

SUMMARY
Doppler-defined pulmonary hypertension (PH) in sickle cell disease (SCD) is associated with 40% mortality at 40 months.
To assess the effect of bosentan in SCD-PH, two randomized, double-blind, placebo-controlled, 16-week studies were initiated. Safety concerns are particularly relevant in SCD due to comorbid conditions. ASSET-1 and -2 enrolled patients with pulmonary arterial hypertension (PAH) and pulmonary venous hypertension (PH), respectively. Hemodynamics and 6-minute walk distance (6MWD) were obtained at baseline and week 16.
The studies were terminated due to slow site initiation and patient enrolment (n=26). Bosentan appeared to be well tolerated. Although sample sizes were limited, in ASSET-1 at baseline, 6MWD correlated with cardiac output (CO; p=0.006) with non-significant correlations between 6MWD and pulmonary vascular resistance (PVR; p=0.07) and between 6MWD and right atrial pressure (p=0.06). In ASSET-2 at baseline, there was a non-significant correlation between 6MWD and CO (p=0.06). Due to limited sample sizes, efficacy endpoints were not analyzed. However, in both studies, non-significant increases in CO were observed with bosentan compared to placebo. Similarly, non-significant decreases in PVR were observed with bosentan.
Limited data in SCD-PH suggest that a low 6MWD predicts a low CO. Standard-dose bosentan appears to be well tolerated. Further investigation is warranted.
Keywords: Sickle cell anemia, pulmonary hypertension, endothelin receptor antagonism, hemodynamics, exercise capacity
INTRODUCTION
Three adult screening studies in sickle cell disease (SCD) have reported a 20% prevalence of mild pulmonary hypertension (PH), defined by Doppler-estimated right ventricular systolic pressure (RVSP) >35 mmHg, and 10% prevalence of at least moderate or greater PH, defined by RVSP >45 mmHg (Ataga, et al 2006, De Castro, et al 2008, Gladwin, et al 2004). Using Doppler echocardiography, RVSP can be estimated by measuring tricuspid regurgitant velocity (TRV) with approximation of right atrial pressure (by inspiratory collapsibility of the vena cava) and subsequent application of the modified Bernoulli equation. Despite estimated RVSP increases that are much lower than those measured in patients with idiopathic pulmonary arterial hypertension (IPAH), the mortality rate associated with mild elevation in estimated RVSP appears to be quite high in adult SCD patients (Ataga, et al 2006, Castro, et al 2003, De Castro, et al 2008, Gladwin, et al 2004, Machado, et al 2006), rising linearly with estimated values between 35-45 mmHg associated with a 4.4-fold mortality (95% confidence interval [CI], 1.6-12.2; p<0.001); an estimated RVSP >45 mmHg is associated with a 10.6-fold mortality (95% CI, 3.3-33.6; p<0.001) (Ataga, et al 2006, De Castro, et al 2008, Gladwin, et al 2004). Left-sided heart disease, i.e. due to left ventricular diastolic dysfunction (LVDD), present in 18% of SCD patients (assessed by echocardiography), has been reported as an independent risk factor in SCD (Sachdev, et al 2007). Additionally, in patients with both Doppler-defined PH, i.e. increased TRV on Doppler echocardiography, and suspected LVDD (by echocardiography), the mortality risk is compounded (12.0-fold mortality; 95% CI, 3.8 to 38.1; p<0.001) (Sachdev, et al 2007).
While SCD is most frequent in African populations, it also affects Mediterranean, Caribbean, South and Central American, Arabian, and East Indian subjects; there are more than 100,000 SCD patients in the United States alone (Minter and Gladwin 2001). However, to date only small, uncontrolled studies have investigated potential treatments in SCD-PH (Castro, et al 2003, Gladwin and Schechter 2001, Little, et al 2009, Machado, et al 2005, Morris, et al 2003, Sullivan, et al 1999, unpublished observations of Jison et al). Given the limitations of these studies, well-controlled phase II/III trials are needed to confirm the safety and efficacy of treatments.
The pathogenesis of SCD-PH appears multi-factorial: vascular occlusion and hemolysis result in intimal hyperplasia and asplenia promotes pulmonary microthrombosis and increases cell-free hemoglobin. Additionally, chronic hypoxaemia due to sleep-disordered breathing, parenchymal and vascular injury resulting from recurrent acute chest syndrome (ACS), and scavenging of nitric oxide by hemoglobin and arginase released during hemolysis also appear to contribute to the pulmonary vasculopathy (Ataga, et al 2004, Gladwin and Vichinsky 2008, Morris, et al 2005, Reiter, et al 2002). Due to high cardiac output (CO) secondary to chronic anemia, SCD-PH has lower pulmonary vascular resistance (PVR) than IPAH (although higher PVR than SCD without PH). Even with mildly increased pulmonary arterial pressure (PAP) and relatively low PVR, adult SCD patients can have clinically significant exercise intolerance and functional limitations (Anthi, et al 2007, Gladwin, et al 2004). Each 10-mmHg increment in mean PAP (PAPm) increases the death rate by 1.7-fold (Castro, et al 2003).
Due to the reported high prevalence of Doppler-estimated PH in SCD and the high associated mortality, PH treatments are needed. Because ET-1 appears important in SCD pathobiology including associated PH, ET-1 receptor antagonism may be efficacious. The objectives of the ASSET Randomized, Placebo-Controlled, Double-Blind, Multicenter, Parallel Group Study to Assess the Efficacy, Safety and Tolerability of Bosentan in Patients With Symptomatic Pulmonary Arterial Hypertension Associated With Sickle Cell Disease)-1 and -2 studies were to evaluate the safety and efficacy of bosentan, a dual ET-1 receptor antagonist, in two different subtypes of SCD-PH patients (see Methods). Given that echocardiography using TRV ≥ 2.5 m/s can overestimate the prevalence of true PH (confirmed by right heart catheterization (RHC), and because co-existent LVDD appears prevalent in SCD (Parent, et al 2009, Sachdev, et al 2007), RHC was required for confirmation of PH for study enrollment. Both ASSET-1 and -2 were stopped prematurely due to slow site initiation and enrolment.
Despite limited power, the ASSET studies provide the largest multi-center cohort of SCD-PH patients to date with hemodynamic and exercise data. Due to the limited sample sizes resulting from premature study discontinuation, safety and efficacy data are presented descriptively.
METHODS
Patient Population and Study Design
For the purposes of these studies, pulmonary arterial hypertension (PAH) and PH were defined as follows:
PAH (precapillary PH) included patients with pulmonary capillary wedge pressure (PCWP) ≤15 mmHg and PVR ≥160 dyn/sec/cm-5. The 160 dyn/sec/cm-5 PVR threshold was chosen rather than the traditional 240 dyn/sec/cm-5 in other PAH subgroups, i.e. IPAH, because even small increases in PVR are associated with adverse functional outcomes in this population (Anthi, et al 2007, Gladwin, et al 2004).
PH (“mixed vasculopathy”) included patients with either PCWP ≤15 mmHg and PVR ≥100 dyn/sec/cm-5 and <160 dyn/sec/cm-5, or PCWP 16 to 25 mmHg and PVR ≥100 dyn/sec/cm-5.
Two concurrent, randomized, double-blind, placebo-controlled, 16-week studies in SCD with PAH (ASSET-1) or PH (ASSET-2) were designed.
Enrollment criteria: male or female ≥12 years with PAH enrolled in ASSET-1, or PH enrolled in ASSET-2. Screening logs were not part of the study. All patients had: SCD (HbSS or HbS/betao genotype; HbA ≤10%), dyspnea with exertion, 6-minute walk distance (6MWD) ≥150 m and ≤450 m, clinical stability ≥3 months with no changes in chronic treatment, i.e. chronic treatment was based on the judgment of the clinical investigator, and PAH or PH confirmed by RHC at the time of enrollment. Exclusion criteria: left ventricular ejection fraction <40%, systolic pressure <85 mmHg, uncontrolled hypertension with systolic pressure >160 mmHg and/or diastolic pressure >100 mmHg, total lung capacity <50% of predicted, hemoglobin <60 g/l, alanine transaminase (ALT) ≥3× upper limit of normal (ULN) or albumin >20% below ULN. Women of childbearing potential had a negative serum pregnancy test and used reliable methods of contraception during, and for 3 months after study completion.
After obtaining informed consent, all patients underwent RHC. Only those patients with confirmed PAPm ≥25 mmHg by RHC were assigned to either ASSET-1 or ASSET-2 based on hemodynamics at randomization. Patients who completed the 16-week study had the option to enter the open-label study (ASSET-3) without a washout period.
Local institutional review committees approved the protocol; written informed consent was obtained from all patients.
Eligible patients were randomized 1:1 ratio to either bosentan or placebo (Figure 1). Patients were started on oral bosentan 62.5 mg b.i.d. or placebo, and up-titrated after 4 weeks to 125 mg b.i.d. or placebo for the remainder of the 16-week study. Safety and efficacy were assessed at Weeks 4, 8, 12, and 16. Enrollment into the trial was stopped early due to slow enrollment.
Fig 1. ASSET-1 and ASSET-2 Study Design.
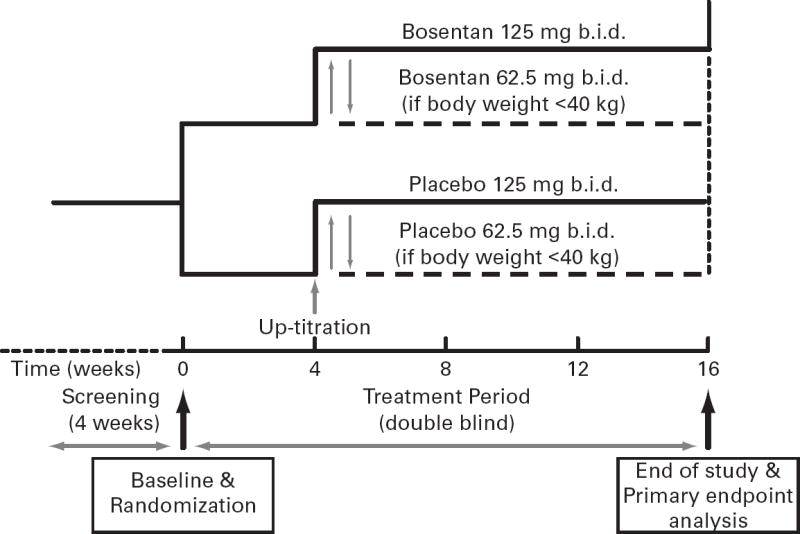
Patients in both ASSET-1 and ASSET-2 were screened and randomized 1:1 to bosentan or matching placebo based upon inclusion/exclusion criteria. After 4 weeks, bosentan dose was increased from 62Æ5 mg b.i.d. to 125 mg b.i.d with a placebo dummy up-titration. At the end of 16 weeks of study drug, patients had the option to enter into the ASSET-3 open-label study.
The investigator could discontinue study treatment due to an adverse event (AE), for administrative reasons (e.g., consent withdrawal), or if continued administration was considered contrary to the patient’s best interests.
Outcome Measures
The primary endpoint in ASSET-1 was change from baseline in PVR at Week 16. The primary endpoint in ASSET-2 was change from baseline in 6MWD at Week 16. The 6MWD was chosen as a functional outcome for ASSET-2 because the PVR was already low, and less likely to be influenced by a vasodilator. Secondary endpoints included change in 6MWD (ASSET-1), and safety of bosentan in all patients. Additionally, correlations between hemodynamics and 6MWD were evaluated. The objectives of the open-label ASSET-3 extension were to collect long-term safety and efficacy data in SCD-PH (PAH and “mixed vasculopathy”) patients.
Statistical Analyses
Hemodynamics and 6MWD correlations were analyzed by Spearman’s rank. Correlation analyses were performed on the treatment-per-protocol populations of ASSET-1 and -2 to determine the potential influence of hemodynamics on 6MWD at baseline. Multivariate regression models were performed to examine hemodynamic predictors of 6MWD at baseline. Pre- and post-treatment parameters were analyzed in both treatment groups in each study by comparing median differences between bosentan and placebo groups. In case of missing values, the last available post-baseline value was carried forward unless a substitution rule applied. For death, the 6MWD was 0 m. Patients without RHC at week 16 were not included in paired analyses. Due to the exploratory nature of the analyses, no correction for multiple comparisons was applied. P<0.05 was considered statistically significant. Actelion Pharmaceuticals, Ltd. sponsored the study. Statistical analyses were performed independently by the authors.
RESULTS
Patients
Twenty-six patients were randomized (14 in ASSET-1; 12 in ASSET-2) before enrollment was stopped due to slow site initiation and enrollment. Enrolled patients remained on study medication, i.e. placebo or bosentan, until their study participation was completed. Baseline demographics of the patients enrolled in the controlled studies were similar to other PAH studies with the exception of race (Table 1). All patients were African-American. Demographics of the two treatment groups were similar, with the exception of body weight (median weight bosentan 83.5 kg vs. placebo 68.9 kg; data not shown).
Table 1.
Baseline Characteristics of ASSET-1 and ASSET-2 Enrolled Patients.
| ASSET-1 | ASSET-2 | |||
|---|---|---|---|---|
| Characteristic | N | Median (IQR)1 | N | Median (IQR)1 |
| Age, years | 14 | 40 (34–53) | 12 | 47.5 (38–54) |
| Female, n (%) | 14 | 10 (71) | 12 | 7 (58) |
| 6MWD, m | 14 | 353 (295–390) | 12 | 370 (292–402) |
| Right heart catheterization | ||||
| mPAP, mm Hg | 14 | 32 (30–39) | 12 | 33 (29–40) |
| mRAP, mm Hg | 14 | 8 (5–10) | 12 | 10 (8–14) |
| PVR, dyn/sec/cm-5 | 14 | 262 (185–320) | 11 | 144 (122–200) |
| mPCWP, mm Hg | 14 | 12 (10–15) | 11 | 17 (15–21) |
| CO, L/min | 14 | 7.3 (6.6–8.1) | 12 | 7.9 (6.8–9.1) |
Interquartile range (25th-75th percentile), unless otherwise noted.
mPAP, mean pulmonary artery pressure; mRAP, mean right arterial pressure; PVR, pulmonary vascular resistance; mPCWP, mean capillary wedge pressure; CO, cardiac output.
The disposition of all patients is shown in Figure 2. Treatment remained blinded for all patients until database lock. Eleven patients continued in the open-label (ASSET-3).
Fig 2. Patient Disposition.

The number of patients from each study that completed each protocol phase.
Two patients in each treatment group had blinded drug discontinued prematurely. The two patients on bosentan were discontinued from ASSET-1 due to an AE; one who had developed sepsis, subsequently died from it. The two patients on placebo were discontinued from ASSET-1 due to investigator withdrawal and patient consent withdrawal. None of the 11 patients who continued in ASSET-3 discontinued prematurely. One death occurred in the controlled studies as mentioned above, and none in the open-label ASSET-3. The patient who died in the controlled study was hospitalized for vaso-occlusive crisis (VOC), at which time study drug (bosentan) was discontinued. While in the hospital, the patient developed sepsis and died 15 days after study treatment discontinuation. The investigator considered the VOC and sepsis unrelated to study treatment. In ASSET-3 (open label extension), the median drug exposure was 24.6 weeks (range 11.0 – 35.6 weeks).
Hemodynamic Parameters
Baseline hemodynamics (Table 1) were consistent with prior studies, i.e. SCD-PAH patients exhibited a moderate PAPm elevation and high CO, with associated modest PVR elevation and decreased 6MWD. The 6MWD was also decreased in the SCD-PH patients with modest increases in PCWP.
Correlations with 6MWD and Hemodynamics at Baseline
Despite the small sample sizes, in ASSET-1, a significant correlation between CO and 6MWD was observed (p=0.006), with potential inverse correlations (not significant) for RAPm and 6MWD (p=0.06), and for PVR and 6MWD (p=0.07; Table 2; Figure 3). In ASSET-2, a potential correlation between CO and 6MWD (p=0.06) and a potential inverse correlation between PCWP and 6MWD (p=0.08) were observed (Table 2; Figure 4).
Table 2.
Correlations of 6MWD at Baseline with Baseline Hemodynamic Parameters.
| ASSET-1 | ASSET-2 | |||
|---|---|---|---|---|
| N | R (p) | N | R (p) | |
| mPAP, mm Hg | 14 | 0.11 (0.7) | 12 | 0.01 (0.9) |
| mRAP, mm Hg | 14 | −0.48 (0.08) | 12 | 0.17 (0.6) |
| PVR, dyn/sec/cm-5 | 14 | −0.50 (0.07) | 11 | 0.05 (0.9) |
| mPCWP, mm Hg | 14 | −0.14 (0.6) | 11 | −0.55 (0.08) |
| CO, L/min | 14 | 0.69 (0.006) | 12 | 0.55 (0.06) |
mPAP, mean pulmonary artery pressure; mRAP, mean right arterial pressure; PVR, pulmonary vascular resistance; mPCWP, mean capillary wedge pressure; CO, cardiac output
Fig 3. ASSET-1 Baseline Measurement Correlations.
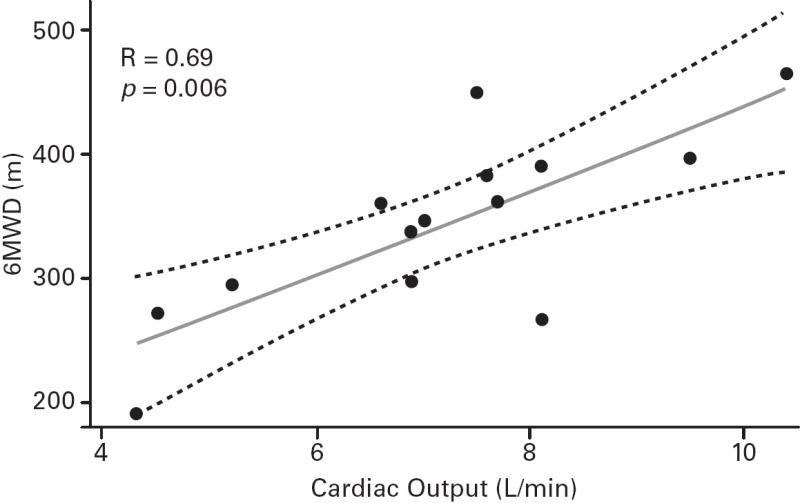
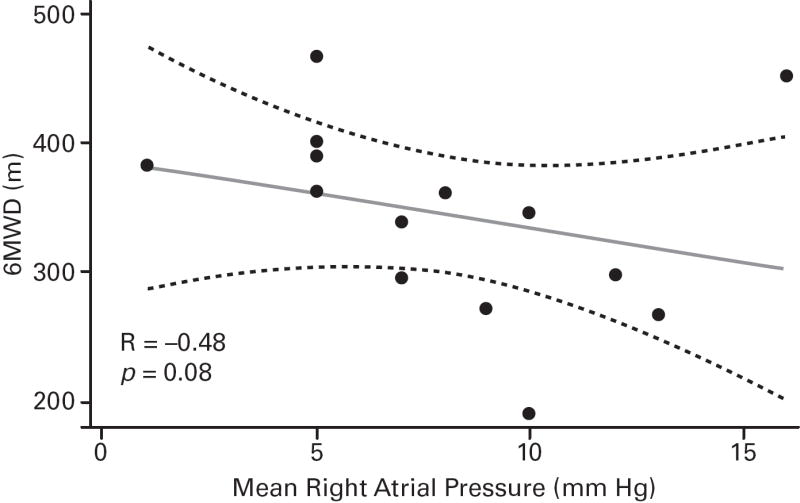

6-minute walk distance (6MWD) and haemodynamic parameter correlations (Spearman’s rank correlation coefficients and P values).
Fig 4. ASSET-2 Baseline Measurement Correlations.


6-minute walk distance (6MWD) and haemodynamic parameter correlations (Spearman’s rank correlation coefficients and P values).
Multivariate regression modeling examining the hemodynamic predictors of 6MWD at baseline demonstrated that 55% of the total variation in 6MWD was explained by the hemodynamic parameters (Table 3A). Table 3B shows that CO remained significant (p=0.0002) in the generated predictive model. No correlations were observed between haemoglobin and 6MWD, or between haemoglobin and CO.
Table 3.
| A. Multiple Regression Analysis of Hemodynamic Predictors of 6MWD Distance at Baseline (ASSET 1 and ASSET 2), full model | |||||
|---|---|---|---|---|---|
| Predictor | Univariate Correlations with 6MWD | Multiple Regression Results | |||
| Spearman r (p) | Regression Coefficient | Standard Error | P1 | Overall Model Fit R2 (p) | |
| mPAP, mm Hg | 0.09 (0.6) | 5.45 | 5.9 | 0.4 | 0.55 (0.007) |
| mRAP, mm Hg | -0.12 (0.6) | 3.56 | 3.9 | 0.4 | |
| PVR dyn/sec/cm-5 | -0.33 (0.1) | -0.55 | 0.5 | 0.3 | |
| mPCWP, mm Hg | -0.10 (0.6) | -10.72 | 5.5 | 0.07 | |
| CO, L/min | 0.65 (0.0003) | 13.60 | 16.1 | 0.4 | |
| B . Multiple Regression Analysis of Hemodynamic Predictors of 6MWD Distance at Baseline (ASSET 1 and ASSET 2), best model | |||||
| Predictor | Univariate Correlations with 6MWD | Multiple Regression Results | |||
| Spearman r (p) | Regression Coefficient | Standard Error | P1 | Overall Model Fit R2 (p) | |
| mPCWP, mm Hg | -0.10 (0.6) | -3.81 | 2.5 | 0.1 | 0.49 (0.0006) |
| CO, L/min | 0.65 (0.0003) | 27.20 | 6.0 | 0.0002 | |
6MWD, 6-minute walk distance; mPAP, mean pulmonary artery pressure; mRAP, mean right arterial pressure; PVR, pulmonary vascular resistance; mPCWP, mean capillary wedge pressure; CO, cardiac output;
From t-test of the null hypothesis that the regression coefficient is zero.
From F-test that all regression coefficients are equal to zero.
Treatment Effect on 6MWD and Hemodynamics
While not analyzed for efficacy, the changes in 6MWD and in hemodynamics from baseline to Week 16 are summarized in Table 4. One patient withdrew consent before a post-baseline assessment was obtained and was excluded from this analysis.
Table 4.
Pre-Treatment and Post-Treatment Results (Non-Parametric)
| ASSET-1 | ||||
|---|---|---|---|---|
| Bosentan | Placebo | |||
| Characteristic | N | Median Difference (IQR)1 | N | Median Difference (IQR)1 |
| 6MWD2 , m* | 6 | -9 (-18,-5) | 7 | 38 (3, 52) |
| PVR, dyn/sec/cm-5 | 5 | -90 (-111,-41) | 4 | -60 (-98, 8) |
| mPAP, mm Hg | 5 | -6 (-6, -5) | 4 | -4 (-8, 1) |
| mRAP, mm Hg | 5 | 0 (-2, 0) | 5 | -2 (-3, 1) |
| mPCWP, mm Hg | 5 | -1 (-2, 1) | 5 | -2 (-3, 1) |
| CO, L/min | 5 | 1.0 (-0.5, 4.0) | 5 | -0.3 (-0.3, 0.5) |
| ALT | 6 | -12(-25,-3) | 7 | -11(-20,-5) |
| Hb | 5 | 0.10(-0.5,0.2) | 7 | -0.6(-1.2,0.5) |
| ASSET-2 | ||||
| Bosentan | Placebo | |||
| Characteristic | N | Median Difference (IQR)1 | N | Median Difference (IQR)1 |
| 6MWD2, m | 5 | 0 (-9, 0) | 7 | 17 (-24, 80) |
| PVR, dyn/sec/cm-5 | 4 | -77 (-116, 23) | 7 | -22 (-41, 40) |
| mPAP, mm Hg | 5 | -4 (-6, -4) | 7 | -5 (-8, 9) |
| mRAP, mm Hg | 5 | 2 (1, 2) | 7 | -4 (-9, 2) |
| mPCWP, mm Hg | 4 | 3 (1, 4) | 7 | -3 (-5, -1) |
| CO, L/min | 5 | 1.0 (-0.8, 1.7) | 7 | -0.9 (-1.6, -0.2) |
| ALT | 4 | 0 (-2,5.5) | 6 | -4(-6,-1) |
| Hb | 4 | -1.0(-1.6,-0.05) | 5 | 0.70(-0.4,0.8) |
1 of the 6 pts in the bosentan group of ASSET-1 died
No imputation rules were applied to missing hemodynamic data
Defined as week 16 visit median value minus baseline visit median value; IQR=Interquartile range (25th-75th percentile)
Last observation carried forward if 6MWD data missing, 0 m used in case of death 6MWD, 6-minute walk distance; mPAP, mean pulmonary artery pressure; mRAP, mean right arterial pressure; PVR, pulmonary vascular resistance; mPCWP, mean capillary wedge pressure; CO, cardiac output; ALT, alanine transaminase; Hb, haemoglobin.
While not significant, in both studies greater increases in median 6MWD were observed in the placebo vs. bosentan groups at study end compared with baseline (Table 4; Figure 5); additionally, there was a non-significant increase in median CO in the bosentan groups and a non-significant decrease in the placebo groups (Table 3). A greater PVR reduction (not significant) also occurred in the bosentan vs. placebo groups in both studies (Figure 6; Table 3).
Fig 5. Changes in 6-Minute Walk Distances (6MWDs).




Changes from baseline to Week 16 are shown for all patients in ASSET-1 and ASSET-2. Black bars indicate median 6MWD values.
Fig 6. Changes in Pulmonary Vascular Resistance (PVR).




Changes from baseline to Week 16 are shown for all patients in ASSET-1 and ASSET-2. Black bars indicate median PVR values.
Safety and Tolerability
Serious adverse events (SAEs) occurred in a similar proportion of patients in each treatment group, and except for VOCs, each event occurred in a single patient per treatment group (Tables 5-6). During the open-label, three of the 11 patients experienced an SAE, which included a VOC in each case.
Table 5.
Incidence of All Adverse Events (ASSET-1 and ASSET-2 Combined).
| Parameter | Bosentan (n=11) | Placebo (n=15) |
|---|---|---|
| AEs | ||
| Total patients with at least one AE, n (%) | 9 (82) | 13 (87) |
| Total number of different AEs, n | 48 | 72 |
| Total patients with an AE that led to premature discontinuation of study treatment, n (%) | 2 (18) | - |
| SAEs | ||
| Deaths, n (%) | 1 (9) | - |
| Total patients with at least one SAE, n (%) | 5 (46) | 6 (40) |
| Total number of different SAEs, | 11 | 15 |
AE, adverse event; SAE, serious adverse event.
Table 6.
Number of AEs, SAEs, and VOC AES by treatment group, ASSET-1 and ASSET-2 – All subjects.
| Treatment Group | |||||
|---|---|---|---|---|---|
| Number of Events | Bosentan N subjects=11 | Placebo N subjects=15 | Wilcoxon’s Exact p | ||
| Mean (std) | Median (min - max) | Mean (std) | Median (min - max) | ||
| AEs | 5.1 (2.5) | 6 (1 - 8) | 5.2 (3.9) | 5 (1 – 13) | 0.9 |
| VOC AEs | 0.8 (1.3) | 0 (0 – 3) | 0.7 (1.4) | 0 (0 – 4) | 0.8 |
| SAEs | 1.5 (1.9) | 1 (0 – 5) | 1.1 (1.9) | 0 (0 – 6) | 0.5 |
| Severe SAEs | 0.7 (1.3) | 0 (0 – 4) | 1.0 (1.9) | 0 (0 – 4) | 1.0 |
AE, adverse event; SAE, serious adverse event; VOC, vaso-occlusive crisis
Bosentan appeared to be well tolerated. In the double-blind studies, AEs were reported for most patients in both treatment groups. No apparent differences between treatments in the number or types of AEs were observed nor evidence of a relationship to treatment in the number or type of events that were graded as severe or serious. Clinically significant decreases in hemoglobin (pre-specified as >15% decrease in hemoglobin to an absolute value <110 g/l) occurred in four patients in each treatment group, with no consequent patient dropout. No increases in liver function tests (ALT >3× ULN) were observed. Asymptomatic decreases in mean systolic (−9.3 vs. −7.3 mmHg on placebo) and diastolic pressures (−7.9 vs. −0.9 mmHg on placebo) and mean increase in body weight (0.8 vs. −0.3 kg on placebo) were observed with bosentan. These differences were neither clinically nor statistically significant. Differences between treatments in laboratory variables, heart rate, or electrocardiography were also not clinically or statistically significant.
In the open-label, no deaths, discontinuations due to an AE, elevations in ALT >3× ULN, or symptoms of liver disease occurred. Three patients experienced an SAE, which included a VOC in each case.
DISCUSSION
The neurohormone ET-1 causes vasoconstriction and promotes fibrosis, cell proliferation, remodeling, and inflammation. In PAH and heart failure, ET-1 concentrations correlate with disease severity and prognosis. Although no data are available in SCD correlating ET-1 levels with PH, elevated ET-1 levels in SCD (Hammerman, et al 1997, Rybicki and Benjamin 1998, Werdehoff, et al 1998) support a pathobiological role of ET-1 in SCD-PH. ET-1 levels rise with VOCs and decline with resolution (Graido-Gonzalez, et al 1998). Sickled erythrocytes induce ET-1 expression (Phelan, et al 1995). ET-1 modulates Gardos channel activity in erythrocytes (Rivera, et al 2002), triggering further erythrocyte sickling. As the ET-1 elevations precede clinical symptoms (Hammerman, et al 1997), ET-1 blockade could also prevent VOCs and ameliorate the disease.
Based on these studies, the ASSET trials were designed to evaluate the safety and efficacy of bosentan, a dual ET-1 receptor antagonist, in SCD-PH. However, due to slow site initiation and enrollment, the studies were stopped prematurely. As no screening logs were kept at the recruiting sites, we were unable to analyze screen failure data. Given the small sample sizes, efficacy endpoints were also not analyzed. Echocardiograms on these patients were not performed simultaneously with RHCs, so we were unable to analyze sensitivity and specificity of echocardiographic indices compared to cardiac catheterization parameters. However, despite the small sample sizes, correlations between hemodynamics and 6MWD were observed in SCD-PH patients, as suggested by a previous study (Anthi, et al 2007). In particular, a potential correlation between 6MWD and CO was observed. These findings were significant in PAH patients in ASSET-1 (consistent with other studies in World Heath Organization Group I PAH), while associations were not significant in PH patients in ASSET-2. Interestingly, a trend towards an increase in CO was noted with bosentan, but this was not borne out by the 6MWD data. Given that the standard error for CO is much smaller than that for 6MWD, and that the 6MWD can be affected by comorbid conditions, these results are not surprising. Similar results have been noted in smaller Phase II studies of other agents for PAH (Barst, et al 2002, Rubin, et al 1990).
Anemia, by decreasing oxygen carrying capacity, reduces systemic oxygen transport. However, in SCD, there is a compensatory increase in CO to maintain tissue perfusion. And although CO is higher in SCD-PH than in non-anemic PH patients (without SCD), it remains lower than in SCD without PH (Anthi, et al 2007). Thus, the observed increase in CO with bosentan in SCD-PH should improve systemic oxygen transport thereby minimizing the adverse cardiovascular effects observed in SCD-PH.
Although bosentan has been approved for other PAH patients, SCD has additional comorbid events (e.g. VOCs, anemia). Safety endpoints in the placebo-controlled studies (using an integrated database of all 26 patients) and the open-label were analyzed, and were consistent with bosentan being well tolerated with no unexpected safety concerns.
The walk-PHaSST trial (double-blind, randomized controlled trial with sildenafil in SCD-PH [Doppler-defined] patients) was recently prematurely terminated due to safety concerns, i.e. increased VOCs in the sildenafil vs. placebo patients (NHLBI Office of Communications, 2009). In contrast, there were no increased episodes of VOCs, other AEs or SAEs in the bosentan patients in ASSET -1 or -2. Although sildenafil is approved for PAH (confirmed by RHC), the early termination of walk-PHaSST highlights the importance of performing well-designed trials to assess safety and efficacy in each specific patient subpopulation. However, it is also important to appreciate that the walk-PHaSST population was not identical to ASSET. Walk-PHaSST included all SCD patients with TRV ≥2.7m/s with only TRV ≥3.0 m/s patients having a RHC at baseline. In contrast, all ASSET patients had PH confirmed by RHC at enrollment.
Minniti et al (2009) recently reported improved exercise capacity in 14 SCD-PH patients after 6 months of treatment with either the dual ET-1 receptor antagonist bosentan or the endothelin type-A selective receptor antagonist ambrisentan. Although these observations were limited by an absence of a placebo arm and all patients were from a single center, these data are consistent with the ASSET data.
Potential safety issues associated with bosentan, i.e. decreased hemoglobin and hepatotoxicity, are relevant in SCD. Although the decrease in hemoglobin was greater with bosentan versus placebo, no patient had study treatment discontinued due to worsening anemia. These findings are similar to observations in much larger populations of patients without SCD. Because SCD patients have elevated levels of aspartate transaminase (AST), indirect bilirubin, and lactate dehydrogenase due to chronic hemolysis, regular monitoring for liver injury relied on ALT and direct bilirubin and not on AST levels. Throughout the studies, no patients had ALT >3× ULN or required a dose change or discontinuation of study drug due to hepatotoxicity.
Recent data suggest that using TRV ≥2.5 m/s to define PH may lead to a false-positive rate as high as 77%, with PH defined as PAPm ≥25 mmHg (Parent, et al 2009). However, it is possible that the increased TRV was observed because echocardiography was performed during a VOC or an ACS episode. As RHCs are usually performed after crisis resolution, this may contribute to a high false-positive rate. Furthermore, whether the increased mortality rate is confined to the subset with hemodynamically confirmed PH (by right heart catheterization), rather than the overall SCD group with TRV ≥2.5 m/s remains unclear. All patients enrolled in ASSET had PH confirmed by RHC before randomization. However, even patients with a TRV ≥2.5 m/s who do not have confirmed PH may have isolated pulmonary systolic hypertension that may be associated with an increased mortality rate, particularly in association with an additional “trigger”, such as a VOC, known to acutely increase pulmonary pressures in SCD (Gladwin and Vichinsky 2008, Machado, et al 2007, Mekontso Dessap, et al 2008).
In conclusion, these limited data suggest that 16 weeks of bosentan in SCD patients with confirmed PH (by right heart catheterization) was well tolerated and continued to be well tolerated in the open-label study. Despite small sample sizes, the correlations between exercise capacity and hemodynamics were consistent with previous reports in other PAH patients, supporting the hypothesis that even mild PH may be clinically significant in SCD. With premature discontinuation of the studies, efficacy analyses were not performed due to the limited sample sizes. With the only other oral class of approved PAH therapies, i.e. phosphodiesterase-5 inhibitors, not appearing to be well tolerated in SCD patients, further investigation appears to be warranted.
Acknowledgments
We gratefully acknowledge all investigators, study coordinators and patients involved in this study. RJ Barst, KK Mubarak, KI Ataga, RL Benza, O Castro, R Naeije, G Simonneau, PS Swerdlow, and MT Gladwin were members of the steering committee responsible for the study protocols. M Hildesheim performed the statistical analyses. ASSET investigators include F Galacteros, CHU Henri Mondor, Créteil, France; G Coghlan, Royal Free Hospital, London, UK; KI Ataga, University of North Carolina, Chapel Hill, NC, USA; R Benza, University of Alabama, Birmingham, AL, USA; A Frost, Baylor College of Medicine, Houston, TX, USA; E Klings, Boston University, Boston, MA, USA; R RF Machado, National Institutes of Health, Bethesda, MD, USA, KK Mubarak, Wayne State University, Detroit, MI, USA; L Muñoz, Alta Bates Medical Center, Berkeley, CA, USA; E Berman-Rosenzweig, Columbia University, New York, NY, USA; W Smith, Virginia Commonwealth University, Richmond, VA, USA; N Sood, Ohio State University, Columbus, Ohio, USA; M Telen, Duke University, Durham, NC, USA.
Funding Actelion Pharmaceuticals Ltd, Allschwil, Switzerland sponsored the studies. The statistical analyses were supported by the National Heart, Lung and Blood Institutes and the University of Pittsburgh (MT Gladwin Funding). MT Gladwin is funded by the Institute of Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
Footnotes
Disclosures: RJ Barst, RL Benza, KK Mubarak, R Naeije, N Sood, KI Ataga and PS Swerdlow have received consultancy fees and research grant funding from Actelion Pharmaceuticals Ltd. M T Gladwin and R Machado received grant support in the form of a Clinical Trials Agreement between the National Institutes of Health and Actelion Pharmaceuticals Ltd. O Castro is a paid consultant at the National Heart, Lung, and Blood Institute. M Hildesheim has no conflicts to disclose.
References
- Anthi A, Machado RF, Jison ML, Taveira-Dasilva AM, Rubin LJ, Hunter L, Hunter CJ, Coles W, Nichols J, Avila NA, Sachdev V, Chen CC, Gladwin MT. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med. 2007;175:1272–1279. doi: 10.1164/rccm.200610-1498OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataga KI, Sood N, De Gent G, Kelly E, Henderson AG, Jones S, Strayhorn D, Lail A, Lieff S, Orringer EP. Pulmonary hypertension in sickle cell disease. Am J Med. 2004;117:665–669. doi: 10.1016/j.amjmed.2004.03.034. [DOI] [PubMed] [Google Scholar]
- Ataga KI, Moore CG, Jones S, Olajide O, Strayhorn D, Hinderliter A, Orringer EP. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol. 2006;134:109–115. doi: 10.1111/j.1365-2141.2006.06110.x. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Rich S, Widlitz A, Horn EM, McLaughlin V, McFarlin J. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot study. Chest. 2002;121:1860–1868. doi: 10.1378/chest.121.6.1860. [DOI] [PubMed] [Google Scholar]
- Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257–1261. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A, Telen MJ. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Am J Hematol. 2008;83:19–25. doi: 10.1002/ajh.21058. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Schechter AN. Nitric oxide therapy in sickle cell disease. Semin Hematol. 2001;38:333–342. doi: 10.1016/s0037-1963(01)90027-7. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359:2254–2265. doi: 10.1056/NEJMra0804411. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1998;92:2551–2555. [PubMed] [Google Scholar]
- Hammerman SI, Kourembanas S, Conca TJ, Tucci M, Brauer M, Farber HW. Endothelin-1 production during the acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med. 1997;156:280–285. doi: 10.1164/ajrccm.156.1.9611085. [DOI] [PubMed] [Google Scholar]
- Little JA, Hauser KP, Martyr SE, Harris A, Maric I, Morris CR, Suh JH, Taylor J, Castro O, Machado R, Kato G, Gladwin MT. Hematologic, biochemical, and cardiopulmonary effects of L-arginine supplementation or phosphodiesterase 5 inhibition in patients with sickle cell disease who are on hydroxyurea therapy. Eur J Haematol. 2009;82:315–321. doi: 10.1111/j.1600-0609.2009.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR, Hunter L, Coles W, Nichols J, Hunter C, Sachdev V, Castro O, Gladwin MT. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol. 2005;130:445–453. doi: 10.1111/j.1365-2141.2005.05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RF, Anthi A, Steinberg MH, Bonds D, Sachdev V, Kato GJ, Taveira-DaSilva AM, Ballas SK, Blackwelder W, Xu X, Hunter L, Barton B, Waclawiw M, Castro O, Gladwin MT. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. 2006;296:310–318. doi: 10.1001/jama.296.3.310. [DOI] [PubMed] [Google Scholar]
- Machado RF, Mack AK, Martyr S, Barnett C, Macarthur P, Sachdev V, Ernst I, Hunter LA, Coles WA, Nichols JP, Kato GJ, Gladwin MT. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol. 2007;136:319–325. doi: 10.1111/j.1365-2141.2006.06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekontso Dessap A, Leon R, Habibi A, Nzouakou R, Roudot-Thoraval F, Adnot S, Godeau B, Galacteros F, Brun-Buisson C, Brochard L, Maitre B. Pulmonary hypertension and cor pulmonale during severe acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med. 2008;177:646–653. doi: 10.1164/rccm.200710-1606OC. [DOI] [PubMed] [Google Scholar]
- Minniti CP, Machado RF, Coles WA, Sachdev V, Gladwin MT, Kato GJ. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol. 2009;147:737–743. doi: 10.1111/j.1365-2141.2009.07906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter KR, Gladwin MT. Pulmonary complication of sickle cell anemia. A need for increased recognition, treatment, and research. Am J Resp Crit Care Med. 2001;164:2016–2019. doi: 10.1164/ajrccm.164.11.2104101. [DOI] [PubMed] [Google Scholar]
- Morris CR, Morris SM, Jr, Hagar W, Van Warmerdam J, Claster S, Kepka-Lenhart D, Machado L, Kuypers FA, Vichinsky EP. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168:63–69. doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHLBI Office of Communications. NHLBI Stops Study of Treatment for Pulmonary Hypertension in Patients with Sickle Cell Disease Due to Safety Concerns. 2009 July 28; http://public.nhlbi.nih.gov/newsroom/home/GetPressRelease.aspx?id=2650.
- Parent F, Bachir D, Lionnet F, Inamo J, Driss F, Savale L, Loko G, Elmazouzi A, Letierce A, Girot R, Galacteros F, Simonneau G. Prevalence and Mechanism of Pulmonary Hypertension in Sickle Cell Disease: A Prospective Multicentre French Study. Am J Respir Crit Care Med. 2009;179:A2646. [Google Scholar]
- Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96:1145–1151. doi: 10.1172/JCI118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- Rivera A, Jarolim P, Brugnara C. Modulation of Gardos channel activity by cytokines in sickle erythrocytes. Blood. 2002;99:357–603. doi: 10.1182/blood.v99.1.357. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Mendoza J, Hood M, McGoon M, Barst R, Williams WB, Diehl JH, Crow J, Long W. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112:485–491. doi: 10.7326/0003-4819-112-7-485. [DOI] [PubMed] [Google Scholar]
- Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood. 1998;92:2594–2596. [PubMed] [Google Scholar]
- Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S, Ernst I, St Peter M, Coles WA, Rosing DR, Blackwelder WC, Castro O, Kato GJ, Gladwin MT. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49:472–479. doi: 10.1016/j.jacc.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KJ, Goodwin SR, Evangelist J, Moore RD, Mehta P. Nitric oxide successfully used to treat acute chest syndrome of sickle cell disease in a young adolescent. Crit Care Med. 1999;27:2563–2568. doi: 10.1097/00003246-199911000-00039. [DOI] [PubMed] [Google Scholar]
- Werdehoff SG, Moore RB, Hoff CJ, Fillingim E, Hackman AM. Elevated plasma endothelin-1 levels in sickle cell anemia: relationships to oxygen saturation and left ventricular hypertrophy. Am J Hematol. 1998;58:195–199. doi: 10.1002/(sici)1096-8652(199807)58:3<195::aid-ajh6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]