SUMF1 by PDI, ERGIC-53 and ERp44. Hum Mol Genet. 2008;17:2610–2621. doi: 10.1093/hmg/ddn161. [DOI] [PubMed] [Google Scholar]
- 84.Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/S0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 85.Appenzeller C, Andersson H, Kappeler F, Hauri HP. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol. 1999;1:330–334. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- 86.Appenzeller-Herzog C, Roche AC, Nufer O, Hauri HP. pH-induced conversion of the transport lectin ERGIC-53 triggers glycoprotein release. J Biol Chem. 2004;279:12943–12950. doi: 10.1074/jbc.M313245200. [DOI] [PubMed] [Google Scholar]
- 87.Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, Sitia R. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 2002;21:835–844. doi: 10.1093/emboj/21.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gilchrist A, Au CE, Hiding J, Bell AW, Fernandez-Rodriguez J, Lesimple S, Nagaya H, Roy L, Gosline SJ, Hallett M, Paiement J, Kearney RE, Nilsson T, Bergeron JJ. Quantitative proteomics analysis of the secretory pathway. Cell. 2006;127:1265–1281. doi: 10.1016/j.cell.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 89.Anelli T, Ceppi S, Bergamelli L, Cortini M, Masciarelli S, Valetti C, Sitia R. Sequential steps and checkpoints in the early exocytic compartment during secretory IgM biogenesis. EMBO J. 2007;26:4177–4188. doi: 10.1038/sj.emboj.7601844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anelli T, Alessio M, Bachi A, Bergamelli L, Bertoli G, Camerini S, Mezghrani A, Ruffato E, Simmen T, Sitia R. Thiol-mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J. 2003;22:5015–5022. doi: 10.1093/emboj/cdg491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mariappan M, Radhakrishnan K, Dierks T, Schmidt B, von Figura K. ERp44 mediates a thiol-independent retention of formylglycine-generating enzyme in the endoplasmic reticulum. J Biol Chem. 2008;283:6375–6383. doi: 10.1074/jbc.M709171200. [DOI] [PubMed] [Google Scholar]
- 92.Settembre C, Annunziata I, Spampanato C, Zarcone D, Cobellis G, Nusco E, Zito E, Tacchetti C, Cosma MP, Ballabio A. Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc Natl Acad Sci USA. 2007;104:4506–4511. doi: 10.1073/pnas.0700382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Settembre C, Arteaga-Solis E, McKee MD, de Pablo R, Al Awqati Q, Ballabio A, Karsenty G. Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification. Genes Dev. 2008;22:2645–2650. doi: 10.1101/gad.1711308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cardone M, Polito VA, Pepe S, Mann L, D’Azzo A, Auricchio A, Ballabio A, Cosma MP. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet. 2006;15:1225–1236. doi: 10.1093/hmg/ddl038. [DOI] [PubMed] [Google Scholar]
- 95.Polito VA, Cosma MP. IDS crossing of the blood–brain barrier corrects CNS defects in MPSII mice. Am J Hum Genet. 2009;85:296–301. doi: 10.1016/j.ajhg.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tessitore A, Faella A, O’Malley T, Cotugno G, Doria M, Kunieda T, Matarese G, Haskins M, Auricchio A. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol Ther. 2008;16:30–37. doi: 10.1038/sj.mt.6300325. [DOI] [PubMed] [Google Scholar]
- 97.Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, Sessa M, Fasano S, Brambilla R, Marchesini S, Bordignon C, Naldini L. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–1129. doi: 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fraldi A, Biffi A, Lombardi A, Visigalli I, Pepe S, Settembre C, Nusco E, Auricchio A, Naldini L, Ballabio A, Cosma MP. SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem J. 2007;405:305–312. doi: 10.1042/BJ20061783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fraldi A, Hemsley K, Crawley A, Lombardi A, Lau A, Sutherland L, Auricchio A, Ballabio A, Hopwood JJ. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet. 2007;16:2693–2702. doi: 10.1093/hmg/ddm223. [DOI] [PubMed] [Google Scholar]
- 100.Freiberg RA, Choate KA, Deng H, Alperin ES, Shapiro LJ, Khavari PA. A model of corrective gene transfer in X-linked ichthyosis. Hum Mol Genet. 1997;6:927–933. doi: 10.1093/hmg/6.6.927. [DOI] [PubMed] [Google Scholar]
- 101.Hernandez-Guzman FG, Higashiyama T, Pangborn W, Osawa Y, Ghosh D. Structure of human estrone sulfatase suggests functional roles of membrane association. J Biol Chem. 2003;278:22989–22997. doi: 10.1074/jbc.M211497200. [DOI] [PubMed] [Google Scholar]
- 102.Bond CS, Clements PR, Ashby SJ, Collyer CA, Harrop SJ, Hopwood JJ, Guss JM. Structure of a human lysosomal sulfatase. Structure. 1997;5:277–289. doi: 10.1016/S0969-2126(97)00185-8. [DOI] [PubMed] [Google Scholar]