

Abstract
Novel synthetic methods for producing an array of chelates for use in “click”-radiolabeling of peptides are described, and their reactivity with regards to subsequent conjugation and radiolabeling is discussed.
Standard techniques for radiolabeling of biomacromolecules use naturally occurring reactive groups which can result in the radionuclide being located at an unpredictable position on the molecule.1 This lack of selectivity is particularly problematic in the case of peptides, because even subtle changes in the structure of a peptide can severely compromise its properties. The current solution for this problem is incorporation of the chelate into the peptide structure during the solid phase synthesis, before cleavage off the resin support and deprotection. This approach requires separate purifications for each synthesized conjugate and therefore complicates the synthesis of conjugate libraries. Conjugation chemistries such as the Huisgen cyclo-addition (“click” reaction) have recently been proposed as a suitable alternative.2–5 Application of “click”-chemistry to peptide radiolabeling calls for convenient synthesis of “clickable” chelates for the radiometals.6,7 Initial studies on the conjugation of the widely used 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) to peptides via “click”-chemistry have been reported, but the value of this method was undermined by the complex (7 steps) synthetic procedures required to make the chelates and the long (12–18 hours) reaction times required for conjugation.8 The full potential of 64Cu-binding (64Cu t1/2 = 12.7 h, ideal for peptide labeling) chelates other than DOTA remain unexplored, despite the fact that 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A) were demonstrated to be superior for use in vivo.9 Desferrioxamine B (DFO), a ligand for radiolabeling with 68Ga (1.1 h)10 and 89Zr (78.4 h),11 has yet to be adopted to click-chemistry also.
We now report a streamlined synthetic approach to the “click” conjugation of chelates and peptides, along with selected members of the library of the “clickable” chelates. We also discuss how this particular conjugation method affects the radiolabeling of the resulting construct with 64Cu.
Scheme 1 illustrates the synthesis of the bifunctional chelators suitable for “click” conjugation. In order to avoid the formation of oligomers, only one reactive group should be present on the chelate. CB-TE2A was derivatised for use in the “click” reaction via a sequence of alkylation reactions. The first step involved reaction with protected bromoacetate,12 followed by chromatographic isolation of the monoalkylated product 1. The second reaction was performed by using similar conditions. This method afforded “clickable” CB-TE2A (2) reproducibly in around 80 to 90% yield. The DOTA analog 3 was synthesized in a similar manner. Commercial availability of monoactivated p-benzyl-SCN-DFO simplifies the synthesis of a respective “clickable” DFO analog. Simple reaction with propargyl amine yielded compound 4 in modest yield (46%) and isolation was achieved by using a C18 Sep-pak cartridge purification method common to radiochemistry.
Scheme 1.

Synthesis of the “clickable” chelates. (i) See ESI† for full details.
The synthesized chelates were tested in “click” conjugation reactions involving a model peptide, which is known to target gastrin-releasing peptide receptors. The peptide was synthesized in accordance with standard Fmoc-protocols, and an azide-containing amino acid was coupled onto the N-terminus (see ESI† for details). Screening of the synthetic conditions for coupling the chelates to the peptide revealed that Cu+ formed in situ by reduction of CuCl2 with ascorbic acid demonstrated the highest catalytic activity. Neither salts of Cu+ nor other reducing agents nor addition of triazole ligands13 showed comparable activity. This system afforded quantitative conversion of the peptide 5 within 5 minutes at room temperature in aqueous solution. Quenching of the reaction with 0.01 M Na2S (used to scavenge the copper catalyst) allowed for isolation of the metal-free conjugate using a C18 cartridge and the analytically pure sample was obtained after HPLC purification. Macrocyclic carboxyl groups were then deprotected with a TFA–iPr3SiH–EDT cleavage cocktail. This protocol allowed the conjugation of both CB-TE2A and DOTA onto the model peptide (Scheme 2).
Scheme 2.

“Click” conjugation onto the model peptide. (i) CuCl2, ascorbic acid in water, RT; (ii) trifluoroacetic acid, iPr3SiH, EDT.
Preliminary radiolabeling experiments showed that both CB-TE2A and DOTA bearing constructs can be efficiently labeled with 64Cu. At pH 6 in ammonium acetate buffer, the DOTA compound 6 forms the copper complex almost instantaneously. Radio-high performance liquid chromatography (HPLC) showed ~84% conversion (>95% purity after HPLC purification). The CB-TE2A conjugate (7) required heating for 5 min at 70 °C and after purification yielded radiolabeled conjugate of the same purity. To estimate the lower achievable limit of the specific activity we gradually increased the amount of 64Cu added to the point of the peptide saturation. The specific activity of the radiolabeled conjugate can be as high as 250 mCi mg−1. Experiments with non-radioactive Cu demonstrated the difference in retention time of the starting peptide and its copper derivative to be greater than 1 min for both chelators. Therefore, the specific activity of the HPLC purified product is primarily determined by the purity of the starting 64CuCl2. The non-radioactive copper derivatives of 6 and 7 co-eluted with the respective radioactive compounds, confirming the identity of 64Cu-6 and 64Cu-7.
The rate of the “click” reaction and the mild conditions of the radiolabeling are at odds with the previously published data. The peptide we employed as a model system for the click conjugation has been previously conjugated with CB-TE2A via the use of a conventional amide linker, but required harsh conditions for radiolabeling with 64Cu up to 1.5 h at 95 °C.14 Similarly, typical experimental conditions for the DOTA radiolabeling include heating at 50 to 65 °C.15,16 This difference indicates that the triazole group is not an innocent linker, and suggests that there is an interaction between the lone pair of the azole ring and the metal ion. This interaction was not observed previously, because we are first to report the construct with a triazole group in the immediate vicinity of the copper ion. However, reports of triazole interaction with Tc do confirm the possibility of this interaction.4 Coordination of copper with the chelating moiety could also facilitate the “click” reaction by bringing the catalyst in close proximity to the triple bond.
In the absence of a crystal structure, we conducted density functional theory (DFT) calculations in order to address whether the triazole ring of the copper labeled CB-TE2A “click” complex 64Cu-5 is capable of forming a coordinate bond to the Cu2+ ion.17 Fig. 1 shows a picture of the DFT [uB3LYP/6-31+G(d,p)] optimized geometry of the model Cu complex 1 (see ESI† for full computational details and analysis).10 The optimized metal-to-ligand bond lengths reveal that the complex is highly distorted and the elongated r(Cu–N7) bond length and r(Cu⋯N3) bonding interaction of 2.299 and 2.559 Å, respectively, are consistent with the expected Jahn–Teller effects from the d9 electron configuration.
Fig. 1.
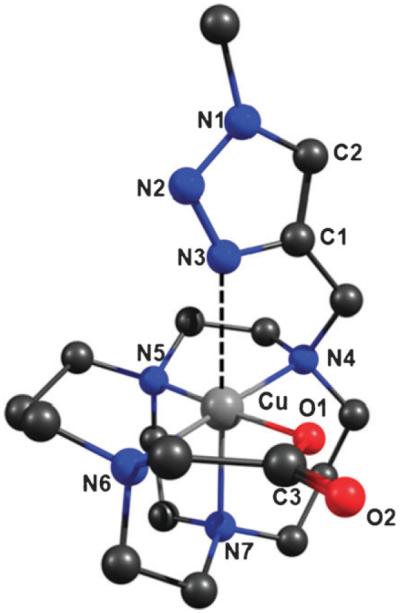
DFT optimized structure of the hypothetical model complex. r(Cu⋯N3) = 2.559 Å; r(Cu–N4) = 2.110 Å; r(Cu–N5) = 2.092 Å; r(Cu–N7) = 2.299 Å; r(Cu–O1) = 1.930 Å.
Natural population analysis (NPA) correctly predicted a net positive charge on the Cu ion of 1.328e, and negative charges on all electronegative donor atoms (Table S1, ESI†). The nitrogen donor atoms N4–N7 of the CB-TE2A chelate have calculated NPA charges in the range −0.604 to −0.627e, and the donor oxygen atom has a net negative charge of −0.863e. Interestingly, the nitrogen atoms N1–N3 of the triazole ring show alternating NPA charges of −0.178, −0.068 and −0.338e, respectively. The negative NPA charge on nitrogen atom N3 is consistent with high electron density on this atom which facilitates a bonding interaction between the lone pair of electrons on atom N3 and the Cu2+ ion.
In order to ascertain if the Cu⋯N3 interaction constitutes a bond, natural bond orbital (NBO) analysis was used to investigate the nature and magnitude of the stabilizing interactions calculated within complex 1. NBO analysis revealed that stabilizing orbital electron density interactions between the ligand donor atoms and the Cu2+ ion contribute 20.2% of the overall stability of complex 1, of which 16.9% arises from ligand-to-metal donation and only 3.3% from metal-to-ligand back-bonding. The strongest bonding interaction occurs between the Cu2+ ion and the donor oxygen atom O1 with a total NBO interaction energy of 98 kcal mol−1 (5.3% of the total interaction). Interactions between the Cu2+ ion and the equatorial donor nitrogen atoms N4–N6 contribute between 62–66 kcal mol−1 each to the overall stability of complex 1. Despite the shorter bond length of r(Cu–N7) of 2.299 Å compared to 2.559 Å from r(Cu⋯N3), NBO analysis indicates that the Cu⋯N3 bond (47 kcal mol−1, 2.6% of the total NBO stabilization energy) is stronger than the Cu–N7 bond (35 kcal mol−1 [1.9%]). The additional stabilization energy for the Cu⋯N3 interaction arises due to an increased contribution from metal-to-ligand back-bonding which accounts for 26.8% of the total bonding interaction (Table S2, ESI†). In contrast, back-bonding only accounts for 16.6% of the Cu–N7 interaction. The strength of the Cu⋯N3 interaction is calculated to be equal to 49.0% of the strength of the Cu–O1 interaction and around 77.0% of the strength of the equatorial Cu–[N4–N6] interactions.
Molecular orbital (MO) analysis also reveals the origins for the difference in bond strength observed between the Cu–N7 and Cu⋯N3 bonds. The highest occupied molecular orbital (HOMO) in the β-spin manifold (βHOMO), and its α-spin spatial counterpart, the αHOMO–1, showed strong metal–ligand σ*-anti-boding interactions between the dz2 orbital of the Cu2+ ion and pσ-orbitals of the donor nitrogen atoms N3 and N7 (Fig. S1, ESI†). The two spin orbitals show pronounced asymmetry with a larger MO coefficient from N7 atom and only a small contribution from the N3 donor. Differential orbital contributions from the N3 and N7 donor orbitals reduce the Cu–N7 bond order and weaken the bond to a greater extent than the Cu⋯N3 bond.
In summary, we have presented versatile methods for the synthesis of novel bifunctional chelates suitable for “click” conjugation to the peptides. “Click” conjugation of these molecules was demonstrated on a model peptide and the resulting conjugates have been labeled with 64Cu, yielding radiolabeled constructs with high chemical purity and specific activity. In contrast to reported conditions for radiolabeling the complexation reaction between 64Cu2+ and the CB-TE2A compound 4 was performed under mild conditions. This change in radiolabeling chemistry may be the result of additional interactions between the Cu2+ ion and the triazole ring. Further studies investigating the kinetics of radiolabeling and complex formation are underway.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Office of Science (BER), U. S. Department of Energy (Awards DE-SC0002456 and DE-SC0002184), for funding. We thank Prof. Jennifer C. Green for access to computational facilities.
Footnotes
Electronic supplementary information (ESI) available: Materials and methods, synthesis and radiolabeling, density functional theory (DFT) calculations, NMR and mass spectra, LC–MS profiles, radio HPLC profiles. See DOI: 10.1039/b924784j
Notes and references
- 1.Brechbiel MW. Q. J. Nucl.Med.Mol. Imaging. 2008;52:166–173. [PMC free article] [PubMed] [Google Scholar]
- 2.de Graaf AJ, Kooijman M, Hennink WE, Mastrobattista E. Bioconjugate Chem. 2009;20:1281–1295. doi: 10.1021/bc800294a. [DOI] [PubMed] [Google Scholar]
- 3.Marik J, Sutcliffe JL. Tetrahedron Lett. 2006;47:6681–6684. [Google Scholar]
- 4.Mindt TL, Struthers H, Brans L, Anguelov T, Schweinsberg C, Maes V, Tourwe D, Schibli R. J. Am. Chem. Soc. 2006;128:15096–15097. doi: 10.1021/ja066779f. [DOI] [PubMed] [Google Scholar]
- 5.Mindt TL, Muller C, Stuker F, Salazar JF, Hohn A, Mueggler T, Rudin M, Schibli R. Bioconjugate Chem. 2009;20:1940–1949. doi: 10.1021/bc900276b. [DOI] [PubMed] [Google Scholar]
- 6.Struthers H, Mindt TL, Schibli R. Dalton Trans. 2010;39:675–696. doi: 10.1039/b912608b. [DOI] [PubMed] [Google Scholar]
- 7.Anderson CJ, Ferdani R. Cancer Biother. Radiopharm. 2009;24:379–393. doi: 10.1089/cbr.2009.0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knor S, Modlinger A, Poethko T, Schottelius M, Wester HJ, Kessler H. Chem.-Eur. J. 2007;13:6082–6090. doi: 10.1002/chem.200700231. [DOI] [PubMed] [Google Scholar]
- 9.Garrison JC, Rold TL, Sieckman GL, Figueroa SD, Volkert WA, Jurisson SS, Hoffman TJ. J. Nucl. Med. 2007;48:1327–1337. doi: 10.2967/jnumed.107.039487. [DOI] [PubMed] [Google Scholar]
- 10.Smithjones PM, Stolz B, Bruns C, Albert R, Reist HW, Fridrich R, Macke HR. J. Nucl. Med. 1994;35:317–325. [PubMed] [Google Scholar]
- 11.Holland JP, Sheh YC, Lewis JS. Nucl. Med. Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boswell CA, Regino CAS, Baidoo KE, Wong KJ, Bumb A, Xu H, Milenic DE, Kelley JA, Lai CC, Brechbiel MW. Bioconjugate Chem. 2008;19:1476–1484. doi: 10.1021/bc800039e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J. Am. Chem. Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 14.Wadas TJ, Eiblmaier M, Zheleznyak A, Sherman CD, Ferdani R, Liang K, Achilefu S, Anderson CJ. J. Nucl. Med. 2008;49:1819–1827. doi: 10.2967/jnumed.108.054502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li WP, Lewis JS, Kim J, Bugaj JE, Johnson MA, Erion JL, Anderson CJ. Bioconjugate Chem. 2002;13:721–728. doi: 10.1021/bc015590k. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Z, Xiong ZM, Subbarayan M, Chen XY, Gambhir SS. Bioconjugate Chem. 2007;18:765–772. doi: 10.1021/bc060306g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frisch MJ. GAUSSIAN03. Gaussian Inc.; (see ESI† for the complete reference) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.