

Abstract
Cerebellar granule (CG) cells generate high-frequency action potentials that have been proposed to depend on the unique properties of their voltage-gated ion channels. To address the in vivo function of Nav1.6 channels in developing and mature CG cells, we combined the study of the developmental expression of Nav subunits with recording of acute cerebellar slices from young and adult granule-specific Scn8a KO mice. Nav1.2 accumulated rapidly at early-formed axon initial segments (AISs). In contrast, Nav1.6 was absent at early postnatal stages but accumulated at AISs of CG cells from P21 to P40. By P40–P65, both Nav1.6 and Nav1.2 co-localized at CG cell AISs. By comparing Na+ currents in mature CG cells (P66–P74) from wild-type and CG-specific Scn8a KO mice, we found that transient and resurgent Na+ currents were not modified in the absence of Nav1.6 whereas persistent Na+ current was strongly reduced. Action potentials in conditional Scn8a KO CG cells showed no alteration in threshold and overshoot, but had a faster repolarization phase and larger post-spike hyperpolarization. In addition, although Scn8a KO CG cells kept their ability to fire action potentials at very high frequency, they displayed increased interspike-interval variability and firing irregularity in response to sustained depolarization. We conclude that Nav1.6 channels at axon initial segments contribute to persistent Na+ current and ensure a high degree of temporal precision in repetitive firing of CG cells.
Introduction
Ten voltage-gated sodium (Nav) channel α subunits (Nav1.1–1.9, Nax) have been identified to date (Goldin, 2001). Each isoform has its own regional, cellular and developmental expression (Trimmer & Rhodes, 2004) but the reasons for this diversity are still largely unknown.
The Nav1.6 channel is highly expressed in the adult cerebellum, where it serves a specialized role in regulating the activity patterns of cerebellar neurons. In Purkinje cells lacking Nav1.6 channels both spontaneous firing activity and high-frequency discharge are impaired (Raman et al. 1997; Khaliq et al. 2003). Nav1.6 channels are particularly important because they are localized at the axon initial segments (AISs), where action potentials (APs) are initiated (Waxman & Quick, 1978; Kaplan et al. 2001; Boiko et al. 2001, 2003; Osorio et al. 2005; Lorincz & Nusser, 2008; Royeck et al. 2008; Hu et al. 2009). Consistently, mice with disrupted Nav1.6 expression exhibited neurological syndromes including ataxia, dystonia and juvenile lethality (Meisler et al. 2004; Meisler & Kearney, 2005), and cognition is impaired in human heterozygotes for a null allele of Nav1.6 (Trudeau et al. 2006), suggesting that Nav1.6 is crucial for impulse propagation at postnatal stages and later in life.
Collectively, these data indicate that Nav1.6 plays a critical role in faithfully transmitting high frequency firing, which could reflect the distinctive biophysical nature of Nav1.6 compared to other Nav isoforms. Particularly, Nav1.6 channels have been widely associated with the presence of resurgent Na+ currents, thereby providing a depolarizing drive following firing of single APs (Raman & Bean, 1997; Swensen & Bean, 2003). Another property that distinguishes Nav1.6 channels is their propensity to generate larger steady-state Na+ currents (Smith et al. 1998; Rush et al. 2005). These ‘persistent’ Na+ currents critically influence AP firing at just-threshold levels of stimulation (Crill, 1996) and are greatly reduced in Purkinje and cortical neurons from Nav1.6-null mice (Raman et al. 1997; Maurice et al. 2001).
Our previous study of rat cerebellar granule (CG) cells has shown that Nav1.2 precedes Nav1.6 at developing AISs in culture (Osorio et al. 2005). The functional benefit of this differential developmental pattern is still unclear, but it might be necessary for the appearance of the complex firing pattern of mature CG cells (D’Angelo et al. 2001; Isope & Barbour, 2002; Chadderton et al. 2004; Isope et al. 2004; Jörntell & Ekerot, 2006). Inactivation of Nav1.6 specifically in cerebellar granule cells resulted in impaired rotarod performance and reduction of evoked firing in Purkinje cells (Levin et al. 2006). However, it is not clear how Nav1.6 subunits contribute to the biophysically distinct Na+ currents of CG cells (Osorio et al. 2005; Magistretti et al. 2006) and set their intrinsic firing properties in vivo.
To test the selective contribution of Nav1.6 channels to CG cell firing properties, we studied the developmental expression pattern of Nav1.2 and Nav1.6 in situ. We found that developmental targeting of Nav1.6 subunits at AISs is complete only into adulthood, making the use of Nav1.6-null mice inappropriate, as they die prematurely. We therefore generated Scn8aflox/flox-Cre+ transgenic mice showing selective inactivation of Nav1.6 in CG cells. We show that Nav1.6 channels at AISs contribute to persistent but not resurgent Na+ currents, and regulate the firing regularity of granule neurons in adult cerebellum.
Methods
Ethical approval
Animal use followed the guidelines established by the European animal care and use committee (N. 86/609/CEE) and complied with The Journal's policies and regulations (Drummond, 2009). Mice older than P14 were anaesthetized with halothane or isoflurane and killed by decapitation. The total number of mice used in this study was 55.
Scn8a null mice, conditional Scn8a KO mice and genotyping
Scn8aflox/flox mice possess ‘floxed’ alleles of the Scn8a gene containing loxP sites flanking the first coding exon (Levin & Meisler, 2004). Scn8aflox/flox mice were crossed with BACα6-Cre transgenic mice, in which Cre expression is under the control of the granule-specific GABAA receptor α6 subunit promoter (Laurie et al. 1992; Aller et al. 2003). The loxP exon is deleted by exposure to Cre recombinase (Cre), resulting in a complete loss of expression of Nav1.6. In that manner, disrupted Nav1.6 expression is restricted to cerebellar granule cells in Scn8aflox/flox, Cre+ mice.
Genotyping used DNA extracted from mouse tails (Truett et al. 2000). Mouse tails were incubated for 20 min at 95°C in a dissociation solution consisting of 25 mm NaOH and 0.2 mm EDTA; the reaction was then stopped with 40 mm Tris-HCl. The resulting solution was centrifugated 5 min at 2000g and used for PCR reactions. PCR amplification of wild-type (WT) and floxed alleles of Scn8a used the F2 (5′-GTG TGT GAT TCT CAA CAG TGG GTT-3′) and R2 (5′-GTC TGT AAG AAG GCC TGA AAG TGA-3′) primers (Levin & Meisler, 2004). Cre transgenes were genotyped with the Cre-specific forward primer 5′-ACT TAG CCT GGG GGT AAC TAA ACT-3′ and the reverse primer 5′-GGT ATC TCT GAC CAG AGT CAT CCT-3′. The reaction consisted of 30 s at 94°C, 30 s at 55°C and 1 min at 72°C (35 cycles) and 5 min at 72°C. PCR products were separated on a 2% agarose gel allowing resolution of a 800 bp product for the WT Scn8a allele, a 900 bp product for the floxed Scn8a allele and a 300 bp product for the Cre transgene. Mice homozygous for the floxed allele of Scn8a produce normal levels of Nav1.6 protein in the absence of Cre (Levin & Meisler, 2004). In addition, Na+ current properties in C57BL/6, Scn8aflox and Scn8aflox/flox mice were indistinguishable and the data were pooled as WT mice.
The med Scn8a-null mutation arose from insertion of a transposable L1 element into exon 2, causing aberrant splicing and disruption in the open reading frame (Kohrman et al. 1996). This mutation was maintained by crossing to strain C3Heb/FeJ. Homozygous med mice developed severe muscle atrophy, weakness and progressive paralysis of the hindlimbs and died by 3 weeks of age. Throughout the manuscript, the term ‘med’ refers to the animals that were homozygous for the recessive null mutation (Burgess et al. 1995; Kohrman et al. 1996).
Reverse transcriptase-PCR
Total RNA was extracted from cerebella and extra-cerebellar brain of Scn8aflox/flox, Cre+ mice with Tri-Reagent (Sigma, Saint-Louis, MO, USA) according to the manufacturer's recommendation. Reverse transcription reactions with oligo(dT)20 primers were performed with the cloned AMV reverse transcriptase following the manufacturer's protocol (Invitrogen, Grand Island, NY, USA), using 3 μg of total RNA. PCR on reverse-transcribed cDNA were performed with a forward primer located in the 5′-UTR (5′-CAC TGA GGT TTG GAC AGG TTG TCA CC-3′) and a reverse primer located in the exon 3 (5′-ATA CAC AGT TGG TCA GGA TGG TGC-3′) (Drews et al. 2005). PCR products were separated on a 2% agarose gel allowing resolution of a 650 bp product for the floxed Scn8a mRNA and a 350 bp product for the truncated Scn8a mRNA.
Antibodies
Primary antibodies used and dilutions were as follows: serine-rich domain of ankyrin-G node (polyclonal, Bouzidi et al. 2002), 1/500; ankyrin-G (monoclonal 4G3F8, Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1/50; Nav1.1 (polyclonal AB5204, Chemicon), 1/150; Nav1.2 polyclonal (ASC-002, Alomone Laboratories, Jerusalem, Israel), 1/100; Nav1.2 monoclonal (K69/3, Upstate Biotechnology, Lake Placid, NY, USA), 1/100; Nav1.6 polyclonal (ASC-009, Alomone), 1/100; Nav1.6 monoclonal (K87A/10, Neuromab, Davis, CA, USA), 1/200. Alexa Fluor488- and Alexa Fluor546-conjugated goat secondary antibodies were used to detect rabbit polyclonal antibodies, and Alexa Fluor555-conjugated goat secondary antibodies were used for visualization of the mouse monoclonal antibodies (all from Molecular Probes, Eugene, OR, USA).
The specificity of the Nav1.6 antibodies was tested on cryosections of cerebella of med mice. Because med mice die before 3 weeks of age, we performed control immunostaining on P19 med mice cryosections. In addition, because WT CG cells did not express Nav1.6 at AISs before P20 (see Figs 1 and 2), comparison were made between WT and med Purkinje cell AISs, which have been shown to cluster Nav1.6 at this developmental stage (Jenkins & Bennett, 2001). Dual labelling with polyclonal anti-ankyrin G antibody (to mark AISs) and monoclonal Nav1.6 antibody or monoclonal anti-ankyrin G antibody and polyclonal anti-Nav1.6 antibody did not reveal staining of med Purkinje cell AISs (not shown), demonstrating that the Nav1.6 antibodies were specific.
Figure 1. Differential expression of Nav1.6 and Nav1.2 at developing CG cell AISs.

A, cryosections of a P21 WT mouse cerebellum were double-labelled with anti-ankyrin G (polyclonal) and anti-Nav1.2 (monoclonal K69/3) antibodies (left panels) or with anti-ankyrin G (monoclonal, 4G3F8) and anti-Nav1.6 (polyclonal ASC-009) antibodies (right panels). Upper panels, virtually all AISs located in the granular layer were stained with the anti-Nav1.2 antibody, whereas Purkinje cell AISs (double arrowheads) and nodes located in the white matter were clearly devoided of Nav1.2 labelling. Purkinje cell AISs as well as nodes in the white matter exhibited Nav1.6 staining. Images are projections of 21 consecutive optical sections spanning 3.3 μm. Bottom panels, expanded deconvolved projections of 11 consecutive optical sections spanning 1.7 μm in the granular layer, in which most CG cell AISs expressed Nav1.2 (arrowheads) and did not express Nav1.6 (arrows) or showed the first signs of Nav1.6 clustering at AISs (dashed circles). B, cryosections of a P60 WT mouse cerebellum were double-labelled with anti-ankyrin G (polyclonal) and anti-Nav1.2 (monoclonal K69/3) antibodies (left panels) and with anti-ankyrin G (monoclonal, 4G3F8) and anti-Nav1.6 (polyclonal ASC-009) antibodies (right panels). Upper panels, most AISs located in the granular layer were stained with the anti-Nav1.2 antibody, whereas Purkinje cell AISs (double arrowheads) and nodes located in the white matter were negative. Purkinje cell AISs and CG cell AISs (arrowheads) exhibited Nav1.6 clustering (see also Fig. 3B for Nav1.6 clustering at P60 Scn8aflox/flox CG cells). Images are projections of 21 consecutive optical sections spanning 3.3 μm. Middle panels, projections of 11 consecutive optical sections spanning 1.7 μm in the granular layer showing Nav1.2-positive or Nav1.6-positive (arrowheads) and Nav1.2-negative (arrows) CG cell AISs. Lower panels, deconvolved images showing fragmented Nav1.2 staining (left) and Nav1.6 labelling (right) at P60 CG cell AISs. Images are deconvolved projection of 7 consecutive optical sections spanning 1 μm. ml, molecular layer; pkj: Purkinje cell layer; gl: granular layer; wm: white matter.
Figure 2. Developmental regulation of Nav1.2 and Nav1.6 clustering at CG cell AISs in mice aged P10–P60.

A, percentage of total ankyrin G-labelled AISs in the granular layer showing staining for Nav1.2 (monoclonal K69/3 antibody) and Nav1.6 (polyclonal ASC-009 and monoclonal K87A/10) in cerebella from mice aged from P10 to P60. Each data point is the mean ±s.e.m. of 200 to 500 ankyrin G positive AISs obtained from at least 3 different animals for each staining condition and developmental stage. B, developmental change in the intensity of Nav1.2 (K69/3 antibody) and Nav1.6 (K87A/10 antibody) immunofluorescence relative to the intensity of ankyrin G immunostaining at the same AIS. Between 30 and 50 AISs were measured for each age from Nav1.2/ankyrin G and Nav1.6/ankyrin G double-labelled confocal images. Inset shows intensity (arbitrary unit) of ankyrin G staining at AISs, which was very much constant over age.
Tissue preparation and immunostaining of cryosections
Mice older than P14 were anaesthetized with halothane and killed by decapitation. Cerebella were rapidly dissected out and frozen in OCT embedding matrix (Cellpath, Hemel Hempstead, UK) bathed in chilled isopentane. Tissues were sagitally cryosectioned at 14–18 μm, transferred to SuperFrostPlus slides (Fisher Scientific, Houston, TX, USA) and stored at −80°C until processed. Slides with cryosections were thawed at room temperature (RT), washed in phosphate-buffered saline (PBS) and then incubated for 40 min at RT in blocking solution containing 3% BSA and 0.1% Triton X-100. Primary antibodies were diluted in blocking solution and applied to sections to be incubated overnight at 4°C in sealed humidified chambers. Slices were then washed three times for 15 min in PBS. Secondary antibodies were applied in 3% BSA-containing PBS for 40 min at RT in the dark. After six washes in PBS, sections were mounted in PBS–50% glycerol.
Confocal imaging and deconvolution
Images were acquired using a TCS SP2 laser-scanning confocal microscope (Leica Microsystems, Mannheim, Germany), initially processed using Leica Confocal Software (LCS Lite) and later exported into Adobe Photoshop for final processing. Images comparing WT and med cerebellar granule cells or WT and granule-specific KO mice cerebella were acquired and digitally processed with identical parameters. No staining was detected in sections or cultures incubated with the secondary antibodies alone.
In order to compare the fluorescence intensities of Nav (Nav1.2 and Nav1.6) and ankyrin G antibodies at AISs over age, images of double labelled sections were acquired using a set of acquisition parameters that was specific for each antibody. LCS Lite software was used to measure intensities of fluorescence on planar projections of confocal raw images spanning 1.6 μm. For each pair of ankyrin G-specific and isotype-specific images of the same field, regions of interest encompassing a single AIS (∼0.4 μm) were defined in the ankyrin G image and the intensities of both ankyrin G- and isotype-specific fluorescences were measured. The measurements were repeated in an adjacent unlabelled area and the resulting background intensities were subtracted from the AIS fluorescence signal. The intensity ratios of Nav1.2 or Nav1.6 signal over ankyrin-G intensity at individual AISs were plotted against age (Fig. 2). Ankyrin G staining intensity was used as reference because it remains stable over age (Fig. 2B).
Images acquired for the deconvolution process were taken with an oil immersion objective 63× NA and pixel sizes were adjusted to 58 nm × 58 nm with a z-sectioning interval of 163 nm. Raw images were transferred to a Silicon Graphics Octane workstation, visualized with Imaris software and deconvolved by the Huygens Professional software (Scientific Volume Imaging, Hilversum, The Netherlands). We determined the microscope's point-spread function from images of spherical beads acquired with the same parameters as raw images of cerebellum and used the Classical Maximum Likehood Estimation as restoration image process.
As indicated in the figures, images show either planar projections of a series of successive confocal images or representative individual confocal sections.
Voltage-clamp experiments: slice preparation and recording
To prepare cerebellar slices for voltage-clamp, patch-clamp experiments, mice (P66–P74) were anaesthetized by inhalation of isoflurane (Merial, Toulouse, France) and decapitated. The procedures followed for extracting the cerebellum, cutting sagittal sections of the cerebellar vermis (220 μm thick), and maintaining the slices before recording were exactly the same as described elsewhere (Magistretti et al. 2006). The apparatus employed and the procedures followed for patch-clamp recordings in the voltage-clamp mode are also described elsewhere (Castelli et al. 2007a). Briefly, the recording chamber was mounted on the stage of an upright microscope (Axioskop 2 FS; Zeiss, Oberkochen, Germany) equipped with differential-contrast optics and a near-infrared charge-coupled device (CCD) camera used to improve cell visualization during the approaching and patching procedures. Slices were perfused with a solution suitable for isolating Na+ currents, containing (in mm): 100 NaCl, 26 NaHCO3, 19.5 tetraethylammonium chloride (TEA-Cl), 3 KCl, 2 MgCl2, 2 CaCl2, 2 BaCl2, 0.5 CdCl2, 4 4-aminopyridine (4-AP), 11 d-glucose (pH 7.4 by saturation with 95% O2–5% CO2). Patch pipettes were fabricated from thick-wall borosilicate glass capillaries (CEI GC 150-7.5; Harvard Apparatus, Edenbridge, UK) and were filled with an intracellular solution containing (in mm): 104 CsF, 50 TEA-Cl, 2 MgCl2, 10 Hepes, 10 EGTA, 2 Na-ATP, and 0.2 Na-GTP (pH adjusted to 7.2 with CsOH). The patch pipettes had a resistance of 5–8 MΩ when filled with the above solution. Liquid junction potential (Vj), estimated as explained in Castelli et al. (2007a), was 5.5 mV. The voltage values specified in the text and figures are uncorrected values (actual membrane potential values can be calculated from nominal command potentials, Vcom, as: Vm=Vcom−Vj).
Tight seals (>5 GΩ) and the whole-cell configuration were obtained by suction according to the standard technique (Hamill et al. 1981). Voltage-clamp recordings of Na+ currents were performed at room temperature (21–22°C) by means of an Axopatch 200B patch-clamp amplifier (Axon Instruments/Molecular Devices, Union City, CA, USA). Series resistance (Rs), which was evaluated on-line and continually monitored during the recording (see Castelli et al. 2007a), averaged 22.1 ± 4.6 MΩ (n= 34), and was always compensated by ∼95%. The program Clampex of the pCLAMP 8.2 software package (Axon Instruments) was used for commanding voltage protocols and digitally acquiring current signals. In all recordings the nominal holding potential was −80 mV. Current signals were low-pass filtered at 5 kHz and digitized at 50 kHz (step protocols) or 10 kHz (ramp protocols). On-line subtraction of leak and capacitive currents was performed using a P/4 routine when step protocols were applied. No on-line subtraction procedure was adopted for ramp protocols. Tetrodotoxin (TTX; Alomone Laboratories) was always applied in the bath with the superfusing solution at the end of the experiment, so as to obtain residual TTX-insensitive currents to be subtracted from control traces and isolate voltage-dependent Na+ currents.
Current-clamp experiments: slice preparation and recording
Parasagittal slices (200 μm) were cut from the cerebellar vermis of mice aged between P40 and P65, as described in Cathala et al. (2003). Briefly, mice were decapitated and the cerebellum was rapidly removed and placed in cold ‘slicing’ (2–4°C) solution of the following composition (mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 25 glucose, 0.5 ascorbic acid and 1 kynurenic acid, pH 7.3 when bubbled with 95% O2–5% CO2. Slices were cut from the dissected cerebellum using a Leica VT1000S (Leica Microsystems, Wetzlar, Germany) or a Vibroslicer (Campden Instrument Ltd) vibrating microtome; these were incubated in slicing solution at 30°C for 45 min and subsequently were maintained without kynurenic acid at room temperature for up to 7 h. Patch-clamp recordings were made at room temperature with an EPC9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany), low-pass filtered at 5 kHz and digitized at 20 kHz. Patch pipettes had a resistance of 8–11 MΩ when filled with the internal solution consisting of (in mm): 110 K-MeSO4, 40 Hepes, 0.15 BAPTA, 6 NaOH, 0.02 CaCl2, 4.6 MgCl2, 4 Na-ATP and 0.3 Na-GTP (adjusted to pH 7.3 with KOH). Capacitance measures were determined directly from the amplifier setting. All voltage recordings were corrected for a measured liquid junction potential of 6 mV.
Data analysis
Data were analysed using pCLAMP 8.2 (Axon Instruments) software. Na+ currents were re-filtered off-line at 3 kHz. Current amplitude was measured at the peak of each tracing. INaR kinetics was studied by fitting the current activation and decay phases with a double exponential function in the form: I=A1exp(−t/τ1) +A2exp(−t/τ2) +C. Na+ permeabilities (PNa) were calculated from current amplitudes (INa) by applying the Goldman equation in the form:
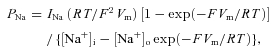 |
in which the nominal intra- and extracellular Na+ concentration values (4.2 and 126 mm, respectively) were introduced. Fittings with the Boltzmann function, PNa=A/{1 + exp[(V−V1/2)/k)]} were carried out using Origin 6.0 (OriginLab Corp., Northampton, MA, USA).
Current-clamp data obtained on cerebellar slices were analysed in the Igor Pro 5.01 environment (Wavemetrics Inc, Lake Oswego, OR, USA) using Neuromatic software (Jason Rothman, University College London, UK; http://www.neuromatic.thinkrandom.com). Measures of AP overshoot, rise time, half-width and AHP amplitude were performed on the first action potential elicited by liminar current injection, unless otherwise stated.
Results are presented as means ±s.e.m., unless otherwise specified (Fig. 4), and n represents the number of cells examined. When data were normally distributed (Shapiro–Wilk test), statistical difference between groups were tested using Student's two-tailed t test for unpaired data (unless otherwise stated). P was considered significant at P < 0.05. *, ** and *** indicate P < 0.05, P < 0.01 and P < 0.001, respectively.
Figure 4. Resurgent and persistent Na+ currents in CG cells from wild-type (left) and Scn8a CG KO (right) mice.

A, currents recorded in response to an INaR activation protocol (upper subpanels) in a representative WT CG cell (cell C8514; a) and a representative Scn8a KO CG cell (cell H8509; b). Transient Na+ currents elicited by the test depolarization at 0 mV have been truncated (∼).The horizontal bars indicate the tracing region over which current data points were averaged to measure the amplitude of the sustained component (INaP), which was subtracted from the peak amplitude of the transient current elicited by repolarizing pulses to obtain a measurement of INaR amplitude. B, average INaR current–voltage (I–V) plots for WT CG cells (a) and Scn8a KO CG cells (b). The plots were obtained from currents recorded 6–9.5 min after break-in. INaR peak amplitudes were normalized for the maximal value observed in each cell, then averaged among cells. Data points and error bars are means ±s.d. (n= 12 and 11 in Ba and Bb, respectively). C, currents recorded in response to a ramp INaP activation protocol (upper subpanels) in a representative WT CG cell (Ca) and a representative Scn8a KO CG cell (Cb) (same cells as shown in Aa and Ab, respectively). In the lower subpanels the currents are shown as ‘instantaneous’I–V relationships. D, voltage dependence of the Na+ permeability underlying the ramp-activated INaP values shown in Ca and Cb. Na+-permeability values were obtained from current values by applying the Goldman equation as explained in Methods. Continuous white lines are best fits obtained by applying a single Boltzmann function. Fitting parameters are specified in each panel.
Results
Nav1.2 and Nav1.6 accumulate differentially at developing axon initial segments of CG cells in situ
We have shown previously that in rat cultured CG cells, Nav1.2 channels are clustered in early-formed AISs (days in vitro (DIV) 7 on), and that during maturation Nav1.6 becomes the predominant Nav channel isoform at these sites (Osorio et al. 2005).
Therefore, we asked whether a similar developmental progression of Nav1.6 expression also occurs in mice CG cell AISs in situ. We examined Nav subunit expression in cryosections of mice cerebella early in postnatal development, starting at P10. By then most CG cells have attained the internal granular layer and show ankyrin G-defined AISs (Supplemental Fig. 1A). Faint Nav1.2 immunoreactivity was observed in ∼80% of these ankyrin G-positive CG cell AISs (Supplemental Fig. 1A; see Fig. 2A for average data). Deconvolved confocal sections, shown in the bottom panel of Supplemental Fig. 1A, revealed that Nav1.2 staining extended through the entire length of the CG cell AISs. In marked contrast, no Nav1.6 immunoreactivity was observed in ankyrin G-positive AISs in the granular layer, whereas Nav1.6 and ankyrin G were co-expressed at Purkinje cell AISs (Supplemental Fig. 1B). At P14, Nav1.6 immunostaining remained undetectable in the granular layer. Nav1.2 was still present and appeared to accumulate at AISs during this period, as evidenced by the increase in the Nav1.2 versus ankyrin G intensity ratio. Nav1.2 expression remained high in most AISs up to P30 (Figs 1A and 2A and B).
The first instances of dim Nav1.6 fluorescence were seen at P21 in about 30% of CG cell AISs (Figs 1A and 2A). From P21 through adulthood, the number of Nav1.6 positive AISs increased sharply, along with an increase in Nav1.6 labelling intensity (Fig. 2A and B). By P60, 90% of ankyrin G-positive AISs in the granular layer were positive for Nav1.6 and Nav1.2 (Figs 1A and 2A). At this stage, Nav1.6 staining appeared regular and extended nearly through the entire ankyrin G-positive AISs (bottom panel in Fig. 1B), while Nav1.2 labelling intensity, although present, was reduced (bottom panel in Figs 1A and 2B). Thus, Nav1.2 channels cluster first at early CG cell AISs, whereas Nav1.6 appears with maturation. It should be noted that neither Nav1.2 nor Nav1.6 (or PanNav immunoreactivities, not shown) was seen in the cell bodies and dendrites of mature CG cells, consistent with the observation that Na+ channel activity was absent from the granule cell somato-dendritic compartment in cerebellar slices (Diwakar et al. 2009).
Maturation of AISs and developmental accumulation of Nav1.2 at AISs are unchanged in CG cells from granule-specific Scn8a KO mice
To investigate Nav1.6 function in CG cells, we used gene targeting to generate granule-specific KO mice deficient in the Nav1.6 subunit. Homozygous mice for the floxed allele of Scn8a (Scn8aflox/flox mice) were crossed with BACα6-Cre transgenic mice, in which expression of the Cre recombinase (Cre) is under the control of the granule-specific GABAA receptor α6 subunit promoter (Aller et al. 2003; Levin & Meisler, 2004; Levin et al. 2006). Deletion of the loxP exon by exposure to Cre in Scn8aflox/flox–Cre+ recombinant mice resulted in the inactivation of the Scn8a gene only in CG cells. Mice homozygous for the deleted Scn8a allele were viable, had normally developing cerebella and had no visible motor impairment, but a slower rate of improvement was observed in 5-month-old mice with the rotarod test (Levin et al. 2006). We refer hereafter to these mice as Scn8a CG KO mice.
Based on the onset of Cre expression in the Cre+ line (Aller et al. 2003), Scn8a inactivation may be expected to occur gradually after P7. Deletion of Scn8a exon 1 genomic DNA was detected in cerebella from P30 Scn8a CG KO mice (Fig. 3A). Scn8a mRNA lacking exon 1 was also detected by RT-PCR in cerebella but not in extra-cerebellar regions of Scn8a CG KO mice (Fig. 3A).
Figure 3. Conditional inactivation of the Scn8a gene in CG cells.

A, PCR amplification of genomic DNA and cDNA (obtained from reverse transcription of mRNA) from the cerebellum and extra-cerebellar brain of a Scn8aflox/flox–Cre+ (Scn8a CG KO) mouse. Flox primers amplified a 0.9 kb product from the floxed Scn8a gene and a 0.3 kb product from the truncated Scn8a, which was detected only in cerebellum. Cre primers amplified a 0.2 kb product. RT-PCR primers amplified a 0.65 kb product from the full-length transcripts and a 0.35 kb fragment from transcripts lacking exon 1. The 0.35 kb amplicon was selectively detected in the cerebellum. RT (−) experiments were done omitting reverse transcriptase. B and C, cryosections of Scn8aflox/flox (B) and Scn8a KO CG (C) mouse cerebella (aged P60) were double-labelled with anti-ankyrin G (polyclonal) and anti-Nav1.6 (monoclonal K87A/10) antibodies. Scn8aflox/flox CG cell AISs showed strong Nav1.6 staining whereas ankyrin G positive AISs in the granular layer were clearly devoid of Nav1.6 in granule-specific Scn8a KO mice (arrows in C). Note that Purkinje cell AISs are still positive for Nav1.6 in granule-specific Scn8a KO mice (double arrowheads). Images are projections of 20 consecutive optical sections spanning 3.1 μm (upper panels) or 10 sections spanning 1.5 μm (bottom panels) in B and C. D, comparison of proportion of ankyrin G positive CG cell AISs that were positive for Nav1.2 or Nav1.6 in WT mice (data from Fig. 2A; open symbols) and Scn8a CG KO mice (filled symbols) at different ages. Data points for Scn8a CG KO mice are means ±s.e.m. of 200–300 AISs obtained from at least 2 animals for each staining condition and developmental stage. E, cryosections of a P60 Scn8a CG KO mouse cerebellum were double-labelled with anti-ankyrin G (monoclonal, 4G3F8) and anti-Nav1.1 (polyclonal) antibodies. No Nav1.1 positive AISs were detected in the granular layer. Images are projections of 21 consecutive optical sections spanning 3.3 μm. Inset: staining of AISs of Purkinje cells (arrowhead) and inhibitory interneurons (arrow) for Nav1.1 (green) and ankyrin G (red) from P19 med cerebellum. Scale bar: 20 μm.
To verify that homologous deletion of the Scn8a allele was associated with a concomitant reduction in protein products, levels of Nav1.6 were examined by in situ immunohistochemical analysis of cerebellar slices at developmental stages where the channel is normally seen at CG cell AISs. At P60, Nav1.6 immunoreactivity was strongly expressed at most CG AISs in Scn8aflox/flox mice, whereas it was absent from most CG AISs in Scn8a CG KO mice (Fig. 3B and C). Nearly 5% of AISs present in the granular layer remained immunoreactive for anti-Nav1.6 antibodies, possibly due to the non-granule cells present in that layer. In contrast, expression and clustering of Nav1.6 at Purkinje cell AISs were not altered in Scn8a CG KO mice (Fig. 3B and C). Figure 3D summarizes the percentage of Nav1.6-positive AISs from P10 to adulthood in Scn8a CG KO mice as compared to control mice (pooled data from Scn8aflox/flox and WT mice). The percentage of Nav1.6-positive AISs in the granular layer of Scn8a CG KO mice decreased to be virtually undetectable by P38. In contrast, neither Nav1.2 subunit clustering nor expression level appeared affected by the Nav1.6 deletion (Fig. 3D). Nav1.2 subunits were sequestered normally at developing CG cell AISs in Scn8a CG KO and expressed in about 80% of AISs, as observed in WT animals (Fig. 3D).
Because retinal ganglion cells have been shown to exhibit compensatory Na+ channel expression in med mice (Van Wart & Matthews, 2006), we also examined whether Nav1.1 was present at CG cell AISs, where it is not normally found (Osorio et al. 2005). Double immunolabelling of P60 Scn8a CG KO cerebella with anti-ankyrin G and anti-Nav1.1 antibodies showed that none of the ankyrin G-positive AISs in the granular layer were immunoreactive for Nav1.1 (Fig. 3E). We confirmed that the Nav1.1 antibody stained AISs of Purkinje cells and inhibitory interneurons in the molecular layer of P19 cerebellum med mice (inset in Fig. 3E), in agreement with Van Wart & Matthews (2006).
Persistent Na+ current is selectively depressed in Scn8a KO mature granule cells
To assess how the loss of Nav1.6 affects the biophysical properties of Na+ currents (INa), we performed a series of voltage-clamp experiments in acute slices of cerebellum from WT and Scn8a CG KO mice aged P66–P74. Voltage-clamp experiments were performed under ionic conditions suitable to isolate Na+ currents (see Methods), and current tracings were always TTX-subtracted to better isolate TTX-sensitive Na+ currents from contaminants. To elicit transient Na+ currents (INaT), an I–V protocol was applied consisting of 20 ms depolarizing step pulses at −75 to +20 mV in 5 mV increments. INaT thus evoked showed evident signs of bad space-clamp control, especially over the ascending part of the I–V relationship (not shown), very similarly to what has been previously observed for INaT of rat CG cells (Magistretti et al. 2006). Such a behaviour is expected for currents generated by channels at least in part located in neuronal processes electrotonically remote from the soma.
The resurgent and persistent Na+ currents, INaR and INaP, were elicited by applying a step repolarizing protocol (Fig. 4A, top) or ramp depolarizing protocol (Fig. 4C, top), respectively, and appeared adequately controlled. Because all Na+-current components showed a tendency to slowly run-up with time, INaT, INaR and INaP activation protocols were repeatedly applied during the course of the experiment, which allowed us to monitor their amplitudes at various moments of the recording (see Fig. 5).
Figure 5. Amplitude of transient, resurgent, and persistent Na+ currents in CG cells from wild-type and Scn8a CG KO mice.

Each panel shows amplitude values for INaT (A), INaR (B), and INaP (C) as determined at various time points after break-in (see the labels at the top of each subpanel). The parameters considered are: INaT absolute amplitude at the peak of its I–V (INaT(peak); Aa); INaT absolute amplitude at a voltage level positive by 20 mV to the I–V peak (INaT(peak+20); Ab); INaR absolute amplitude at the peak of its I–V (Ba); INaR peak amplitude relative to INaT(peak+20) amplitude (as a percentage; Bb); absolute, peak amplitude of ramp-evoked INaP (Ca); peak amplitude of ramp-evoked INaP relative to INaT(peak+20) amplitude (as a percentage; Cb). Note in C that both 0.1 mV ms−1 and 0.05 mV ms−1 ramps were applied to measure INaP amplitude (left and right parts of each subpanel, respectively). Open circles are individual values; filled squares indicate mean values ±s.e.m.
Figure 4A shows currents recorded in response to the INaR activation protocol in representative CG cells from WT and Scn8a KO mice. An evident INaR was observed in 18 out of 19 CG cells from WT mice, and 14 out of 15 CG cells from KO mice. In both cases, INaR amplitude increased, with increasing repolarizations, up to a peak at −35 to −40 mV, and then decreased for further repolarizations and became particularly fast-decaying at potentials negative to −50 mV. Such a behaviour is characteristic of INaR and has been observed in several other neuronal populations (Raman & Bean, 1997; Do & Bean, 2003; Afshari et al. 2004; Cummins et al. 2005; Enomoto et al. 2006; Magistretti et al. 2006; Castelli et al. 2007a,b;).
To measure INaR amplitude, the amplitude of the ‘persistent’ component observed at the end of the 100 ms repolarizing pulse (horizontal bars in Fig. 4A) was subtracted from the peak amplitude of the decaying component (see Magistretti et al. 2006). The voltage dependence of INaR amplitude was very similar in WT and Scn8a KO CG cells (Fig. 4B). Absolute INaR amplitude, measured at the peak of this current's I–V relationship, was, on average, only slightly and not significantly smaller in Scn8a KO CG cells as compared to WT CG cells (Fig. 5Ba; P > 0.18 in all cases). INaR amplitude was also evaluated relative to INaT amplitude. The amplitude of INaT at the peak of its I–V relationship (which in different cells occurred at widely variable voltage values, i.e. −35 to −10 mV) was not significantly different in Scn8a KO and WT CG cells (Fig. 5Aa; P > 0.20 in all cases). However, it can be expected that measurements of this parameter are strongly influenced by the poor clamp control of INaT. To attenuate this problem, we considered, as a more significant parameter, INaT amplitude at a voltage level positive by 20 mV to the I–V peak (INaT(peak+20)). Indeed, the currents recorded in the descending portion of INaT's I–V relationship are expected to be less affected by clamp artifacts than those recorded in the ascending and peak regions (discussed in Castelli et al. 2007b). INaT(peak+20) was also not significantly different in Scn8a KO and WT CG cells (Fig. 5Ab; P > 0.12 in all cases). Expressing INaR peak amplitude relative to INaT(peak+20) (the temporally closest pairs of INaR and INaT recordings were always considered to calculate this ratio) further reduced the slight, non-significant differences observed for INaR absolute amplitude (P > 0.10 at 1.5–4 min, P > 0.4 at the other time points; Fig. 5Bb).
INaR kinetics was also compared between WT and KO CG cells. For these measurements, INaR recordings obtained in individual cells at different time points were averaged, so as to improve the signal-to-noise ratio and the reliability of measures. First, the activation kinetics of INaR was evaluated by measuring the current's time to peak (TTP) at the different voltage levels examined. As shown in Fig. 6A, TTP progressively increased from ∼3.5 ms at −15 mV to ∼5.5 ms at −30 to −40 mV, and decreased back sharply at potentials negative to −50 mV. At potentials negative to −60 mV, INaR activation was so fast that no current peak clearly separated from the foregoing tail currents and/or unsubtracted artifacts by a rising phase was distinguishable, and TTP could not be measured. It can be seen that TTP absolute values and TTP voltage dependence were extremely similar in WT and KO CG cells (Fig. 6A). INaR kinetics was further characterized by performing exponential fittings of the current rising and decay phases. Both phases could be consistently fitted jointly by applying a double exponential function (Fig. 6B, upper and middle tracings). Increasing the number of the exponential components did not improve the goodness of fit. At potentials negative to −50 mV, INaR rising phase was too short or indistinguishable, and a single exponential function was only applied to fit INaR decay phase (Fig. 6B, lower tracing). The voltage dependence of the time constant of INaR activation (τact) closely paralleled that of TTP (Fig. 6Ca). The time constant of INaR decay (τdecay) displayed a behaviour very similar to that previously reported for INaR of rat CG cells (Magistretti et al. 2006), including a nearly plateau level at −30 to −50 mV, a sharp decrease at more negative voltages, and a trend to increase at potentials positive to −25 mV (Fig. 6Cb). Moreover, τdecay turned out to be very similar between WT and KO CG cells over the whole voltage range explored (Fig. 6Cb). Hence, KO of the Nav1.6 subunit has no apparent effects on the kinetics of the resurgent sodium current expressed by CG cells.
Figure 6. INaR activation kinetics is not affected by Scn8a knockout.

A, voltage dependence of INaR time to peak (TTP) in WT (open circles) and Snc8a KO (filled circles) CG cells (n= 15 and 13 for WT and KO, respectively). B, currents recorded in response to the INaR activation protocol (see Fig. 4A) at three different repolarisation potentials (−20, −40 and −70 mV) in a representative WT neuron (cell A8514), and biexponential fittings (enhanced lines) of INaR activation and decay phases. Fitting parameters are: A1= 37.6 pA, τ1= 1.33 ms, A2=−50.1 pA, τ2= 21.3 ms, C=−13.2 pA (−20 mV); A1= 130.0 pA, τ1= 3.25 ms, A2=−166.3 pA, τ2= 20.2 ms, C=−15.4 pA (−40 mV); A2=−24.0 pA, τ2= 2.88 ms, C= 0.0 pA (−70 mV). C, voltage dependence of INaR activation time constant, τact (equivalent to panel B's τ1) (Ca), and INaR decay time constant, τdecay (equivalent to panel B's τ2) (Cb), in WT (open circles) and Snc8a KO (filled circles) CG cells (n= 15 and 13 for WT and KO, respectively).
The persistent Na+ current, INaP, was evoked by applying slow depolarizing ramps with a slope of 0.1 mV ms−1 (Fig. 4C) and 0.05 mV ms−1 (not shown), which allowed us to obtain ‘instantaneous’I–V relationships for INaP. The currents evoked by 0.05 mV ms−1 ramps were consistently smaller in amplitude than those evoked by 0.1 mV ms−1 ramps (Fig. 5Ca), indicating the development of INaP inactivation during slow depolarizations (see Fleidervish & Gutnick, 1996; Magistretti & Alonso, 1999; Osorio et al. 2005). Plots of INaP voltage dependence of activation were obtained from ‘instantaneous’I–V relationships by deriving Na+ permeability values (PNaP) from Na+ currents as explained in Methods (Fig. 4D). Fitting PNaP(V) plots with a single Botzmann function (Fig. 4D) returned average half-activation potential (V1/2) and slope factor (k) values that were not significantly different in Scn8a KO and WT CG cells (Table 1). However, INaP peak amplitude was significantly smaller, by a factor of ∼1.5 to 2.2, in Scn8a KO CG cells as compared to WT CG cells (Fig. 5Ca). Peak INaP amplitude relative to INaT(peak+20) was also significantly smaller (by ∼1.35–1.9 times) in Scn8a KO CG cells (Fig. 5Cb).
Table 1.
INaP activation parameters
| Time point: 3.5–7.5 min |
Time point: 10–14.5 min |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT |
Scn8a KO |
WT |
Scn8a KO |
|||||||
| mean ±s.d. | n | mean ±s.d. | n | P | mean ±s.d. | n | mean ±s.d. | n | P | |
| 0.1 V s−1 | ||||||||||
| V1/2 | −55.9 ± 3.4 | 15 | −56.0 ± 4.5 | 6 | 0.96 | −59.7 ± 2.8 | 15 | −60.6 ± 3.9 | 6 | 0.59 |
| k | −3.6 ± 0.7 | −3.0 ± 0.4 | 0.08 | −3.5 ± 0.6 | −3.0 ± 0.6 | 0.15 | ||||
| 0.05 V s−1 | ||||||||||
| V1/2 | −56.7 ± 2.7 | 15 | −56.5 ± 3.2 | 6 | 0.87 | −59.7 ± 2.8 | 15 | −60.7 ± 3.2 | 6 | 0.49 |
| k | −3.9 ± 0.8 | −3.5 ± 0.9 | 0.33 | −3.9 ± 1.1 | −3.1 ± 0.6 | 0.11 | ||||
The table shows INaP activation parameters (V1/2 and k, in mV) returned by Boltzmann fittings of PNaP(V) plots. Statistical comparisons were made applying Student's two-tail t test for unpaired data.
Scn8a KO CG cells have impaired firing regularity
To test how the loss of Nav1.6 affects the firing properties of CG cells, we performed a series of current-clamp experiments in acute slices of cerebellum from WT and Scn8a CG KO mice aged P40–P65. To characterize intrinsic membrane properties, we examined the response of CG cells to constant current injection (Fig. 7A). Passive properties (input capacitance, input conductance and resting membrane potential) of WT and Scn8a KO CG cells were not significantly different (Table 2). Subthreshold I–V relationships of both WT and Scn8a KO CG cells exhibited moderate inward rectifications, typical of mature CG cells (Fig. 7A; Cathala et al. 2003). The current required to reach firing threshold was also not significantly different between the two conditions (Table 2 and Fig. 7B). Figure 7D shows superimposed representative action potentials obtained from WT and Scn8a KO cells. Action potential threshold and overshoot (Table 2 and Fig. 7D) were identical in the WT and Scn8a KO CG cells. However, both AP rise time and half-width were significantly reduced in Scn8a KO CG cells (WT CG cells: rise time = 0.42 ± 0.02 ms, half-width = 0.67 ± 0.02 ms; Snc8a KO CG cells: rise time = 0.33 ± 0.01 ms, half-width = 0.51 ± 0.02 ms; Table 2). Moreover, a significant enhancement of the postspike AHP was typically observed in Scn8a KO CG cells compared to WT CG cells (Fig. 7G and Table 2). When considering the AHP that followed the first spike obtained in response to the minimum injected current which evoked firing, AHP peak was at −74.9 ± 0.8 mV in Scn8a KO CG cells vs.−71.6 ± 0.7 mV in WT CG cells (P < 0.05; Table 2). Similar differences and statistical significance were observed when comparing the postspike AHP at 40 or 100 Hz firing frequency.
Figure 7. Passive and firing properties of CG cells from wild-type and Scn8a CG knockout mice.

A, characteristic responses of CG cells from WT (P37) and Snc8a CG KO (P59) mice to current injection (2 pA step increments). Shown are passive electrotonic responses to hyperpolarizing current pulse injections and responses up to the first current step that induced spiking in each cell. B, relationship between input conductance and firing threshold (the minimal current required to evoke action potentials) in WT (n= 21) and Scn8a KO CG cells (n= 15; P > 0.05). Circles indicate individual values and squares indicate mean values ±s.e.m. (WT: filled symbols; Snc8a CG KO: open symbols.) D, characteristic action potentials from WT (black line, P49) and Snc8a CG KO (grey line, P41) mice. Shown are the first recorded action potentials aligned at threshold. The WT action potential has a rise time of 0.35 ms, a peak amplitude of 31.3 mV and a half-width of 0.64 ms while the Snc8a CG KO action potential has a rise time of 0.29 ms, a peak amplitude of 32.0 mV and a half-width of 0.50 ms. E, action potential peak in CG cells from WT mice (+22.5 ± 2.0 mV, n= 23) and Snc8a CG KO mice (+21.2 ± 2.0 mV, n= 17) (P > 0.05). Open circles indicate individual values; filled squares indicate mean values ±s.e.m. F, action potential rise time (filled squares) in CG cells from WT mice (0.42 ± 0.02 ms, n= 23) and Snc8a CG KO mice (0.33 ± 0.01 ms, n= 16) and half-width (open squares) in CG cells from WT mice (0.67 ± 0.02 ms, n= 23) and Snc8a CG KO mice (0.51 ± 0.02 ms, n= 16). Open circles indicate individual values; filled squares indicate mean values ±s.e.m. G, postspike AHP amplitude in CG cells from WT mice (−71 ± 0.7 mV, n= 17) and Snc8a CG KO mice (−74.9 ± 0.8 mV, n= 23). Open circles indicate individual values; filled squares indicate mean values ±s.e.m. C, representative responses of CG cells from a WT (P45, left) and an Snc8a CG KO (P41, right) mice to increasing current injections. H, cumulative plot showing the difference in the distribution of the interspike intervals in WT (black line) and Snc8a KO CG cells (grey line) measured following a +40 pA current step (Kolmogorov–Smirnov test: P < 0.001). I, average mean firing rate plotted against current intensity for WT CG cells (filled squares, n= 23) and Snc8a KO CG cells (open squares, n= 17). Straight lines are linear regression fits to data points, giving slope coefficients of 1.72 and 1.16 Hz pA−1 for WT and Scn8a KO CG cells, respectively.
Table 2.
Intrinsic membrane properties and action potential (AP) characteristics measured in WT and Scn8a KO CG cells
| WT |
Scn8a KO |
||||
|---|---|---|---|---|---|
| mean ±s.e.m. | n | mean ±s.e.m. | n | P | |
| Input capacitance (pF) | 3.9 ± 0.2 | 23 | 3.9 ± 0.3 | 17 | >0.05 |
| Input conductance (pS) | 0.7 ± 0.1 | 21 | 0.8 ± 0.1 | 15 | >0.05 |
| Resting membrane potential (mV) | −92.5 ± 1.6 | 11 | −94.1 ± 1.3 | 13 | >0.05 |
| AP threshold (mV) | −42.4 ± 1 | 23 | −42.8 ± 0.9 | 17 | >0.05 |
| AP overshoot (mV) | 22.5 ± 2 | 23 | 21.2 ± 2 | 17 | >0.05 |
| AP rise time (ms) | 0.42 ± 0.02 | 23 | 0.33 ± 0.01 | 16 | <0.05 |
| AP half-width (ms) | 0.67 ± 0.02 | 23 | 0.51 ± 0.02 | 16 | <0.05 |
| Postspike AHP (mV) | −71.6 ± 0.7 | 23 | −74.9 ± 0.8 | 17 | <0.05 |
Statistical comparisons were made applying Student's two-tail t test for unpaired data.
In agreement with previous studies, mature CG cells supported high-frequency firing in response to sustained depolarization. Figure 7C shows representative examples of firing responses to injection of increasing amounts of current in WT and Scn8a KO CG cells. There was a tendency toward reduction in the mean firing frequency of Scn8a KO CG cells (Fig. 7I), resulting in a reduction in the gain of the frequency–stimulation relationship (slopes of the frequency–current plots: 1.72 ± 0.04 in WT vs. 1.17 ± 0.03 Hz pA−1 in Scn8a KO CG cells). AP firing was more variable in Scn8a KO than in WT, as shown by the difference in the shape of interspike interval (ISI) distribution (Kolmogorov–Smirnov test, P < 0.001; Fig. 7H). The slope of the cumulative plot for small ISI durations was higher in Scn8a KO CG cells than in WT cells, whereas it was lower for longer ISI durations (Fig. 7H). These results indicate that firing was less regular in Scn8a KO CG cells, thus implying that Nav1.6 is necessary to maintain regular spiking.
Discussion
Developmental regulation of Nav isoforms in mouse cerebellar granule cells in situ
The present study provides evidence that expression of Nav1.2 and Nav1.6 subunits in mouse CG cells in situ is developmentally regulated. Nav1.2 subunits were detected at most CG cell AISs from the early stages of maturation to adulthood with a sparser distribution in mature CG cell AISs as compared to the first postnatal stages. In contrast Nav1.6 expression at the AISs appears much later, once CG cells have attained their mature electrophysiological phenotype (D’Angelo et al. 1997; Cathala et al. 2003). Our data also suggest that the mechanisms responsible for selective targeting of Nav1.6 to the AISs do not exclude Nav1.2 from this site. Thus, as with AISs of retinal ganglion cells (Boiko et al. 2003), but unlike nodes of Ranvier in the myelinating optic nerve (Boiko et al. 2001), Nav1.2 expression does not disappear from CG cell AISs as Nav1.6 expression increases, indicating that in CG cells both subunits participate to define the characteristics of mature Na+ currents.
Contribution of Nav1.6 and Nav1.2 to transient, resurgent and persistent Na+ currents
The biophysical properties of CG cell INaR were similar to those described for INaR in rat CG cells in situ (Magistretti et al. 2006) and in acutely dissociated mice CG cells (Afshari et al. 2004). It displayed a bell-shaped voltage dependence with a peak at −35/−40 mV. In Purkinje cells, INaR has been primarily ascribed to the activity of Nav1.6 channels (Raman et al. 1997; Levin et al. 2006; Royeck et al. 2008). However, it is becoming clear that other Na+ channel subunits, including Nav1.2 (Rush et al. 2005) and Nav1.1 (Grieco & Raman, 2004), may also contribute to INaR in some neurons (Do & Bean, 2004; Enomoto et al. 2007). Consistent with this view, we found that in Scn8a KO CG cells, INaR amplitude levels were unchanged with respect to WT cells. Interestingly, INaR activation and decay kinetics were also indistinguishable in Scn8a KO CG cells as compared to WT cells. This is different from what has been reported for Purkinje cells, in which a slowing of activation and decay kinetics of residual INaR has been observed in Nav1.6-null mice (Aman & Raman, 2007). Our results clearly show that the Nav1.2 subunit is capable of generating a resurgent current which appears fully normal in terms of both amplitude and kinetic properties, at least in particular conditions. They also may imply that Nav1.6 channels normally do not contribute to INaR, or, alternatively, that they can be fully substituted by Nav1.2 channels, as far as INaR generation is concerned, when Nav1.6 expression is impaired. Which one of these two possibilities is correct cannot be inferred on the basis of the present findings.
Our data raise another open question regarding the basis of the resurgence mechanism itself. Recently, it has been shown that in Purkinje cells the cytoplasmic tail of the Na+ channel auxiliary β4 subunit can act as the ‘resurgent particle’ by competing with the inactivation ball domain of the Na+ channel α subunit (Grieco et al. 2005). However, the auxiliary β4 subunit is expressed at detectable levels neither in rat (Yu et al. 2003) nor in mouse (Allen Brain Atlas, http://www.brain-map.org) CG cells, raising the question as to which molecule acts as the blocking particle in CG cells. However, because we did not specifically look at the expression of β4 subunits, we cannot rule out a contribution of β4 subunits in CG cell KO mice. Whatever the identity of the blocking particle, our data show that Nav1.2 can undergo the resurgence process at least as well as Nav1.6 in CG cells.
While showing that loss of Nav1.6 does not significantly affect INaR in CG cells, our data revealed that the same condition results in a significant reduction of INaP. Thus, Nav1.2 channels that remain in CG cells of Scn8a KO animals are not capable of passing steady-state current as well as Nav1.6 channels. This is consistent with previous data showing that INaP is markedly reduced in brain neurons from Nav1.6-null mice (Raman et al. 1997; Maurice et al. 2001; Enomoto et al. 2007; Royeck et al. 2008), and that Nav1.1 and Nav1.2 yield smaller persistent currents in heterologous expression systems (Smith & Goldin, 1998; Rush et al. 2005). Thus, our data add to the previous results in suggesting that Nav1.6 subunits are more effective than other isoforms in generating persistent Na+ currents.
Functional evidences suggest a difference in the propensity of different neuronal compartments to generate INaP (Osorio et al. 2005; Yue et al. 2005; Astman et al. 2006). Our previous experiments using focal application of TTX onto the soma of CG cells indicated that channels responsible for INaP are located on axonal processes but not in the cell body plasma membrane (Osorio et al. 2005). In addition, somatic and dendritic Nav channels in layer 5 neocortical neurons have been shown to play a minimal role in generating INaP, which appears to be almost entirely carried by channels located in the proximal axon (Astman et al. 2006). These observations together with our findings that somato-dendritic compartments of CG cells are not immunoreactive for Nav antibodies indicate that persistent Nav1.6 current is generated in the AISs where it is segregated.
Nav1.6 channels regulate action potential waveform and spike timing
Functional consequences of Nav1.6 elimination on CG cell excitability and firing pattern were analysed in mature cerebellar slices. Our data indicate that the absence of Nav1.6 in granule cells had no significant effects on the rheobase and threshold for AP generation. The observed reduction in INaP amplitude would, instead, be expected to increase the rheobase of AP discharge (Magistretti et al. 2006). Adaptive rearrangements in the balance between the conductances active in the subthreshold voltage range could explain this unexpected finding. On the other hand, AP duration was found to be significantly shortened in Scn8a KO granule cells, and AHP was significantly enhanced. This could be due to an acceleration of INaT kinetics. Indeed, in particular conditions (although not always: Do & Bean, 2004; Enomoto et al. 2007) neuronal Na+ currents produced in the absence of Nav1.6 channels inactivate more quickly than those due to Nav1.6 channels (Raman et al. 1997; Mercer et al. 2007). If inactivation of Na+ channels is more complete in Scn8a KO cells than in WT cells, the tail current flowing through non-inactivated channels during the AHP will be smaller. Consequently, the balance between inward currents and K+ currents will move towards the latter, and a larger hyperpolarization will result.
In CA1 hippocampal pyramidal cells it has been shown that INaP contributes to amplify the post-spike AHP, and that its absence results in AHP decrease (Vervaeke et al. 2006). This is because INaP reduced the current injection required to elicit any given level of firing activity, whereas AHP peak occurred at potential levels more negative than those at which INaP is significantly activated. As a consequence, at the peak of the AHP the total depolarizing current was larger in the absence of INaP, and AHP amplitude was reduced. Our observation that in Scn8a KO granule cells the AHP was enhanced even for equal levels of firing frequency indicates that, if the above mechanism is also active in CG cells, it is overcome, after abolition of Nav1.6 expression and the consequent INaP reduction, by additional mechanism(s) that tend to enhance the AHP.
Previous work suggested that INaP plays critical roles in other aspects of neuronal functions, including subthreshold oscillations and repetitive firing (Do & Bean, 2003; Magistretti et al. 2006; Kang et al. 2007; Mercer et al. 2007). We found that Scn8a KO CG cells exhibited a mild – but not significant – impairment of frequency–current relationship compared with their WT counterparts. We showed that this subtle firing deficit in Scn8a KO CG cells was not the consequence of compensatory changes in expression of other Nav isoforms, since neither Nav1.1 nor Nav1.2 was upregulated during development nor took the place normally occupied by Nav1.6. It is not yet clear, however, whether other K+ or Ca2+ currents exert compensatory effects (Swensen & Bean, 2005) and influence the overall electrophysiological phenotype of CG cells in a manner to favour high frequency firing. Nevertheless, it may not be surprising that high frequency capability was so little affected in Scn8a KO CG cells, given that these cells showed normal Na+ resurgent current, which is critical to fast spiking (Mercer et al. 2007).
One major difference between WT and Scn8a KO CG cells was a propensity for Scn8a KO CG cells to display increased interspike interval variability and firing irregularity. This observation may appear at odds with the observation of AHP enhancement, because spike afterhyperpolarization is a mechanism that improves temporal precision during AP trains (de Ruyter van Steveninck et al. 1997; Berry & Meister, 1998; Vervaeke et al. 2006). Why then in the presence of a more profound AHP does the ISI interval variability not decrease, but, if anything, increase? This could be the result of a combination of factors. Per se, the enhanced AHP would tend to make firing precision higher: indeed, a more profound AHP promotes Nav channel repriming, and combined with resurgent gating it may allow membrane potential to rise back to spike threshold quickly, thus increasing spiking precision. On the other hand, the smaller INaP of Scn8a KO CG cells could make the slope of the interspike depolarizing ramp less steep, which would have the opposite effect on firing precision. Indeed, from Fig. 7H it can be seen that the slope of the cumulative plot at smaller ISI durations was higher (greater precision) in Scn8a KO CG cells than in WT cells, and somewhat lower (smaller precision) for longer ISI durations. This would mean that when the interspike phase lingers a little longer around the threshold, AP firing becomes less regular, perhaps because the depolarizing contribution of INaP is partially lacking.
In conclusion, our results demonstrate that in CG cells, Nav1.2 channels, clustered in the AISs in the absence of Nav1.6 channels, can generate normal levels of transient and resurgent Na+ currents, and sustain a high-frequency firing that appears normal in many respects. However, Nav1.6 channels seem to be required for normal expression of persistent Na+ current and to warrant a high degree of temporal precision in repetitive firing of CG cells.
Acknowledgments
We thank Dr G. Alcaraz for the rabbit polyclonal anti-ankyrin G antibody. We thank A. Fernandez and S. Roffe-Vidal and acknowledge the late J. C. Reynaud for excellent technical assistance and M. P. Blanchard for expert assistance with the confocal imaging. The monoclonal antibody anti-Nav1.6 K87A/10 was developed by and obtained from the UC Davis/NINDS/NIMH NeuroMab Facility, supported by NIH grant (U24NS050606) and maintained by the Department of Pharmacology, School of Medicine, University of California, Davis. This work was supported by the Centre National pour la Recherche Scientifique and by grants from the Agence Nationale de la Recherche (ANR-05-NEURO, ANR-05-PCOD, ANR-08-MNP, ANR-08-GENOPAT, ANR-09-MNP), Fondation Schlumberger, ARCInca-2006, UPSA, IRME, the Fondation pour la Recherche Médicale (équipe FRM 2007) (to P.D.), and the United States Public Health Service (NS34509 to M.H.M.). N.O. was the recipient of the grant from the Fondation pour la Recherche Médicale.
Glossary
Abbreviations
- AHP
afterhyperpolarization
- AIS
axon initial segment
- AP
action potential
- BSA
bovine serum albumin
- CG
cerebellar granule
- DIV
days in vitro
- P
post-natal days
- ISI
inter-spike interval
- KO
knock-out
- RT
room temperature
Author contributions
N.O., L.C., J.M. and P.D. contributed to the conception, design, analysis and interpretation of the data presented in this paper. The immunohistochemical experiments were designed, carried out and analysed by N.O. The electrophysiological experiments were designed, carried out and analysed by L.C. and J.M. The manuscript was written by N.O., L.C., J.M. and P.D. and revised with the critical appraisal of L.C., M.H.M., M.C. and J.M. All authors approved the final version for publication.
Supplemental material
Supplementary Figure 1
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Afshari FS, Ptak K, Khaliq ZM, Grieco TM, Slater NT, McCrimmon DR, Raman IM. Resurgent Na currents in four classes of neurons of the cerebellum. J Neurophysiol. 2004;92:2831–2843. doi: 10.1152/jn.00261.2004. [DOI] [PubMed] [Google Scholar]
- Aller MI, Jones A, Merlo D, Paterlini M, Meyer AH, Amtmann U, Brickley S, Jolin HE, McKenzie AN, Monyer H, Farrant M, Wisden W. Cerebellar granule cell Cre recombinase expression. Genesis. 2003;36:97–103. doi: 10.1002/gene.10204. [DOI] [PubMed] [Google Scholar]
- Aman TK, Raman IM. Subunit dependence of Na channel slow inactivation and open channel block in cerebellar neurons. Biophys J. 2007;92:1938–1951. doi: 10.1529/biophysj.106.093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astman N, Gutnick MJ, Fleidervish IA. Persistent sodium current in layer 5 neocortical neurons is primarily generated in the proximal axon. J Neurosci. 2006;26:3465–3473. doi: 10.1523/JNEUROSCI.4907-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MJ, 2nd, Meister M. Refractoriness and neural precision. J Neurosci. 1998;18:2200–2211. doi: 10.1523/JNEUROSCI.18-06-02200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci. 2003;23:2306–2313. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzidi M, Tricaud N, Giraud P, Kordeli E, Caillol G, Deleuze C, Couraud F, Alcaraz G. Interaction of the Nav1.2a subunit of the voltage-dependent sodium channel with nodal ankyrinG. In vitro mapping of the interacting domains and association in synaptosomes. J Biol Chem. 2002;277:28996–29004. doi: 10.1074/jbc.M201760200. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Cathala L, Brickley S, Cull-Candy S, Farrant M. Maturation of EPSCs and intrinsic membrane properties enhances precision at a cerebellar synapse. J Neurosci. 2003;23:6074–6085. doi: 10.1523/JNEUROSCI.23-14-06074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli L, Biella G, Toselli M, Magistretti J. Resurgent Na+ current in pyramidal neurones of rat perirhinal cortex layer II: Axonal location of channels and contribution to depolarising drive during repetitive firing. J Physiol. 2007a;582:1179–1193. doi: 10.1113/jphysiol.2007.135350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli L, Nigro MJ, Magistretti J. Analysis of resurgent sodium-current expression in rat parahippocampal cortices and hippocampal formation. Brain Res. 2007b;1163:44–55. doi: 10.1016/j.brainres.2007.05.065. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005;579:2166–2170. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- D’Angelo E, De Filippi G, Rossi P, Taglietti V. Synaptic activation of Ca2+ action potentials in immature rat cerebellar granule cells in situ. J Neurophysiol. 1997;78:1631–1642. doi: 10.1152/jn.1997.78.3.1631. [DOI] [PubMed] [Google Scholar]
- D’Angelo E, Nieus T, Maffei A, Armano S, Rossi P, Taglietti V, Fontana A, Naldi G. Theta-frequency bursting and resonance in cerebellar granule cells: experimental evidence and modelling of a slow K+-dependent mechanism. J Neurosci. 2001;21:759–770. doi: 10.1523/JNEUROSCI.21-03-00759.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruyter van Steveninck RR, Lewen GD, Strong SP, Koberle R, Bialek W. Reproducibility and variability in neural spike trains. Science. 1997;275:1805–1808. doi: 10.1126/science.275.5307.1805. [DOI] [PubMed] [Google Scholar]
- Diwakar S, Magistretti J, Goldfarb M, Naldi G, D’Angelo E. Axonal Na+ channels ensure fast spike activation and back-propagation in cerebellar granule cells. J Neurophysiol. 2009;101:519–532. doi: 10.1152/jn.90382.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP. Sodium currents in subthalamic nucleus neurons from Nav1.6-null mice. J Neurophysiol. 2004;92:726–733. doi: 10.1152/jn.00186.2004. [DOI] [PubMed] [Google Scholar]
- Drews VL, Lieberman AP, Meisler MH. Multiple transcripts of sodium channel SCN8A (NaV1.6) with alternative 5′- and 3′-untranslated regions and initial characterization of the SCN8A promoter. Genomics. 2005;85:245–257. doi: 10.1016/j.ygeno.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Chandler SH. Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J Neurophysiol. 2007;98:710–719. doi: 10.1152/jn.00292.2007. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Wu N, Chandler SH. Participation of sodium currents in burst generation and control of membrane excitability in mesencephalic trigeminal neurons. J Neurosci. 2006;26:3412–3422. doi: 10.1523/JNEUROSCI.5274-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleidervish IA, Gutnick MJ. Kinetics of slow inactivation of persistent sodium current in layer V neurons of mouse neocortical slices. J Neurophysiol. 1996;76:2125–2130. doi: 10.1152/jn.1996.76.3.2125. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Raman IM. Production of resurgent current in Nav1.6-null Purkinje neurons by slowing sodium channel inactivation with β-pompilidotoxin. J Neurosci. 2004;24:35–42. doi: 10.1523/JNEUROSCI.3807-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- Isope P, Barbour B. Properties of unitary granule cell–>Purkinje cell synapses in adult rat cerebellar slices. J Neurosci. 2002;22:9668–9678. doi: 10.1523/JNEUROSCI.22-22-09668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isope P, Franconville R, Barbour B, Ascher P. Repetitive firing of rat cerebellar parallel fibres after a single stimulation. J Physiol. 2004;554:829–839. doi: 10.1113/jphysiol.2003.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H, Ekerot CF. Properties of somatosensory synaptic integration in cerebellar granule cells in vivo. J Neurosci. 2006;8:11786–11797. doi: 10.1523/JNEUROSCI.2939-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Saito M, Sato H, Toyoda H, Maeda Y, Hirai T, Bae YC. Involvement of persistent Na+ current in spike initiation in primary sensoryneurons of the rat mesencephalic trigeminal nucleus. J Neurophysiol. 2007;97:2385–2393. doi: 10.1152/jn.01191.2006. [DOI] [PubMed] [Google Scholar]
- Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of Ranvier. Neuron. 2001;30:105–119. doi: 10.1016/s0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modelling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrman DC, Harris JB, Meisler MH. Mutation detection in the med and medJ alleles of the sodium channel Scn8a. Unusual splicing due to a minor class AT-AC intron. J Biol Chem. 1996;271:17576–17581. doi: 10.1074/jbc.271.29.17576. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin SI, Khaliq ZM, Aman TK, Grieco TM, Kearney JA, Raman IM, Meisler MH. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (Nav1.6) in cerebellar Purkinje neurons and granule cells. J Neurophysiol. 2006;96:785–793. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- Levin SI, Meisler MH. Floxed allele for conditional inactivation of the voltage-gated sodium channel Scn8a (NaV1.6) Genesis. 2004;39:234–239. doi: 10.1002/gene.20050. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;31:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti J, Alonso A. Biophysical properties and slow-voltage dependent inactivation of a sustained sodium current in entorhinal cortex layer-II principal neurons: A whole-cell and single-channel study. J Gen Physiol. 1999;114:491–509. doi: 10.1085/jgp.114.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti J, Castelli L, Forti L, D’Angelo E. Kinetic and functional analysis of transient, persistent and resurgent sodium currents in rat cerebellar granule cells in situ: an electrophysiological and modelling study. J Physiol. 2006;573:83–106. doi: 10.1113/jphysiol.2006.106682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Plummer NW, Burgess DL, Buchner DA, Sprunger LK. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica. 2004;122:37–45. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- Mercer JN, Chan CS, Tkatch T, Held J, Surmeier DJ. Nav1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. J Neurosci. 2007;27:13552–13566. doi: 10.1523/JNEUROSCI.3430-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio N, Alcaraz G, Padilla F, Coste B, Delmas P, Crest M. Differential targeting and functional specialization of sodium channels in cultured cerebellar granule cells. J Physiol. 2005;569:801–816. doi: 10.1113/jphysiol.2005.097022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Royeck M, Horstmann M-T, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal Nav1.6 sodium channels in action potential initiation of CA1 Pyramidal neurons. J Neurophysiol. 2008;100:2361–2380. doi: 10.1152/jn.90332.2008. [DOI] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol. 2005;564:803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Goldin AL. Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci. 1998;18:811–820. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen A, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Robustness of burst firing in dissociated purkinje neurons with acute or long-term reductions in sodium conductance. J Neurosci. 2005;25:3509–3520. doi: 10.1523/JNEUROSCI.3929-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66:477–519. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:52–54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking Nav1.6 sodium channels. J Neurosci. 2006;26:7172–7180. doi: 10.1523/JNEUROSCI.1101-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Hu H, Graham LJ, Storm JF. Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron. 2006;49:257–270. doi: 10.1016/j.neuron.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Quick DC. Functional architecture of the initial segment. In: Waxman SG, editor. Physiology and Pathobiology of Axons. New York: Raven Press; 1978. pp. 125–130. [Google Scholar]
- Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel β4, a new disulfide-linked auxiliary subunit with similarity to β2. J Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Remy S, Su H, Beck H, Yaari Y. Proximal persistent Na+ channels drive spike afterdepolarizations and associated bursting in adult CA1 pyramidal cells. J Neurosci. 2005;25:9704–9720. doi: 10.1523/JNEUROSCI.1621-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.