

Abstract
The dimerization of HIV reverse transcriptase (RT), required to obtain the active form of the enzyme, is influenced by mutations, non-nucleoside reverse transcriptase inhibitors (NNRTIs), nucleotide substrates, Mg ions, temperature, and by specifically designed dimerization inhibitors. In the present study, we have utilized NMR spectroscopy of the [methyl-13C] methionine labeled enzyme and small angle X-ray scattering (SAXS) to investigate how several of these factors influence the dimerization behavior of the p51 subunit. The 1H-13C HSQC spectrum of p51 obtained at micromolar concentrations indicates that a significant fraction of the p51 adopts a “p66-like” conformation. SAXS data obtained on p51 samples were used to determine the fractions of monomer and dimer in the sample, and to evaluate the conformation of the fingers/thumb subdomain. All of the p51 monomer observed was found to adopt the compact, “p51C” conformation observed for the p51 subunit in the RT heterodimer. The NMR and SAXS data indicate that the p51 homodimer adopts a structure that is similar to the p66/p51 heterodimer, with one p51C subunit and a second p51 subunit in an extended, “p51E” conformation that resembles the p66 subunit of the heterodimer. The fractional dimer concentration and the fingers/thumb orientation is found to depend strongly on the experimental conditions, and exhibits a qualitative dependence on nevirapine and ionic strength (KCl) that is similar to the behavior reported for the heterodimer and the p66 homodimer. The L289K mutation interferes with p51 homodimer formation as it does with formation of the heterodimer, despite its location far from the dimer interface. This effect is readily interpreted in terms of a conformational selection model, in which p51L289K has a much greater preference for the compact, p51C conformation. Reduced dimer formation then results from the reduced ratio of the p51EL289K to p51CL289K monomers.
Keywords: HIV-1 Reverse transcriptase, p51 subunit of reverse transcriptase, SAXS, [methyl-13C] methionine RT, NMR spectroscopy
HIV-1 reverse transcriptase (RT) has emerged as a central target for drug intervention in the treatment of AIDS (1, 2). It plays a pivotal role in HIV replication by converting single-stranded genomic RNA into double-stranded proviral DNA. The enzyme is composed of two subunits, p66 and p51, with the polymerase and RNase H active sites located in the p66 subunit. The p51 subunit is identical in sequence to p66, but lacks the C-terminal RNase H domain and adopts a very different conformation in the heterodimer. Studies utilizing an RT mutation that interferes with dimerization support the sequential processing of an initially formed p66 homodimer, followed by proteolytic processing to yield the mature, p66/p51 heterodimer (3). The dimerization of HIV reverse transcriptase has been of interest since it can be significantly influenced by non-nucleoside RT inhibitors (NNRTI) and is therefore presumably related to their mechanism of action (4–6). Because drug-resistant strains of RT continue to evolve, the development of dimerization inhibitors has been an active area of research (7–14).
The dimerization of HIV reverse transcriptase is influenced by mutations (15–25), nucleotide substrates (26), temperature, magnesium, and other solution conditions (8, 26–29). For residues located at or near the dimer interface, the effect of the mutation on dimer stability is readily interpreted, while for other mutations, the effect appears to be more indirect. Among the dimerization-interfering mutations, L289K is one of the most counterintuitive (15). The L28951 residue is positioned at the dimer interface and makes multiple hydrophobic contacts with residues on the p66 subunit, while the L28966 residue is a surface-exposed residue located at the tip of the thumb subdomain, far from the interface. Unexpectedly, the L289K66 mutation strongly interferes with dimerization, while the L289K51 mutation is benign (15). If the structure of the RT heterodimer is used as a model for the p51 homodimer, neither of the L289 residues is positioned near the dimer interface. In addition to the surprising effects of the L289K mutation on dimerization behavior, results for the p51 homodimer dissociation constant (5, 27) and for the effects of the NNRTI nevirapine on dimer formation (4, 30, 31) have been inconsistent. The p51 homodimer has been reported to exhibit approximately half the activity and processivity of the RT heterodimer (32), however it is well known that the primer/template substrate used for activity measurements significantly promotes dimerization (8, 26, 29, 33), and p51 activity shows a particular sensitivity to the nature of the substrate used.
In the present study, we have utilized biophysical techniques that directly probe the solution behavior of p51. We have extended our use of [methyl-13C] methionine labeling to understand conformational aspects of the dimerization process for the p51 subunit. The dependence of the methionine methyl resonances on subunit conformation provides a useful basis for characterizing the conformational heterogeneity of the p51 homodimer. Application of the SAXS technique to the characterization of the p51 solutions has been found to be particularly valuable, both for quantifying the monomer/dimer ratio, and for evaluating the open/closed conformational preferences of the fingers/thumb subdomains. Previous evaluations of this ratio required considerably more involved and invasive procedures, such as spin-labeling of the protein (34). Based on these studies, we have also developed a conformational selection model that provides a more physically intuitive basis for understanding how some mutations and NNRTIs can influence the dimerization process. This approach allows analysis of effects that arise not from a direct perturbation of the dimer interface, but rather from a change in the conformational preferences of the p51 monomer.
Materials and Methods
Materials
Oligonucleotides used as PCR primers for site-directed mutagensis were purchased from Integrated DNA Technologies. Unlabeled amino acids, Q-Sepharose FF and the ssDNA cellulose matrix were purchased from Sigma-Aldrich. [ε-13C] Methionine was obtained from Cambridge Isotope Laboratories. Isopropyl thio-D-galactoside (IPTG) was from Invitrogen. HiLoad 26/60 Superdex 200 column was from Amersham Pharmacia Biotech AB.
The plasmids pET21a (+) p66 and pET30a (+) p51 were a gift from the laboratory of Dr. Sam Wilson, NIEHS. The p51 construct terminates at W426. Mutagenesis studies were carried out by using the QuickChange XL site-directed mutagenesis kit (Stratagene). The desired mutated gene sequences were confirmed by DNA sequence analysis. The two expression plasmids were transformed into E. coli BL21 (DE3) codon plus RIPL, and the protein expression was induced for 4 hours at 37 °C by addition of 0.5 mM IPTG into the culture, when the OD600 value reached 0.8~1. The mutants constructed for these studies included: M16L, M230L, M230L/M357L, C280S, and L289K.
Sample Preparation
For the purpose of NMR studies, each mutant of p51 was expressed at 37 °C using enriched medium (PAG) (35) containing 17 unlabeled amino acids (no C, Y and M), plus [ε-13C] methionine, which is expected to repress the endogenous synthesis of methionine by the bacteria (36). The purification of the samples was identical to that previously described (37, 38). Briefly, no affinity tags were used so three columns were used for purification: cation-exchange followed by an ssDNA cellulose column and finally size exclusion chromatography.
The final samples were exchanged into NMR buffer (10mM Tris-HCl-d11, pD7.6, 200 mM KCl, 1.5 mM sodium azide, 4 mM MgCl2, and 100 μM 2,2-dimethylsilapentane-5-sulfonic acid (DSS) as an internal chemical shift standard, in D2O) using a PD-10 desalting column (Pharmacia), and further concentrated to approximately 50 μM. The concentration of each sample was determined by uv absorbance. The use of at least 200 mM KCl was determined to be important for the long term stability of the samples required for the NMR investigations. Use of lower salt concentrations resulted in gradual precipitation of the protein.
NMR spectroscopy
All NMR experiments were performed at 25 °C using a Varian UNITY INOVA 500 MHz NMR spectrometer, equipped with a 5 mm Varian (500 MHz) 1H{13C, 15N} triple-resonance cryogenically cooled probe, with actively shielded Z-gradients. We used the Varian gChsqc experiment included in Biopack with the phase-cycling option. The acquisition parameters for all experiments were 64 transients, 64 ms acquisition, 1 s relaxation delay with 1024 points and sweep width of 14 ppm. In the indirect dimension, 128 points were acquired with a sweep width of 11 ppm, the 13C offset was set to 17 ppm. All NMR data were processed using NMRPipe (39) and analyzed with NMRviewJ (40).
Size-exclusion FPLC analysis
The SEC-FPLC was performed at room temperature on an Akta FPLC system using a SuperdexTM 200 10/300 GL column (Amersham Bioscience). Protein samples (100 μL) were loaded and eluted with running buffer (50mM Tris-HCl, 200mM NaCl, pH 8.0) at a flow rate of 0.75 ml/min. Data is presented for the following samples: 43μM p51, 43μM p51 in the presence of nevirapine, 80μM p51L289K and 50μM p51L289K plus nevirapine, and 15μM RT, used as a control. The elution profiles were recorded at 280 nm after the column had been pre-equilibrated with running buffer.
SAXS data and analysis
SAXS spectra were obtained at the National Synchrotron Light Source Beamlines X21 and X9 (Brookhaven National Laboratory). The wavelengths of the beam were 1.24 or 0.855 Å respectively. For the SAXS analysis, protein was dialyzed into 50 mM Tris-HCl, pH 8.0, 200 mM KCl buffer with either no magnesium or 4 mM MgCl2, as indicated. The recorded intensity of each sample was circularly averaged and scaled to obtain a relative scattering intensity I(q) as a function of momentum transfer vector, q = 4πsinθ/λ, after subtraction of buffer scattering contributions. SAXS data were collected on 1 – 6 mg/ml protein concentrations. Background-subtraction, averaging, and scaling were conducted with Primus (41).
CRYSOL was used to compare the SAXS-based intensity data with theoretical intensity data derived from the atomic structures obtained from X-ray crystallographic studies (42). Additionally, the scattering data was analyzed according to the method of Svergun and coworkers, using OLIGOMER to compute the fraction of structures present with known scattering (41). This procedure assumes that the macromolecule(s) present in the cell can adopt a limited number of pre-determined structures or conformations, and seeks to optimize the fit of the experimental scattering curve by varying the fractional contributions of the scattering curves calculated for each component structure. The success of this approach is dependent on how well the assumed conformational ensemble covers the set of conformations present in solution. Three models were considered for isolated p51, as shown in Figure 1. In the first, p51 adopts a compact globular conformation identical to its conformation in the RT heterodimer (Fig. 1a). The isolated p51 subunit was also allowed to adopt two alternate, extended conformations similar to those observed for the first 426 residues of the p66 subunit in the heterodimer, referred to here as p51Eopen and p51Eclosed. For the first model, the fingers and thumb were positioned in an open conformation, as observed in RT-NNRTI complexes such as 2VG5 (43) (Fig. 1b), while in the second, the thumb and fingers were positioned in a closed conformation, as observed in structure 3DLK (44) (Fig. 1c). Two structures for the p51 homodimer were also included in the structural ensemble. The p51 homodimer was modeled by truncating the p66 subunit at residue 426, and allowing the fingers/thumb conformation to be either open, p51Eopen/p51C (Fig. 1d) or closed p51Eclosed/p51C (Fig. 1e). All structures lack the RNase H domain of p66/p51 RT (Fig. 1f). The theoretically predicted SAXS curves are shown in Figure 1g. The intensity and inflection of these contributing contributions differs sufficiently to support the feasibility of a conformational deconvolution of the SAXS data. The lines are color coded with respect to the structures shown in 1a–e.
Figure 1.
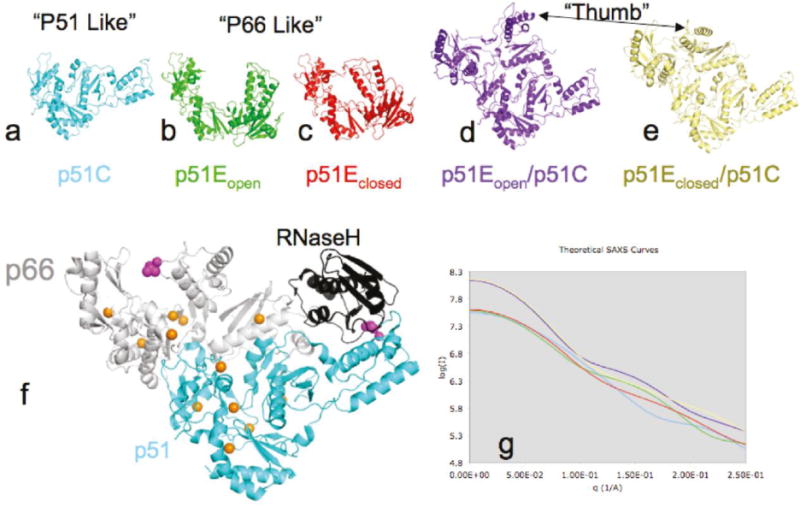
Structures of RT and predicted SAXS parameters. The structures in panels a–e were used to predict the SAX scattering profiles in panel g. The color coding of the SAXS curves in panel g is identical to the coloring of the structures in panels a–e. A detailed discussion of the component structures is contained in the text. Panel f shows 3DLK with p51 colored cyan and p66 colored white except for the RNase H domain that is colored black. Orange spheres indicate the position of the methyl group of the methionine residues. The side chain of L289 is shown with magenta spheres.
The main assumptions made in the above analysis are: 1) the actual structural ensemble present in solution can be described as a mixture of these five component structures only; 2) we neglect any dynamic aspects of these structures; 3) the p51E and p51E/p51C dimer structures can be modeled by dropping the RNase H domain from the corresponding p66/p51 heterodimer structures. Removal of the RNase H domain to create the p51E/p51C homodimer structure eliminates the RNase H interfaces with the connection domain in p51E, and with the thumb domain of p51C. The loss of these interactions might lead to some additional destabilization and conformational heterogeneity. The experimental support for this approach is based on: 1) the low χ2 values obtained for the analyses under a broad range of experimental conditions, and 2) the qualitative consistency of the results with the effects of concentration, nevirapine, acetonitrile and Mg predicted from the literature describing the behavior of p51, p66 and p66/p51.
Equilibrium modeling
Numerical evaluation of the sets of equilibrium equations presented in this study was performed using Mathematica (Wolfram Research, Champaign, IL), and following an approach similar to that described by Venezia et al. (5). Briefly, solutions of the simultaneous algebraic equations describing the equilibria were obtained using the Solve and Evaluate commands.
Nomenclature
Subscripts have been used to denote the subunit involved when there is any possibility of ambiguity, e.g., M18466 refers to residue M184 in the p66 subunit. We have also introduced the nomenclature p51C, to describe the p51 monomer in that compact, globular conformation which it adopts in heterodimeric RT, and p51E to describe the extended, p66-like conformation that it can adopt when it forms a p51 homodimer. We thus refer to the p51 homodimer structure as p51E/p51C, analogous to the p66/p51 nomenclature used for the heterodimer. When a specific conformation of the fingers/thumb subdomain is also indicated, the structure is further identified as p51Eopen, p51Eclosed, p51Eopen/P51C, and so forth.
Results
NMR Characterization of RT and p51 in Solution
Figure 2 compares the 1H-13C HSQC spectra of HIV-1 RT containing [methyl-13C] methionine in either the p66 or the p51 subunits, with the spectra obtained for p51 alone. The resonances for four of the six methionine methyl groups have been assigned previously (38), while the resonances for two residues with very low solvent exposure, M41 and M164, are not readily observed as a result of greater dipolar broadening. The enzyme contains two active site methionine residues, M184, a component of the active site YMDD motif, and M230, located on the primer grip and positioned to make direct contact with the primer terminus of the DNA substrate. Based on crystal structures of RT, the environment of these two residues is expected to be very different in the two subunits, so that the NMR parameters are expected to show a corresponding subunit dependence. The M184 resonances show the expected dependence (Figure 2a,b), with M18451 not readily visible as a result of the shorter T2 value characterizing the buried residue, while the M230 resonances appeared to be similar in both subunits. We note that segments of the protein containing M230 were disordered in three of four p51 subunits and one p66 subunit in crystal structures of the apo enzyme, so that the observation may be biased towards the observation of the disordered species (38).
Figure 2.
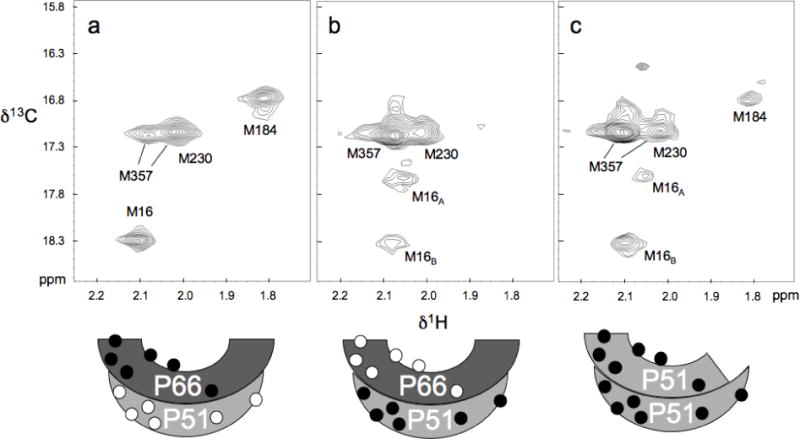
1H-13C HSQC spectra of a) 57 μM [methyl-13C] methionine66 RT, b) 50 μM [methyl-13C] methionine51 RT. and c) 30 μM [methyl-13C] p51 subunit. All samples were dissolved in the NMR buffer: 10 mM Tris-HCl-d11, pD 7.6, 200 mM KCl, 1.5 mM sodium azide, 4 mM MgCl2, and 100μM DSS as an internal chemical shift standard, in D2O. Spectra were obtained at 25°C. Schematic figures at the bottom indicate the subunit labeling pattern, with filled circles indicating [methyl-13C] methionine labeling. The spectra of the p66/p51 heterodimer preparations shown in panels a and b are reproduced from Figure 2a and 2b of Zheng et al. (38). Preparation of the selectively labeled RT heterodimer is described in that reference.
The other assigned methionine resonances, corresponding to residues M16 and M357, exhibit two resonances each, with characteristics that are also subunit-dependent. Confirmation of the M16 assignments is provided by spectra of the mutant p51M16L shown in Figure 3a, in which both M16 resonances have been eliminated. In the p66 subunit the intensity of M16A >> M16B, while p51, the intensities of the two component resonances are more similar, Figure 2. Finally, M357 also was found to produce two resonances, with M357A much more intense in the p51 subunit than in p66 (38). This difference, as well as the two observed component resonances, appears to be related to the proximity of M35766 to the subunit interface. The segment containing M35751 is disordered in two of the reported crystal structures of the apo enzyme, and the segment containing this residue is probably highly mobile in the p51 subunit, explaining its high intensity.
Figure 3.
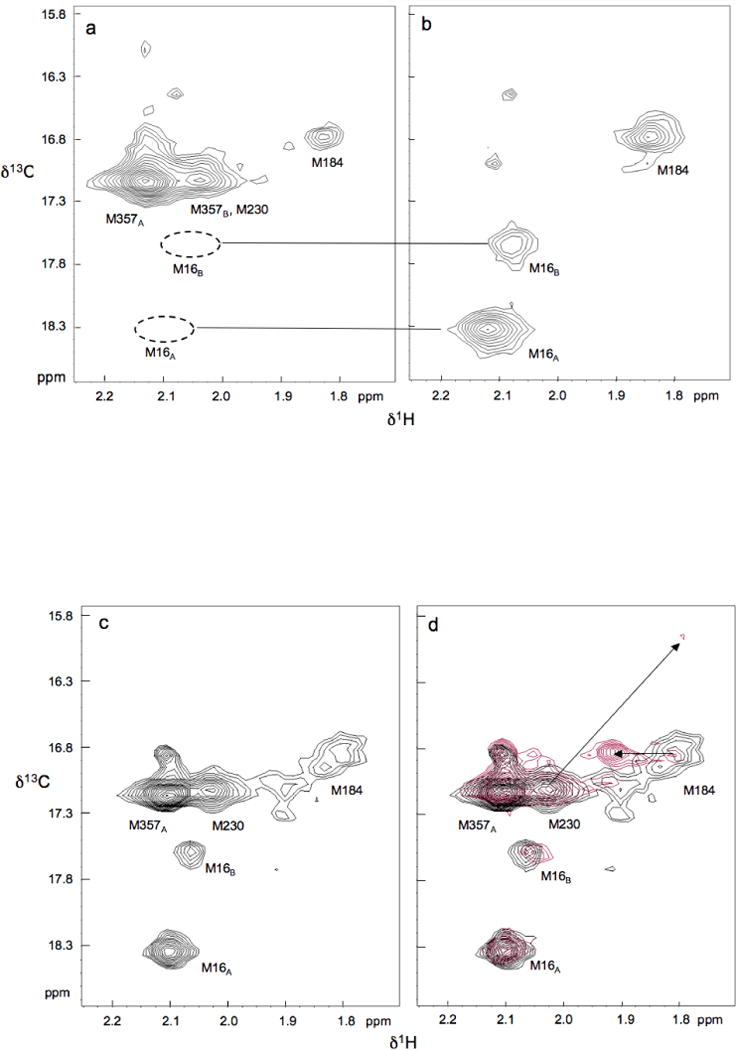
1H-13C HSQC spectra of [methyl-13C] methionine p51 mutants: a) M16L; b) M230L, M357L; c) L289K; d) L289K (black) and L289K + nevirapine (red). The arrows in (d) indicate nevirapine-induced shifts. Residue M16 produces two resonances, labeled M16A and M16B, with the M16A/M16B ratio considerably higher in p66 and p51E subunits than in the p51C subunit. The M357 residue in the p51E subunit also produces two resonances, one of which, M357B, overlaps M230. The intensity of the M357B resonance appears to be related to the proximity of M357 to the dimer interface. Temperature and buffer conditions were as in Figure 2.
The above assignments provide a basis for interpretation of the spectrum obtained for the isolated [methyl-13C] methionine p51 subunit, which can exist in solution as either a monomer or a dimer. Based on a recently determined p51 homodimer dissociation constant of 230 μM measured at 5 °C, we initially estimated that at 30 μM p51 this sample should be ~ 90 % monomer, 10 % dimer (5). The most striking feature of the HSQC spectrum obtained for the [methyl-13C] p51 sample is the appearance of a reasonably intense resonance with 1H and 13C resonance shifts identical to those observed for M18466 in the heterodimer (cf. Fig. 2a,c). In combination with the homodimer dissociation constant given above, this observation supports the conclusion that a substantial fraction of the p51 monomer adopts an extended, p66-like conformation, rather than the compact globular structure observed for p51in the RT heterodimer. In contrast, the strong intensity of the M357 resonance is more similar to that observed for the p51 subunit of RT, supporting the opposite conclusion. However, a quantitative examination of the relative resonance intensities of the p51 sample, normalized using the total M16 resonance intensity, indicates that they approximate an average of the intensities observed for equal concentrations of the p66 and p51 subunits (Table 1). Thus, the evidence of the HSQC spectrum supports the conclusion that even at a 30 μM concentration, most of the sample exists as a p51 homodimer that is a conformational heterodimer. In particular, the intensities of the M184 and M16 resonances closely approximate the average values expected if the p51 subunit adopts the two different tertiary structures with equal probability.
Table 1.
Normalized Intensities of p51 resonances
| p66/p51a | p51 | L289K p51 | L289K p51+NVP |
|
|---|---|---|---|---|
| M16 | 1.0 | 1.0 | 1.0 | 1.0 |
| M41 | – | – | – | – |
| M164 | – | – | – | – |
| M184 | 0 | 0.45 | 0.21 | 0.50 |
| M230 | 1.4 | 0.89 | 0.69 | 0.52 |
| M357 | 4.1 | 3.24 | 2.8 | 3.0 |
Resonance intensities for [methyl-13C]methionine51 RT are normalized relative to the total intensity of both M16 resonances.
As a related technical point, we wish to address the potential spectral consequences of contaminant unfolded or partially unfolded protein. One limitation on the use of [methyl-13C] methionine as an NMR probe is the frequent appearance of a significant resonance or group of resonances with shift values near δ1H, 13C = 2.05, 17.0 ppm, that results from the presence of some soluble denatured protein. Such resonances can be observed in many of the reported spectra of methionine-labeled enzymes. As a result of the high internal mobility of the methionine residues and the tendency of all of the methionine residues of the denatured protein to resonate at similar shifts, the corresponding resonance intensity of a relatively small fraction of denatured protein can be significant. Since the M357 resonance, which has a high degree of solvent exposure, is near this position, we also obtained a spectrum of the doubly mutated [methyl-13C] methionine p51M230L, M357L (Fig. 3b). As is apparent from Figure 3, the only significant resonances that remain correspond to M184 in a p66-like environment, and to M16, presumably arising from the p51 subunits in both the p66-like and p51-like conformations. Hence, this observation supports the conclusion that the M357 resonance contains at most a very small contribution from unfolded protein.
SAXS analysis of p51
SAXS data were obtained on p51 solutions under various conditions, as described in Methods. Our analysis assumes that in all cases the sample contained an undetermined mixture of five structures – three monomers and two dimers as described in the methods section and illustrated in Figure 1. Figure 4 shows two examples of the fit of the linear combinations of the predicted scattering profiles with the experimental data using the program OLIGOMER (41). In both cases shown here, the fit is very good (χ2 = 4.8 and 3.1) in the range of long vectors and somewhat poorer in the range the highest resolution data, which is typical for SAXS fits.
Figure 4.
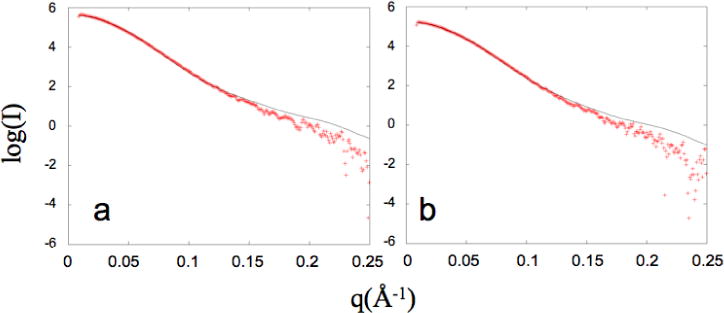
SAXS scattering profiles analyzed with OLIGOMER. The experimental SAXS data is indicated with plus signs and the fit of a linear combination of predicted scattering profiles is drawn with a black line for a) 60 μM p51 and b) 40 μM p51. The χ2 of the fit for a) and b) is 4.8 and 3.1, respectively. All other SAXS fits are displayed in Supporting Information.
Figure 5 summarizes the fractional percent of each component structure present in solution under the conditions indicated based on the analysis of the SAXS data. For clarity, the color coding of the structures in Figure 1 is the same as the bars in Figure 5. The fits indicate that a sample containing 60 μM p51 has a monomer:dimer ratio of ~ 1:3, and that all of the dimer present adopts a closed fingers/thumb subdomain orientation. No significant contributions were observed for each of the three other structures allowed by the modeling. At slightly lower concentration (40 μM p51) the monomer:dimer ratio increases to ~1:2 and the fingers/thumb orientation remains closed. Using a simple monomer/dimer equilibrium analysis described by the relation:
| [1] |
the data in columns 1 and 2 of Figure 5 correspond to KDapp ~ 4 μM, while the data in columns 3 and 4 for p51L289K correspond to KDapp ~ 36 μM, a 9-fold increase. The qualitative effects of concentration, nevirapine, and the L289K mutation on conformation and on the monomer/dimer ratio, discussed in greater detail below, are all consistent with expectations based on the related dimerization literature. These results confirm the interpretation of the NMR spectra, which imply that a substantial amount of dimer was present. These KDapp values are substantially lower than a recent reported value of 230 μM (5), reflecting the hypersensitivity of the dimerization process experimental conditions.
Figure 5.
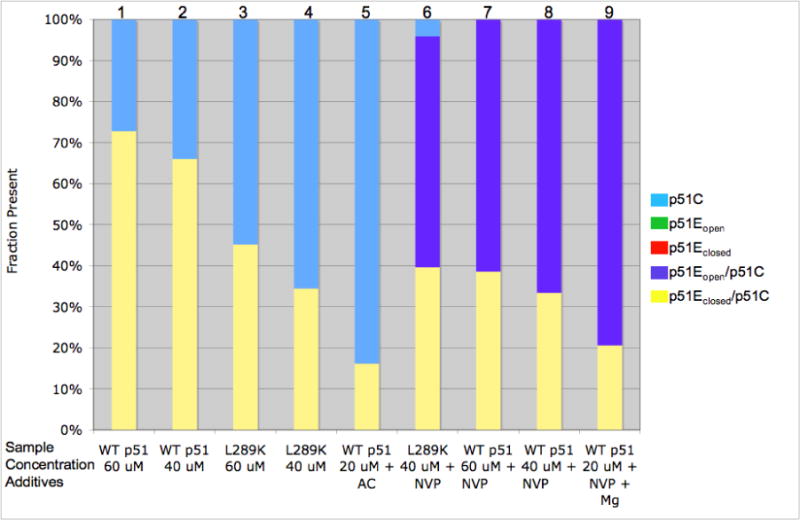
Fraction of p51 models present in solution. Based on OLIGOMER analysis of SAXS data the fraction of each p51 model present is shown in a bar graph, with the total set to 100%. Results of the analysis for each sample are summarized in a Table included as Supporting Information. Each column is a different sample indicated at the bottom of the figure. The bars are colored to be similar to the structures shown in figure 1. No p51E monomer, either open (green) or closed (red) was found in the analysis. The additives indicated at the bottom of the columns are: Mg – 4 mM MgCl2; NVP - 0.5 mM nevirapine, AC – 10 % acetonitrile. All samples were dissolved in 50 mM Tris-HCl, 200 mM KCl, pH 8.0 buffer.
Effect of L289K mutation, acetonitrile on p51 monomer-dimer equilibrium
Many studies have identified mutations that influence heterodimer stability (15–25). The effects of the L289K mutation were of particular interest, since this mutation is located far from the dimer interface, particularly in the case of the p51 homodimer (Supporting Information). The effects of the L289K mutation on the behavior of p51 were examined using NMR, SAXS, and size exclusion chromatography. The HSQC spectrum of [methyl-13C] methionine p51L289K shows many of the features seen for wt p51, however the M184 resonance is broader and weaker than observed for the p51 homodimer (Figures 2c and 3c). In addition, the high intensity of the M357 resonance is more similar to the spectrum of the p51 subunit of the RT heterodimer (38). These results are consistent with a greater fraction of the p51 monomer, as predicted for the dimerization deficient L289K mutant, although we emphasize that in this case, the interpretation of the spectral characteristics is not unique. The OLIGOMER analysis of the SAXS data summarized in Figure 5 yields results that are generally consistent with the analysis of the NMR spectra. The fractional percentages and the χ2 of each fit are given as Supporting Information. As is apparent from a comparison of columns 1,2 with 3,4 of Figure 5, p51L289K is significantly more monomeric than wt p51 at equivalent concentrations. Again, there are no detectable conformations that resemble a p51E monomer in either the open or closed conformation or p51 dimer with an open thumb conformation. The conclusions derived from size-exclusion chromatographic analysis are generally consistent with the expected results, demonstrating that L289K p51 elutes at the same time as monomeric p51 (Figure 6). Under the conditions of the chromatography study, the addition of nevirapine produced only a small increase in the fraction of p51 homodimer. The chromatographic results generally indicated lower dimer fractions, presumably as a consequence of sample dilution.
Figure 6.
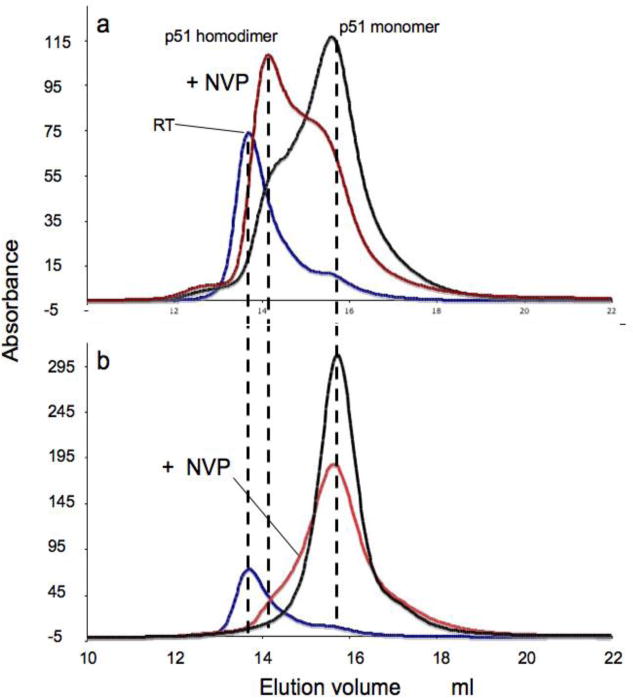
Size exclusion chromatography profiles. a) p51 in the absence (black) or presence (red) of nevirapine; b) p51L289K in the absence (black) or presence (red) of nevirapine. For reference, the RT elution profile is indicated with a blue line. Maximum elution positions for the p51 monomer, dimer, and RT heterodimer are indicated by dotted lines.
It also has been reported that acetonitrile is capable of producing subunit dissociation without producing a significant degree of unfolding (27, 29). We assessed the effect of acetonitrile on p51 structure by both NMR and SAXS. For the NMR studies, the M357B resonance which we have previously found to be sensitive to dimer formation, is unfortunately degenerate with that of M230. For this reason, we used [methyl-13C] methionine p51M230L to eliminate the overlap, and better evaluate its position. A reduction in the intensity of the M357B resonance was observed, consistent with this type of dissociation (spectra included as Supporting Information). There is also an observed broadening and some loss of intensity of the M184 resonance, which would be consistent with a reduction in the amount of p51E conformation present as either a monomer or dimer. The fact that a significant M184 resonance remains evident even in the presence of 10% acetonitrile suggests that there is still some homodimer present, a result also consistent with the SAXS analysis (Figure 5, column 5). In conclusion, both the NMR and SAXS analyses support the conclusions that L289K and acetonitrile reduce dimer stability.
Effect of Nevirapine and KCl on dimerization and thumb subdomain orientation
Addition of the NNRTI nevirapine strongly favors the open conformation of the fingers/thumb subdomains (46, 47), while inconsistent results have been obtained for the effect of nevirapine on dimerization (4, 30, 31). As demonstrated previously (38), the addition of nevirapine to the RT heterodimer results in large shift perturbations for the two active site methionine residues, M18466 and M23066 in the p66 subunit, while not perturbing any of the methionine residues in the p51 subunit. This result is reproduced in Figure 8a for [methyl-13C] methionine51 RT (38). In the present study, we found that nevirapine produces analogous changes when added to the isolated p51 subunit, interacting with the p51E subunit of the homodimer, and possibly with the p51E monomer, if this is present in solution (Fig. 8b). Nevirapine also produces qualitatively similar but weaker effects on the spectrum of [methyl-13C] methionine p51L289K (Figure 3d). We note that much of the M230 resonance intensity remains unshifted after the addition of the nevirapine (Fig. 8b), consistent with the main contribution to this resonance from M230 in the compact, p51-like subunit of the heterodimer. In addition, there is probably also some resonance intensity from M357B at this position. These results demonstrate the close similarity in the active site structures and NNRTI interactions between heterodimeric p66/p51 and the p51 homodimer.
Figure 8.
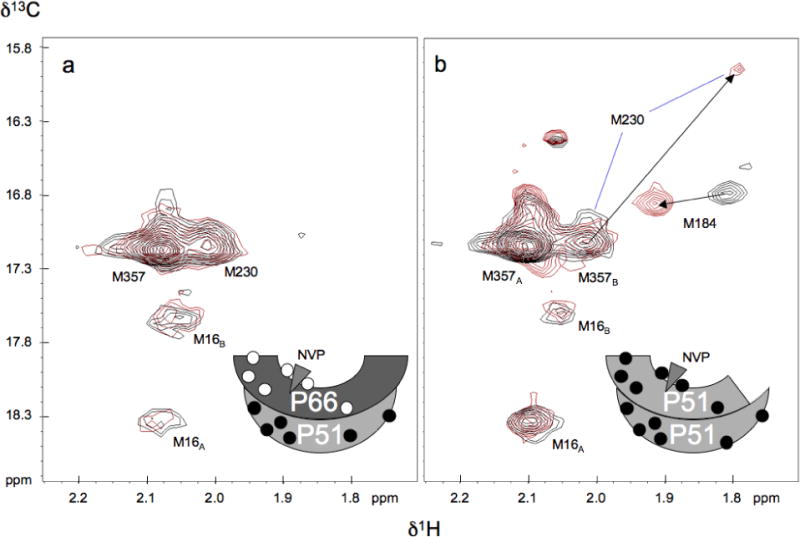
Effect of nevirapine on p51 NMR spectra. a) 1H-13C HSQC spectra of uncomplexed [methyl-13C] methionine51 RT (50 μM) in the absence (black) and presence (red) of 200 μM nevirapine; b) 1H-13C HSQC spectra of uncomplexed [methyl-13C] methionine p51 (30 μM) in the absence (black) and presence (red) of 100 μM nevirapine. The arrows indicate resonance shifts in response to the nevirapine. NMR parameters and buffer as in Figure 2. Schematic figures at the bottom indicate the subunit labeling pattern, with filled circles indicating [methyl-13C] methionine labeling; the triangle at the active site represents nevirapine. The spectrum of the p66/p51 heterodimer preparation shown in panel a is reproduced from Figure 7c of Zheng et al. (38). Preparation of the selectively labeled heterodimer is described in that reference.
The effect of nevirapine, which can be evaluated by comparing columns 7 and 8 with columns 1 and 2 in Figure 5, is both to increase the total dimer fraction, and also to stabilize the open conformation of the fingers/thumb subdomains. At 60 μM p51, 500 μM nevirapine, no monomer was present, and more than 60% of the dimer was in the open conformation, as expected from crystal structures with NNRTI’s bound (47). In addition, the nevirapine also counteracts much of the effect of the L289K mutation (compare columns 4 and 6), when the monomer concentration is reduced from 66 % to 4 %.
We also have evaluated the effects of ionic strength (KCl) on p51 dimerization. The SAXS data were well described using a mixture of only two component species: the p51C monomer and the p51Eclosed/p51C dimer, with the dimer fraction showing a monotonic increase with KCl concentration (New Figure 7). This dependence is similar to results obtained for p66 homodimer formation (26, 48) and also is consistent with the large hydrophobic interface between the two subunits (29, 45).
Figure 7.
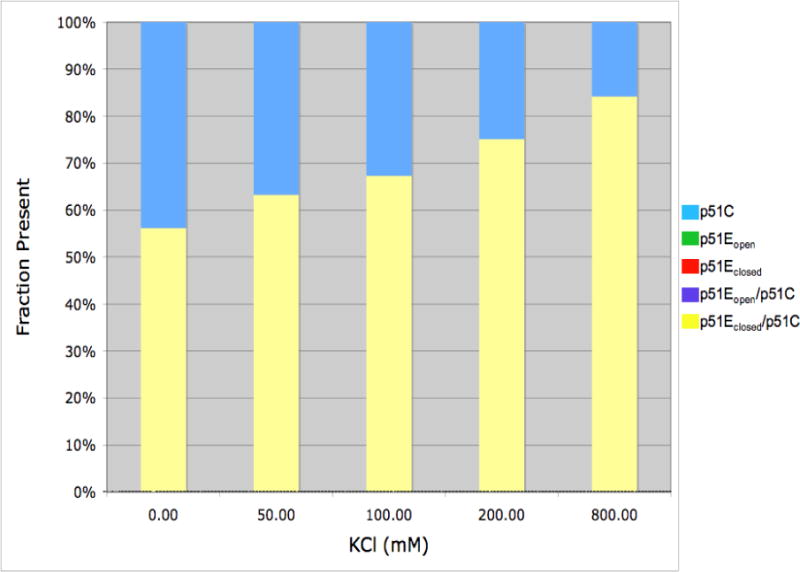
Effect of KCl on p51C monomer/dimer ratio. Samples contained 50 μM p51C280S in 50 mM Tris-HCl, pH 8.0, 4 mM MgCl2, and the concentrations of KCl indicated. Fractional component concentrations were determined using the OLIGOMER analysis of the SAXS data, as in Figure 5. Only two species were observed under the conditions of the study: the cyan bars represent the compact monomer, p51C, and the yellow bars correspond to the dimer with a closed fingers/thumb conformation, p51Eclosed/p51C.
Equilibrium modeling of the effect of the L289K mutation on dimerization
The surprising effects of the L289K mutation on p51 homodimer formation can be understood on the basis of a conformational selection model outlined below. This model is based on the results summarized above showing that p51 samples contain predominantly p51C, a variable level of concentration- and condition-dependent homodimer, and no other significantly populated species. We assume that the p51 monomer can exist in either of two conformations, p51C and p51E, and the only stable dimer that can exist is the conformational heterodimer, p51E/p51C. The dimerization process is thus described by the following two equilibrium equations:
| [2] |
Figure 9a shows a representative calculation for the fractions of the three molecular species, p51C, p51E, and p51E/p51C, as a function of the equilibrium constant KC. The calculation sets KD = 1 μM and the total concentration of p51 (p51T) = 50 μM in order to reproduce the large amount of dimer that is observed experimentally. This value is much lower than the recently reported p51 homodimerization KD of 230 μM (5) due to the different definition used and to the differences in experimental conditions. The studies of Venezia et al. (5) were performed at 5° C in 50 mM Tris (pH 7.5), 10 % (v/v) glycerol, and 25 mM NaCl, and it has been shown that the association rate constant for the heterodimer is strongly dependent on both temperature and Mg concentration (28, 29), both of which are substantially higher in the present study. To summarize, the above calculation introduces an explicit dependence of the dimerization process on the relative stabilities of the two p51 conformations that constitute the p51 homodimer.
Figure 9.
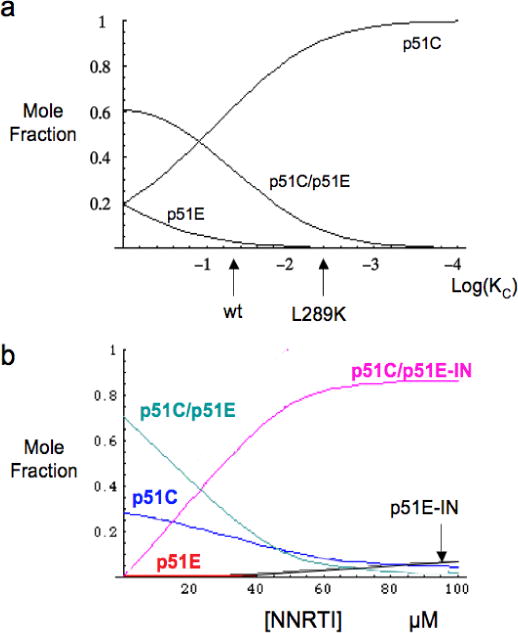
Illustrative numerical calculations of molecular species concentrations based on a conformational selection dimerization model. a) The fractional p51C, p51E, and dimeric p51E/p51C concentrations are calculated as a function of KC for parameters KD = 2 μM, p51T = 50 μM. The KC values that would approximately correspond to wt and to L289K mutant p51 are indicated. b) Molecular species determined as a function of NNRTI concentration for the model shown in Scheme 1. The calculation corresponds to the parameters p51T = 100 μM, KC = 0.03, KD = KD2 =0.2 μM, KI = KI2 = 1.0 μM. Thus, for the calculation shown, the binding of the inhibitor does not contribute directly to the affinity of the monomers, but only indirectly, through the effect on the conformational distribution of p51 monomers.
According to the conformational selection model outlined above, a pre-existing p51E conformation is stabilized by the interaction with p51C. The explicit temperature dependence of the equilibrium ratio of p51E/p51C is given by:
| [3] |
The complete absence of any p51E species under a range of experimental conditions used in the SAXS studies summarized in Figure 5 indicates that ΔG is at least several kcal/mol, and very possibly much larger. A larger ΔG would in general be consistent with slower association kinetics, although in principle, such effects can also result from a large activation energy barrier required for the p51C to p51E transition. Higher ΔG values, leading to lower KC values, require correspondingly lower KD values in order to explain the observed dimer concentrations. Low KD values are qualitatively consistent with the large buried surface area of 3000 Å2 calculated for the p51 homodimer (45). For example, setting the total p51 concentration p51T = 50 μM, KC = 0.1, KD = 2 μM, one obtains 55% dimer, 41% p51C and 4 % p51E, while lowering KC to 10−4 requires a KD value of 1 nM to obtain a similar dimer fraction. As noted above, no p51E was detected in any of the SAXS studies (Figure 5).
Within the context of the above model, we suggest that the effect of the L289K mutation is to further stabilize the p51C conformation by replacing a solvent-exposed leucine residue with a lysine, thus further lowering the value of KC = [p51E]/[p51C] (Supporting Information). Consistent with the above discussion, we propose that this mutation would have little direct effect on KD, while it would significantly stabilize p51C, resulting in a lower KC value. The qualitative effect of the L289K mutation on the equilibrium distribution of p51C, p51E, and p51C/p51E is indicated by the arrow in Figure 9a.
Equilibrium Modeling the interaction of p51 with an NNRTI
We have generalized the above equilibrium model to evaluate the effects of NNRTI binding, and in principle the effect of any agent whose affinity shows a strong conformational dependence. As in the model developed in the preceding section, p51 is allowed to adopt two conformations: p51E and p51C. The equilibrium constants KC and KD have the same definition as in the preceding discussion. Since, according to this model, p51 is capable of adopting a p66-like conformation, it is also capable of forming the NNRTI binding pocket and interacting with NNRTIs. In addition, p51C and p51E±NNRTI are allowed to dimerize (Scheme 1). The equilibrium condition requires that the corresponding equilibrium constants are related according to (Supporting Information):
| [4] |
Scheme 1.
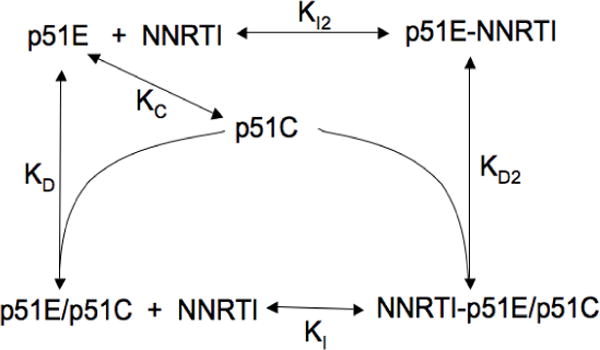
An illustrative calculation of the dependence of the molecular species on the NNRTI concentration is shown in Figure 9b. In the absence of the inhibitor, three species are present: p51C, p51E, and dimeric p51E/p51C. Addition of the NNRTI results in the formation of two additional species: p51E-NNRTI and NNRTI-p51E/p51C, and to compensatory decreases in p51C, p51E, and p51E/p51C (unliganded dimer). In the calculation shown in Figure 9b, we have set KC = 0.03, so that p51C >> p51E, consistent with the results of the SAXS experiments (Figure 5). We also set KI = KI2, so that the affinity of the NNRTI for p51E is independent of whether the interaction is with the p51E monomer or the p51E/p51C dimer. Based on the equilibrium constraint (Equation 4), this also requires that the affinity of p51E for p51C to form the dimer is equal to the affinity of p51E-NNRTI for p51C. Thus, the entire effect on dimer formation shown in Figure 9b results from the effect of the NNRTI on the conformational distribution of the p51 monomer, and not from a preference for binding to the dimer.
According to the model of Scheme 1, the NNRTI can influence p51 homodimer formation in two ways: 1) it can stabilize (or for some NNRTIs, destabilize) the dimer by altering the structure of the subunit interface, and thus KD; 2) it can influence dimerization indirectly by altering the conformational distribution of the monomer through an effect on KC. Formation of the p51E-NNRTI complex results in a second pathway favoring dimer formation. In this way, the NNRTI is predicted to exert an effect that opposes the effect of the L289K mutation, consistent with the SAXS data (Figure 5). The affinity of Mg for the active site aspartyl residues of p51E suggests that it should promote homodimer formation in a manner that is analogous to the effect of the NNRTI. This effect is consistent with the reported effects of Mg on dimerization (8), although we note that Mg can also influence this process as a result of a change in ionic strength, similar to the effect shown for KCl (Figure 7). Another way of considering this effect is that the subunit binding energy that would be required for maturation of the dimer complex does not have to be expended to the extent that a preformed p51E-NNRTI complex is available which more closely approximates the p51E conformation present in the mature complex.
Discussion
Dimerization of HIV reverse transcriptase is an essential step required for obtaining mature, active enzyme. It can be influenced by mutations (15–25), non-nucleoside reverse transcriptase inhibitors (4–6), solution conditions (8, 27–29) as well as small molecule dimerization inhibitors developed specifically for this purpose (7, 9–14, 28, 49). The p51 subunit represents a useful model system for the behavior of the RT heterodimer since it exhibits a significant level of homodimer-dependent polymerase activity (32, 50) as well as analogous dimerization behavior. Further, the observation of significant polymerase activity has been used to infer that one of the subunits of the p51 homodimer must adopt a p66-like conformation (50). Alternatively, multiple studies have demonstrated that the primer/template substrate used for activity measurements can significantly lower the apparent KD for both homo- and heterodimer formation (8, 26, 29, 33). Thus, these activity-dependent measurements do not necessarily indicate that the p51 homodimer is present in the absence of primer/template substrates. Comparison of the KCl effects on the activity of the p51 homodimer (32) with the KCl-dependent dimerization observed in the SAXS studies indicates that increased dimerization is not necessarily associated with increased activity, presumably as a consequence of the interference of high ionic strength with primer/template binding. On the basis of a radiation-target analysis study, Sluis-Cremer et al. (33) determined that the p51 homodimer could only be observed in the presence of a DNA primer/template. This conclusion is, however, at odds with other studies based on size exclusion chromatography (27) and sedimentation equilibrium (5), according to which p51 dimer was observed in the absence of nucleotide substrates.
In the present study, the NMR spectra of [methyl-13C] methionine p51 studied at 30 μM total monomer concentration in 200 mM KCl and 4 mM MgCl2 shows several features that are unequivocally associated with a p66-like, p51E conformation (e.g. Figure 1c). The conclusion that a p66-like conformation is present – even in the absence of primer/template - is further strengthened by the observation that the spectral perturbations induced by nevirapine in the sample containing only p51 are essentially identical with those observed in the methionine-labeled p66/p51 heterodimer (Figure 7). The NMR spectra therefore demonstrate the presence of a mixture that contains both the p51C and p51E species, but provides limited evidence on the degree of dimer formation. The SAXS analysis resolves this issue, providing a satisfactory fit for a binary mixture of the p51C monomer and a p51Eclosed/p51C dimer in which the fingers/thumb subdomains adopt a closed conformation. Importantly, there is no evidence for a significant concentration of the p51E monomer under any of the conditions studied. These observations do not completely rule out the presence of some p51E monomer or other oligomers as long as these correspond to only a small fraction of the molecular species present.
Although we cannot offer a precise explanation for the differences among these reported studies, it is clear that the high sensitivity of the dimerization process to the experimental conditions is the major reason for these apparent discrepancies. For example, Divita et al. have previously identified temperature (28, 29) and Mg (8) as critical factors for p66/p51 heterodimerization. These results are consistent with the substantially lower apparent KD for p51 homodimer formation indicated by our observations at 25° C compared with the recent KD value determined at 5° C and 10 % glycerol based on equilibrium sedimentation (5). Cabodevilla et al. (26) found that p66 homodimer formation is strongly favored at higher ionic strength, similar to our SAXS results for p51 homodimer formation (Figure 7). Although it is unclear why no evidence for a p51 homodimer was obtained in the previously reported radiation target analysis study (33), the lower temperature (20° C), lower salt concentration (100 mM NaCl), and apparent absence of Mg (which was present for the activity measurements) would have contributed to a reduced fractional dimer concentration.
Conclusions regarding the effects of the NNRTI nevirapine on homdimer formation have also been inconsistent (4, 30, 31). The SAXS studies presented here demonstrate a significant nevirapine-induced enhancement of p51/p51 homodimer formation under the conditions of the studies (Figure 5). In addition to the effects on wt enzyme, nevirapine is able to significantly counteract the effects of the L289K mutation on dimerization (Figure 5).
The validity of the SAXS analysis is supported primarily by the qualitative consistency of the effects of different experimental conditions with expectations based on the limited literature for p51 and the more extensive literature for p66 and the p66/p51 dimer. The high fractional concentration of p51C observed is consistent with the analysis of Wang et al. (45), who determined that formation of the compact p51C structure eliminates an extensive hydrophobic solvent interface, so that the monomeric forms of p51, as well as p66, are expected to have the structure observed for the p51 subunit of the heterodimer, i.e. p51C. The effect of nevirapine in selecting the open conformation of the fingers/thumb subdomains is also qualitatively consistent with multiple crystal structures of the nevirapine-p66/p51 complex (46, 47, 51–53). Alternatively, the preference of uncomplexed p51E/p51C dimer for a closed fingers/thumb orientation agrees with the results of ESR studies of spin-labeled RT (34), and indicates that the SAXS methodology provides a useful approach for this type of conformational analysis of RT. The qualitative effect of the L289K mutation on reducing p66 homodimerization and p66/p51 heterodimerization (15) is similarly supported by the SAXS analysis. Thus, despite uncertainties about the details of the component structures used for the deconvolution, the qualitative consistency of the results with a large amount of experimental data provides strong support for the validity of the SAXS analysis.
The SAXS and NMR results demonstrating significant levels of both p51C and p51E/p51C in solution provide important insights into the possible mechanism of homodimer formation, supporting a conformational selection model in which the predominant p51C species is able to “select” the small concentration of p51E that is present to form a homodimer. The model provides an extremely attractive basis for understanding the effect of the L289K mutation on dimer formation; it is apparent that this substitution will alter the equilibrium constant KC, but unclear why it should influence KD. We also have generalized the model to include the effects of NNRTI binding, which can be considered to result from a combination of direct effects exerted at the subunit interface, and indirect effects resulting from perturbation of the p51E p51C conformational distribution. The acute effect of temperature on homodimer formation is more readily understood based on a temperature-dependent monomer conformation, rather than an effect on the dimerization reaction. In general, we suggest that the hypersensitivity of the apparent dimerization equilibrium to temperature and Mg concentration results primarily from an effect on the KC equilibrium.
The experimental data do not necessarily rule out several related alternative models in which an initial p51C/p51C homodimer might form, followed by selection or perhaps even induction of the p51E conformer:
| [5] |
For such a model to be valid, the p51C/p51C conformer would need to be sufficiently stable to persist long enough for one of the p51C subunits to undergo a major conformational rearrangement. At this point, we find no evidence for the presence of such p51C/p51C homodimers, however the presence of a small percentage of such species would not be inconsistent with the SAXS analysis. Although most previous treatments of RT dimerization favor the initial formation of a weak complex followed by a slow induced fit to progress to the mature dimer (e.g. (28)), Venezia et al. (54) have recently found that the association rate constant is slower than previously indicated, and concluded that a conformational selection model provides the most attractive explanation for the dimerization kinetics of RT.
Finally, our results support the analysis presented by Wang et al. (45) indicating that the p51 monomer should adopt a p51C conformation, and are thus also consistent with the prediction that p66 monomer should have a similar structure. Based on the stability of the isolated RNase H domain (55), the p66 monomer would thus exist primarily as two globular domains, p51C and RNase H, connected by a flexible linker, a result which appears to be consistent with limited proteolysis studies (56). Such a model also provides a basis for understanding the inhibition of heterodimer formation by the L289K66 mutation, analogous to the effect described here for p51 homodimerization.
Supplementary Material
Acknowledgments
This research was supported by Research Project Number Z01-ES050147 to REL in the Intramural Research Program of the National Institutes of Health. The authors would like to Dr. Lin Yang of the X9 beamline, at the National Synchrotron Light Source at Brookhaven National Laboratory, for assistance with data collection. Use of the X9 beamline is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-98CH10886.
This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health. Dr. DeRose’s contribution was funded in whole with Federal funds from NIH/NIEHS, under Delivery Order HHSN273200700046U to SRA International, Inc.
Abbreviations
- RT
reverse transcriptase
- IPTG
Isopropyl thio-D-galactoside
- HSQC
heteronuclear single-quantum coherence
- DSS
2,2-dimethylsilapentane-5-sulfonic acid
- NNRTI
non-nucleoside RT inhibitors
- NMR
nuclear magnetic resonance
- SAXS
small angle X-ray scattering
Footnotes
Supplemental Information. Supplemental materials may be accessed free of charge online at http://pubs.acs.org. Supplemental materials include: fits of all the SAXS data presented in Figures 5 and 7, tablulated SAXS data results, apparent KD values, 1H-13C HSQC spectra showing the effects of acetonitrile on p51, ribbon diagrams indicating the positions of the L289 residues in the p66/p51 heterodimer and the modeled p51/p51 homodimer, Analytical solutions for Equation 2 used to generate Figure 9a, and a proof of Eq. 4
References
- 1.Autran B, Carcelain G, Li TS, Blanc C, Mathez D, Tubiana R, Katlama C, Debre P, Leibowitch J. Positive effects of combined antiretroviral therapy on CD4(+) T cell homeostasis and function in advanced HIV disease. Science. 1997;277:112–116. doi: 10.1126/science.277.5322.112. [DOI] [PubMed] [Google Scholar]
- 2.Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Stanford J, Stryker R, Johnson P, Labriola DF, Farina D, Manion DJ, Ruiz NM. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. Study 006 Team. N Engl J Med. 1999;341:1865–1873. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 3.Sluis-Cremer N, Arion D, Abram ME, Parniak MA. Proteolytic processing of an HIV-1 pol polyprotein precursor: insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int J Biochem Cell Biol. 2004;36:1836–1847. doi: 10.1016/j.biocel.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 4.Tachedjian G, Orlova M, Sarafianos SG, Arnold E, Goff SP. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci U S A. 2001;98:7188–7193. doi: 10.1073/pnas.121055998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venezia CF, Howard KJ, Ignatov ME, Holladay LA, Barkley MD. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry. 2006;45:2779–2789. doi: 10.1021/bi051915z. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava S, Sluis-Cremer N, Tachedjian G. Dimerization of human immunodeficiency virus type 1 reverse transcriptase as an antiviral target. Curr Pharm Des. 2006;12:1879–1894. doi: 10.2174/138161206776873590. [DOI] [PubMed] [Google Scholar]
- 7.Morris MC, Robert-Hebmann V, Chaloin L, Mery J, Heitz F, Devaux C, Goody RS, Divita G. A new potent HIV-1 reverse transcriptase inhibitor. A synthetic peptide derived from the interface subunit domains. J Biol Chem. 1999;274:24941–24946. doi: 10.1074/jbc.274.35.24941. [DOI] [PubMed] [Google Scholar]
- 8.Divita G, Restle T, Goody RS. Characterization of the dimerization process of HIV-1 reverse transcriptase heterodimer using intrinsic protein fluorescence. FEBS Lett. 1993;324:153–158. doi: 10.1016/0014-5793(93)81383-b. [DOI] [PubMed] [Google Scholar]
- 9.Divita G, Restle T, Goody RS, Chermann JC, Baillon JG. Inhibition of human immunodeficiency virus type 1 reverse transcriptase dimerization using synthetic peptides derived from the connection domain. J Biol Chem. 1994;269:13080–13083. [PubMed] [Google Scholar]
- 10.Camarasa MJ, Velazquez S, San-Felix A, Perez-Perez MJ, Bonache MC, De Castro S. TSAO derivatives, inhibitors of HIV-1 reverse transcriptase dimerization: recent progress. Curr Pharm Des. 2006;12:1895–1907. doi: 10.2174/138161206776873563. [DOI] [PubMed] [Google Scholar]
- 11.Sluis-Cremer N, Hamamouch N, San Felix A, Velazquez S, Balzarini J, Camarasa MJ. Structure-activity relationships of [2′,5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]- 3′-spiro-5″-(4″-amino-1″, 2″-oxathiole-2″, 2″-dioxide)thymine derivatives as inhibitors of HIV-1 reverse transcriptase dimerization. J Med Chem. 2006;49:4834–4841. doi: 10.1021/jm0604575. [DOI] [PubMed] [Google Scholar]
- 12.Depollier J, Hourdou ML, Aldrian-Herrada G, Rothwell P, Restle T, Divita G. Insight into the mechanism of a peptide inhibitor of HIV reverse transcriptase dimerization. Biochemistry. 2005;44:1909–1918. doi: 10.1021/bi0484264. [DOI] [PubMed] [Google Scholar]
- 13.Grohmann D, Corradi V, Elbasyouny M, Baude A, Horenkamp F, Laufer SD, Manetti F, Botta M, Restle T. Small molecule inhibitors targeting HIV-1 reverse transcriptase dimerization. Chembiochem. 2008;9:916–922. doi: 10.1002/cbic.200700669. [DOI] [PubMed] [Google Scholar]
- 14.Agopian A, Gros E, Aldrian-Herrada G, Bosquet N, Clayette P, Divita G. A new generation of peptide-based inhibitors targeting HIV-1 reverse transcriptase conformational flexibility. J Biol Chem. 2009;284:254–264. doi: 10.1074/jbc.M802199200. [DOI] [PubMed] [Google Scholar]
- 15.Goel R, Beard WA, Kumar A, Casas-Finet JR, Strub MP, Stahl SJ, Lewis MS, Bebenek K, Becerra SP, Kunkel TA, et al. Structure/function studies of HIV-1(1) reverse transcriptase: dimerization-defective mutant L289K. Biochemistry. 1993;32:13012–13018. doi: 10.1021/bi00211a009. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh M, Jacques PS, Rodgers DW, Ottman M, Darlix JL, Le Grice SF. Alterations to the primer grip of p66 HIV-1 reverse transcriptase and their consequences for template-primer utilization. Biochemistry. 1996;35:8553–8562. doi: 10.1021/bi952773j. [DOI] [PubMed] [Google Scholar]
- 17.Wohrl BM, Krebs R, Thrall SH, Le Grice SF, Scheidig AJ, Goody RS. Kinetic analysis of four HIV-1 reverse transcriptase enzymes mutated in the primer grip region of p66. Implications for DNA synthesis and dimerization. J Biol Chem. 1997;272:17581–17587. doi: 10.1074/jbc.272.28.17581. [DOI] [PubMed] [Google Scholar]
- 18.Tachedjian G, Aronson HE, Goff SP. Analysis of mutations and suppressors affecting interactions between the subunits of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci U S A. 2000;97:6334–6339. doi: 10.1073/pnas.97.12.6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sluis-Cremer N, Tachedjian G. Modulation of the oligomeric structures of HIV-1 retroviral enzymes by synthetic peptides and small molecules. Eur J Biochem. 2002;269:5103–5111. doi: 10.1046/j.1432-1033.2002.03216.x. [DOI] [PubMed] [Google Scholar]
- 20.Auwerx J, Van Nieuwenhove J, Rodriguez-Barrios F, de Castro S, Velazquez S, Ceccherini-Silberstein F, De Clercq E, Camarasa MJ, Perno CF, Gago F, Balzarini J. The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes of anti-HIV-1 drugs. FEBS Lett. 2005;579:2294–2300. doi: 10.1016/j.febslet.2005.02.077. [DOI] [PubMed] [Google Scholar]
- 21.Balzarini J, Auwerx J, Rodriguez-Barrios F, Chedad A, Farkas V, Ceccherini-Silberstein F, Garcia-Aparicio C, Velazquez S, De Clercq E, Perno CF, Camarasa MJ, Gago F. The amino acid Asn136 in HIV-1 reverse transcriptase (RT) maintains efficient association of both RT subunits and enables the rational design of novel RT inhibitors. Mol Pharmacol. 2005;68:49–60. doi: 10.1124/mol.105.012435. [DOI] [PubMed] [Google Scholar]
- 22.Mulky A, Sarafianos SG, Jia Y, Arnold E, Kappes JC. Identification of amino acid residues in the human immunodeficiency virus type-1 reverse transcriptase tryptophan-repeat motif that are required for subunit interaction using infectious virions. J Mol Biol. 2005;349:673–684. doi: 10.1016/j.jmb.2005.03.057. [DOI] [PubMed] [Google Scholar]
- 23.Mulky A, Vu BC, Conway JA, Hughes SH, Kappes JC. Analysis of amino acids in the beta7-beta8 loop of human immunodeficiency virus type 1 reverse transcriptase for their role in virus replication. J Mol Biol. 2007;365:1368–1378. doi: 10.1016/j.jmb.2006.10.089. [DOI] [PubMed] [Google Scholar]
- 24.Wapling J, Moore KL, Sonza S, Mak J, Tachedjian G. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J Virol. 2005;79:10247–10257. doi: 10.1128/JVI.79.16.10247-10257.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Figueiredo A, Zelina S, Sluis-Cremer N, Tachedjian G. Impact of residues in the nonnucleoside reverse transcriptase inhibitor binding pocket on HIV-1 reverse transcriptase heterodimer stability. Curr HIV Res. 2008;6:130–137. doi: 10.2174/157016208783885065. [DOI] [PubMed] [Google Scholar]
- 26.Cabodevilla JF, Odriozola L, Santiago E, Martinez-Irujo JJ. Factors affecting the dimerization of the p66 form of HIV-1 reverse transcriptase. European Journal of Biochemistry. 2001;268:1163–1172. doi: 10.1046/j.1432-1327.2001.01939.x. [DOI] [PubMed] [Google Scholar]
- 27.Restle T, Muller B, Goody RS. Dimerization of human immunodeficiency virus type 1 reverse transcriptase. A target for chemotherapeutic intervention. J Biol Chem. 1990;265:8986–8988. [PubMed] [Google Scholar]
- 28.Divita G, Rittinger K, Geourjon C, Deleage G, Goody RS. Dimerization kinetics of HIV-1 and HIV-2 reverse transcriptase: a two step process. J Mol Biol. 1995;245:508–521. doi: 10.1006/jmbi.1994.0042. [DOI] [PubMed] [Google Scholar]
- 29.Divita G, Rittinger K, Restle T, Immendorfer U, Goody RS. Conformational stability of dimeric HIV-1 and HIV-2 reverse transcriptases. Biochemistry. 1995;34:16337–16346. doi: 10.1021/bi00050a014. [DOI] [PubMed] [Google Scholar]
- 30.Sluis-Cremer N, Dmitrienko GI, Balzarini J, Camarasa MJ, Parniak MA. Human immunodeficiency virus type 1 reverse transcriptase dimer destabilization by 1-[Spiro[4″-amino-2″,2″ -dioxo-1″,2″ -oxathiole-5″,3′-[2′, 5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]]]-3-ethylthy mine. Biochemistry. 2000;39:1427–1433. doi: 10.1021/bi991682+. [DOI] [PubMed] [Google Scholar]
- 31.Tachedjian G, Moore KL, Goff SP, Sluis-Cremer N. Efavirenz enhances the proteolytic processing of an HIV-1 pol polyprotein precursor and reverse transcriptase homodimer formation. FEBS Lett. 2005;579:379–384. doi: 10.1016/j.febslet.2004.11.099. [DOI] [PubMed] [Google Scholar]
- 32.Bavand MR, Wagner R, Richmond TJ. HIV-1 reverse transcriptase: polymerization properties of the p51 homodimer compared to the p66/p51 heterodimer. Biochemistry. 1993;32:10543–10552. doi: 10.1021/bi00091a003. [DOI] [PubMed] [Google Scholar]
- 33.Sluis-Cremer N, Kempner E, Parniak MA. Structure-activity relationships in HIV-1 reverse transcriptase revealed by radiation target analysis. Protein Sci. 2003;12:2081–2086. doi: 10.1110/ps.03130503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kensch O, Restle T, Wohrl BM, Goody RS, Steinhoff HJ. Temperature-dependent equilibrium between the open and closed conformation of the p66 subunit of HIV-1 reverse transcriptase revealed by site-directed spin labelling. Journal of Molecular Biology. 2000;301:1029–1039. doi: 10.1006/jmbi.2000.3998. [DOI] [PubMed] [Google Scholar]
- 35.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 36.Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol. 1989;177:44–73. doi: 10.1016/0076-6879(89)77005-1. [DOI] [PubMed] [Google Scholar]
- 37.Hou EW, Prasad R, Beard WA, Wilson SH. High-level expression and purification of untagged and histidine-tagged HIV-1 reverse transcriptase. Protein Expr Purif. 2004;34:75–86. doi: 10.1016/j.pep.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Zheng X, Mueller GA, Derose EF, London RE. Solution characterization of [methyl-(13)C]methionine HIV-1 reverse transcriptase by NMR spectroscopy. Antiviral Res. 2009 doi: 10.1016/j.antiviral.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Johnson BA, Blevins RA. Nmr View - a Computer-Program for the Visualization and Analysis of Nmr Data. Journal of Biomolecular Nmr. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 41.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–1282. [Google Scholar]
- 42.Svergun D, Barberato C, Koch MHJ. CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography. 1995;28:768–773. [Google Scholar]
- 43.Spallarossa A, Cesarini S, Ranise A, Ponassi M, Unge T, Bolognesi M. Crystal structures of HIV-1 reverse transcriptase complexes with thiocarbamate non-nucleoside inhibitors. Biochem Biophys Res Commun. 2008;365:764–770. doi: 10.1016/j.bbrc.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 44.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Research. 2008;36:5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Smerdon SJ, Jager J, Kohlstaedt LA, Rice PA, Friedman JM, Steitz TA. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc Natl Acad Sci U S A. 1994;91:7242–7246. doi: 10.1073/pnas.91.15.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smerdon SJ, Jager J, Wang J, Kohlstaedt LA, Chirino AJ, Friedman JM, Rice PA, Steitz TA. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1994;91:3911–3915. doi: 10.1073/pnas.91.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren J, Esnouf R, Garman E, Somers D, Ross C, Kirby I, Keeling J, Darby G, Jones Y, Stuart D, et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat Struct Biol. 1995;2:293–302. doi: 10.1038/nsb0495-293. [DOI] [PubMed] [Google Scholar]
- 48.Rowley GL, Ma QF, Bathurst IC, Barr PJ, Kenyon GL. Stabilization and activation of recombinant human immunodeficiency virus-1 reverse transcriptase-P66. Biochem Biophys Res Commun. 1990;167:673–679. doi: 10.1016/0006-291x(90)92078-e. [DOI] [PubMed] [Google Scholar]
- 49.Andreola ML. Therapeutic potential of peptide motifs against HIV-1 reverse transcriptase and integrase. Curr Pharm Des. 2009;15:2508–2519. doi: 10.2174/138161209788682244. [DOI] [PubMed] [Google Scholar]
- 50.Dufour E, El Dirani-Diab R, Boulme F, Fournier M, Nevinsky G, Tarrago-Litvak L, Litvak S, Andreola ML. p66/p51 and p51/p51 recombinant forms of reverse transcriptase from human immunodeficiency virus type 1–interactions with primer tRNA(Lys3), initiation of cDNA synthesis, and effect of inhibitors. Eur J Biochem. 1998;251:487–495. doi: 10.1046/j.1432-1327.1998.2510487.x. [DOI] [PubMed] [Google Scholar]
- 51.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal-Structure at 3.5 Angstrom Resolution of Hiv-1 Reverse-Transcriptase Complexed with an Inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 52.Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S, Stuart DI, Stammers DK. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J Mol Biol. 2001;312:795–805. doi: 10.1006/jmbi.2001.4988. [DOI] [PubMed] [Google Scholar]
- 53.Chamberlain PP, Ren J, Nichols CE, Douglas L, Lennerstrand J, Larder BA, Stuart DI, Stammers DK. Crystal structures of zidovudine- or lamivudine-resistant human immunodeficiency virus type 1 reverse transcriptases containing mutations at codons 41, 184 and 215. Journal of Virology. 2002;76:10015–10019. doi: 10.1128/JVI.76.19.10015-10019.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venezia CF, Meany BJ, Braz VA, Barkley MD. Kinetics of association and dissociation of HIV-1 reverse transcriptase subunits. Biochemistry. 2009;48:9084–9093. doi: 10.1021/bi9010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pari K, Mueller GA, DeRose EF, Kirby TW, London RE. Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry. 2003;42:639–650. doi: 10.1021/bi0204894. [DOI] [PubMed] [Google Scholar]
- 56.Lowe DM, Aitken A, Bradley C, Darby GK, Larder BA, Powell KL, Purifoy DJ, Tisdale M, Stammers DK. HIV-1 reverse transcriptase: crystallization and analysis of domain structure by limited proteolysis. Biochemistry. 1988;27:8884–8889. doi: 10.1021/bi00425a002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.