

Abstract
Heterozygous LIS1 mutations and males with loss of the X-linked DCX result in lissencephaly, a neuronal migration defect. LIS1 regulates nuclear translocation and mitotic division of neural progenitor cells, while the role of DCX in cortical development remains poorly understood. Here, we uncovered novel neuronal migration and proliferation defects in the Dcx mutant embryonic brains. Although cortical organization was fairly well preserved, Dcxko/Y neurons displayed defective migration velocities similar to Lis1+/ko neurons when characterized by time-lapse video-microscopy of embryonic cortical slices. Dcxko/Y migrating neurons displayed novel multidirectional movements with abnormal morphology and increased branching. Surprisingly, Dcxko/Y radial glial cells displayed spindle orientation abnormalities similar to Lis1+/ko cells that in turn lead to moderate proliferation defects both in vivo and in vitro. We found functional genetic interaction of the two genes, with the combined effects of Lis1 haploinsufficiency and Dcx knock-out leading to more severe neuronal migration and proliferation phenotypes in the Lis1+/ko;Dcxko/Y male double mutant compared with the single mutants, resulting in cortical disorganization and depletion of the progenitor pool. Thus, we provide definitive evidence for a critical role for Dcx in neuronal migration and neurogenesis, as well as for the in vivo genetic interaction of the two genes most commonly involved in human neuronal migration defects.
Introduction
Proper development of the mammalian cerebral cortex relies on the integrated control of neurogenesis and neuronal migration. Proliferation of neuronal progenitor cells during early stages of brain development is critical to expand the progenitor pool at the ventricular surface and later mitotic divisions result in the generation of postmitotic neural precursors, which then migrate to the cortical plate (Gupta et al., 2002; Götz and Huttner, 2005). Defective neurogenesis or neuronal migration leads to brain malformations, and are often associated with different forms of mental retardation or cognitive disabilities and severe epilepsy Guerrini et al., 2008). For example, classical lissencephaly (or “smooth brain”) is due to a reduced number or absence of gyri and sulci of the cortical surface, resulting in severe mental retardation, seizures and early death (Kato and Dobyns, 2003). Mutations in two genes, LIS1 (Reiner et al., 1993; Lo Nigro et al., 1997) and DCX (Gleeson et al., 1998; des Portes et al., 1998), are responsible for most cases of classical lissencephaly (for review, see Dobyns et al., 1993; Kato and Dobyns, 2003).
The study of murine Lis1 by genetic knock-out or knockdown by RNAi revealed important roles in both neuronal migration and proliferation of neuronal progenitors. Reduction of LIS1 protein leads to neuronal migration defects in a dose-dependent manner (Hirotsune et al., 1998; Gambello et al., 2003). The translocation of the nucleus during migration toward the tip of the leading process is generated through dynein motor activity that is ultimately regulated by LIS1 via its binding to phosphorylated NDEL1 (Sasaki et al., 2000; Niethammer et al., 2000) and perhaps NDE1 (Feng and Walsh, 2004), which binds to 14-3-3ε for protection against phosphatase activity (Toyo-oka et al., 2003). The LIS1-NDEL1 complex directly associates with cytoplasmic dynein heavy and light chains to promote dynein motor function in a positive manner (for review see references: Gupta et al., 2002; Wynshaw-Boris, 2007; Vallee and Tsai, 2006). The LIS1/dynein complex at the cell periphery also regulates the orientation of the spindle of dividing neuronal precursors at the ventricular surface, and reduction of LIS1 protein levels lead to asymmetric positioning of the spindle of dividing cells at the ventricular surface causing cell death of neuroepithelial stem cells and depletion of radial glial progenitor cells (Yingling et al., 2008).
In contrast to LIS1 function, less is known about the role of DCX during brain development. Similar to LIS1, DCX is a microtubule binding protein that has a strong bundling activity that promotes microtubule polymerization and stability during neuronal migration (Gleeson et al., 1999). The germ line ablation of the Dcx gene in mouse resulted in disorganization of the hippocampus but did not lead to abnormalities in radial movements nor cerebral cortex disorganization (Corbo et al., 2002; Kappeler et al., 2006), although Dcx knock-down by RNAi resulted in neuronal migration defects in rat (Bai et al., 2003). Nuclear translocation defects have been reported in Dcx mouse mutants for tangential movements related to the migration of interneurons in the rostral migratory stream (RMS) (Koizumi et al., 2006a) or in the cortex (Kappeler et al., 2006). These migration defects resulted from frequent stalling of migrating cells that were unable to maintain proper bipolar morphology during movement, resulting in disorganized migration secondary to increased branching and persistence of multipolar morphology. Similar results were seen after Dcx knock-down in tangentially migrating neurons from the ganglionic eminence to the cerebral cortex (Friocourt et al., 2007). Genetic redundancy might partially explain the discrepancy existing between the cortical malformations affecting human patients with DCX mutations and the mild or no phenotypic effects of the mutated gene in Dcx mouse knock-outs, since there are two other Dcx mutants in mammals: doublecortin-like (DCL) and Dcx-like kinase (DCLK) (Koizumi et al., 2006b; Shu et al., 2006). Additionally, it is unknown whether Dcx expression and function is restricted to postmitotic neurons and whether it plays any role in neurogenesis. DCL is highly expressed during early stages of neocortical development and DCLK does play a role during neurogenesis (Shu et al., 2006; Vreugdenhil et al., 2007). DCLK ensures the correct transition from prometaphase to metaphase in dividing cells by regulating the structural formation of the bipolar spindles (Shu et al., 2006). Whether Dcx plays a role during neurogenesis remains unknown.
We analyzed the combined effects of Lis1 and Dcx mutations on mouse cortical development, to understand whether the contemporaneous ablation of Lis1 and Dcx leads to a more severe phenotype than single mutants in both neuronal migration and neurogenesis. In addition to finding more severe defects in double mutants, we surprisingly found neuronal migration and proliferation defects in the Dcx knock-out brain. Our study provides strong evidence for a functional genetic interaction of the two genes during brain development as well as for neuronal migration and proliferation defects in the cortex of the Dcx single knock-out.
Materials and Methods
Mice and matings.
Dcx+/ko female mice (Corbo et al., 2002) were mated to Lis1+/ko male mice (Hirotsune et al., 1998) to produce Lis1+/ko, Dcx+/ko female and Lis1+/ko; Dcxko/Y male mice in a mixed 129SvJ × NIH Black Swiss background. The thy-1-YFP mice [strain name: B6.Cg-TgN(Thy1-YFP-H)2Jrs] were obtained from The Jackson Laboratory. Mice were housed in standard cages in Association for Assessment and Accreditation of Laboratory Animal Care-approved facilities at University of California, San Diego and University of California, San Francisco (UCSF). All experiments followed the guidelines of University's animal care and use committee. Mice were maintained on a 12:12 h light/dark cycle at 22°C. All testing occurred during the light portion of the cycle.
Histologic, immunohistochemical, and spindle orientation analysis.
All analyses were performed on experimental animals with littermate-matched controls. Brains were fixed in 4% paraformaldehyde and cryo-protected in 30% sucrose for freezing or treated with 70% ethanol for paraffin embedding. At least three brains were analyzed for each experiment and matched sections were stained. Sections were roughly matched by counting the number of coronal sections starting at the rostral-most edge of the brain, and then the sections were more closely matched by anatomical landmarks. Cresyl-violet/Nissl staining was done according to standard protocols. Cortical thickness was measured by marking a line around the whole cortex of the brain sections (n ≥ 25 per genotype) followed by quantification of the total area with ImageJ (http://rsbweb.nih.gov/ij/).
Immunohistochemical staining with guinea pig anti-testis-1 antibody provided cortical layer V-specific staining in the postnatal cortex. Paraffin sections were dewaxed in xylenes, rehydrated, and reacted with primary antibody overnight at 4°C. Primary antibody was diluted 1:1000 in blocking buffer. Secondary antibody (biotinylated goat anti-guinea pig) was diluted 1:200 in blocking buffer and incubated for 2 h at RT. The Vector ABC detection system (Vector Laboratories) was used for development with DAB plus metal enhancement (Sigma) as the chromogen. Cortical layer markers Cux1 (rabbit; Santa Cruz Biotechnology), Foxp1 and Foxp2 (rabbit; Abcam) were diluted 1:200 in blocking agent (PBS, 0.2% Triton X-100, 5% Normal Goat Serum) at incubated overnight. Nestin (mouse; Millipore Bioscience Research Reagents), Pericentrin (rabbit; Covance), TuJ (mouse; Sigma), Dcx (rabbit; kind gift from Dr. J. G. Gleeson, University of California, San Diego, School of Medicine) and phospho-H3 (PH3) (rabbit; Sigma) were used at 1:200 dilution. Secondary antibodies were diluted 1:500 and incubated for 1 h. For BrdU (mouse; Sigma), Ki-67 (rat; Dako) and Tbr2 (rabbit; kind gift from Dr. R. F. Hevner, University of Washington School of Medicine) double and triple staining, pregnant dams were injected 30 min prior embryos collection with 100 μg of BrdU per g of mouse weight. Brain sections were pretreated with antigen-retrieval (10 mm Sodium Citrate pH6) and denaturing solutions (2N HCl, 0.5% Triton-X). Primary antibodies were used together at 1:100 dilution (BrdU and Ki-67) and 1:2000 for Tbr2, while secondary antibodies at 1:500 dilution. Quantification of the single-, double- and triple-stained cells was done manually by counting all the cells present in the images taken from the dorsolateral cortex (n ≥ 20 per genotype for PH3; n ≥ 30 per genotype for the double staining; n ≥ 20 for the triple staining). Acquisition was done at the level of the dorsolateral cortex using the Nikon Spectral C1 confocal microscope with 60× objective (UCSF Nikon Center, Parnassus).
Embryonic day 9.5 (E9.5) and E14.5 brains sections were prepared at 12 μm thickness and counter-stained with DAPI to see telophase mitotic cells at the ventricular surface. Captured images were analyzed with ImagePro v.6 to determine the angles of the spindle orientation by drawing a line parallel to the ventricular surface and a line parallel to the axis of the dividing cells (see Fig. 5A; n > 150 cells per genotype) and average angle values were calculated from these measurements (Yingling et al., 2008).
Figure 5.
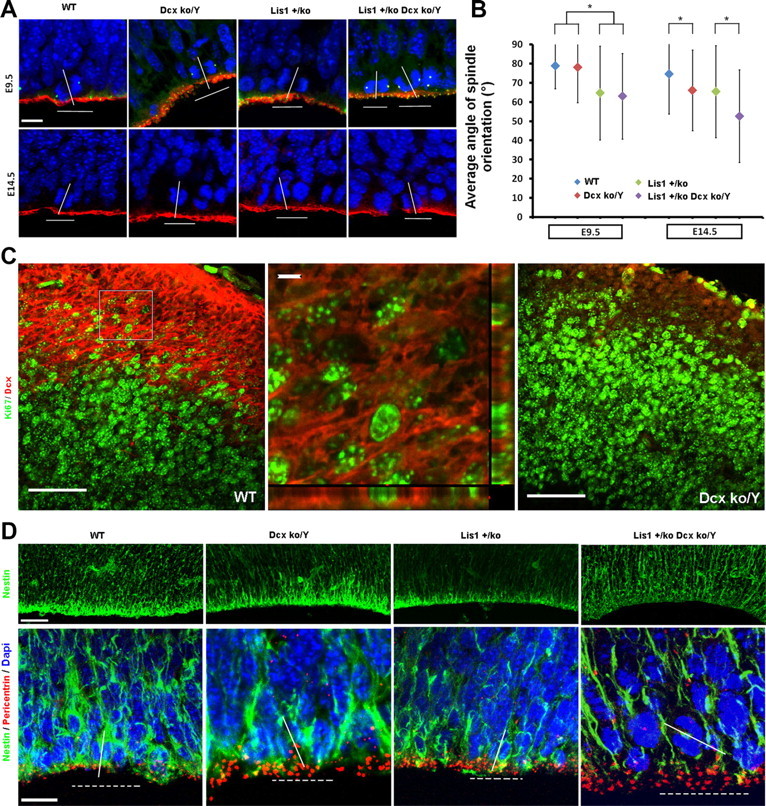
Dcxko/Y and Lis1+/ko;Dcxko/Y male mutants display randomized spindle positioning at the VZ. Analysis of spindle orientation and cellular organization at the ventricular surface. A, Spindle orientation analysis of cells in telophases and immunostaining of aPKC/γ-Tubulin at E9.5 (top; red/green) and aPKC E14.5 (bottom; red). The localization of atypical PKC (red) is normal indicating that apical polarity is preserved. Scale bar, 10 μm. B, Quantification of the average angle of spindle positioning during neuroepithelial and radial glial mitotic divisions. At E9.5 the average spindle orientation is affected only by Lis1 mutation. At E14.5 both Lis1 and Dcx mutants display spindle orientation defects with a more severe randomization in the Lis1+/ko;Dcxko/Y double mutant (*p < 0.05 by two-tailed t test). Error bars indicate SD. C, Double staining with Dcx/Ki67 markers in WT and Dcxko/Y mutant (negative control). DCX expression is highly expressed at the IZ/SVZ in dividing and nondividing cells. In the orthogonal view (second panel, zoom-in), Dcx is localized around the cell. Scale bars: (first and third panel) 50 μm; (second panel) 5 μm. D, Nestin immunostaining suggests that cellular organization is disrupted in the Lis1+/ko;Dcxko/Y male mutant with signal reduction at the VZ and more fragmented fibers. Scale bars, 50 and 10 μm. Pericentrin was used to delineate the apical surface and to define mitotic cells by helping to orient the spindle.
Organotypic slice cultures and movie analysis.
These experiments were performed as previously described (Nadarajah et al., 2001; Youn et al., 2009) and three brains for each genotype were analyzed. Briefly, E14.5 brains were collected and sectioned coronally with a tissue slicer (MX-TX, Siskiyou). Cortical slices were transferred to culture medium with 1% N2 supplement in F12/MEM medium containing 10 μg/ml Oregon green (488 BAPTA-1, Invitrogen). Slices were incubated for 1 h at 37°C in 5% CO2. Individual slices were washed and then placed in a 60 mm culture dish and covered with neutralized collagen (Rat tail, BD biosciences), which was allowed to solidify for 2–3 h. Oregon green-stained cells were imaged using a 2-photon confocal microscope (Olympus FV300) with a 20× dipping objective. The same medium with the addition of HEPES was used during the recording. Time-lapse capture was performed every 7 min using the FLUOVIEW program and slices were imaged for 6–8 h in a temperature controlled chamber. The files generated were merged and projected movies were analyzed using ImagePro v.6. The quantification of the angle changes was done using the same software (n > 25 for WT; n > 80 for Dcx+/ko; n > 100 for Dcxko/Y). The number of branches was quantified in at least 50 moving cells per genotype. All movies have been scaled and calibrated according to the acquisition parameters before the tracking analysis. All cells that displayed a migration of at least 3 cell-body distance was quantified, while the remaining cells were considered stationary (n > 200 for WT, n > 100 for single mutants; n = 10 for double mutant). The percentage of moving cells was estimated within three arbitrary areas from each time-lapse video-microscopy movie at the level of the IZ (Youn et al., 2009).
Neurosphere culture and 3-(4,5-dimethylthazol-2-yl)-5-3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay.
Primary embryonic cortical cultures were established from E14.5 dissociated brains and cultured to generate neurospheres using a modification (Youn et al., 2009) of previously described methods (Reynolds et al., 1992; Rietze and Reynolds, 2006; Ishii et al., 2008). In brief, papain-dissociated brains were passed through a 70 μm nylon cell-strainer (BD Falcon) and medium-containing cells were homogenized until single cells were visible under light microscope. EGF and bFGF (10 ng/ml) were added to the medium (DMEM/F12 with N2 supplement) to stimulate the growth of neurospheres in suspension. No significant differences among the genotypes were seen in the number of neurospheres generated at postnatal day 0 (P0). These neurospheres were collected and trypsinized to obtain P1 single cells. P1 single cells (5000) were seeded per each well (12 well plates) and cultured for 13 days in vitro (13 DIV). Usually floating neurospheres appeared after 3–5 DIV. For the 3-(4,5-dimethylthazol-2-yl)-5-3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (CellTiter 96 AQueus One solution cell proliferation kit, Promega) (Learish et al., 2000; Bantubungi et al., 2008), 5000 P1 single cells were seeded in triplicates in a 96 well plate. MTS solution was added to the medium and after 2 h MTS reduction was recorded at 490 nm using an automated microplate reader (Biotek Synergy 2).
Results
Lis1 and Dcx interact during the development of the neocortex and hippocampus
To study whether Lis1 and Dcx genetically interact during mouse brain development, we generated double mutant Lis1+/ko;Dcxko/Y mice by mating single mutant Lis1+/ko males with Dcx+/ko females. Due to the lethality of most male Lis1+/ko;Dcxko/Y mouse mutants shortly after birth, we analyzed cresyl violet-stained sections of adult (P21) female wild-type (WT), Lis1+/ko, Lis1+/ko;Dcx+/ko mice. Both Lis1+/ko and Lis1+/ko;Dcx+/ko mutants displayed an increased volume of the ventricles, disorganized layering and decreased size of the hippocampus with disorganization and loss of other brain areas such as the caudate and putamen (Fig. 1A). Double mutant Lis1+/ko;Dcx+/ko females displayed more severe defects than single Lis1 mutants, especially in the hippocampus (Fig. 1A). Analysis of WT, Lis1+/ko, Lis1+/ko;Dcx+/ko mice containing the thy1-YFP transgene that is expressed in a subset of cerebral cortical layer V pyramidal cells, hippocampal pyramidal cells and cells of the fascia dentate confirmed this graded severity of disruption (Fig. 1A). A few Lis1+/ko;Dcxko/Y males survived to P21. These mice displayed more severe disorganization of the hippocampal structure, as demonstrated by cresyl violet staining and Testis-1 immunochemistry (Fig. 1B). The organization of the cerebellum of double mutant males was also severely disrupted to a greater extent than in female Dcx+/ko and Lis1+/ko;Dcx+/ko mice (Fig. 1B).
Figure 1.
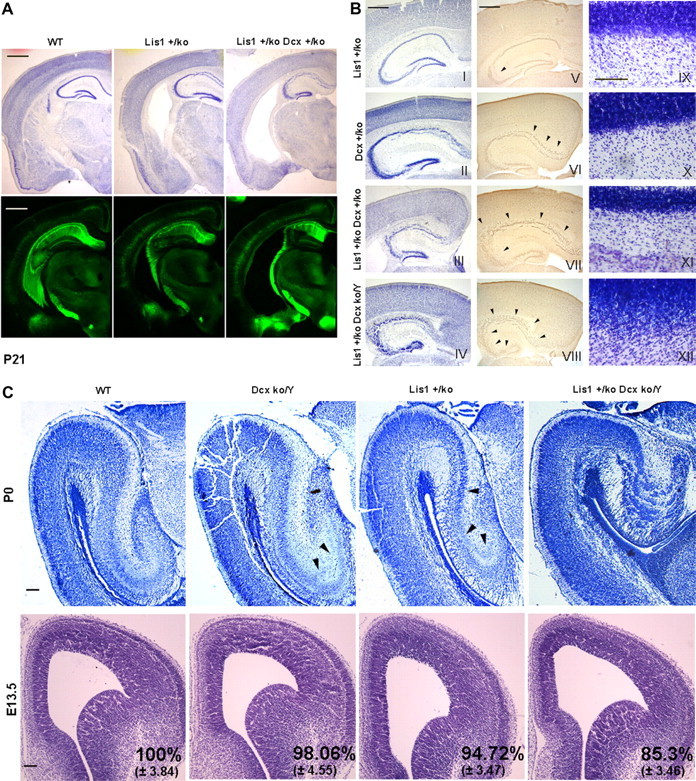
Lis1 and Dcx double mutants have severe disorganization of the hippocampus, cerebellum, and reduction of the cortical area during development. Morphological analysis at P21, P0, and E13.5 of WT and mutant brain sections. A, Cresyl violet staining and thy1-YFP transgene expression revealed severe morphological abnormalities in the Lis1+/ko;Dcx+/ko female mutants with enlargement of the ventricles, hippocampal disorganization and loss of other brain areas. Scale bar, 1 mm. B, Cresyl violet (I–IV) and Testis-1 staining (V–VIII) of the hippocampus demonstrated an increased severity of disorganization across the mutants. Scale bar, 500 μm. Cerebellar organization (IX–XII) was also severely compromised in the Lis1+/ko;Dcxko/Y male mutant compared with the other genotypes. Scale bar, 100 μm. C, Nissl staining of the hippocampus at P0 confirmed the severe developmental disorganization of the Lis1+/ko;Dcxko/Y male double mutant. Scale bar, 200 μm. Cresyl violet staining of E13.5 brain sections revealed a reduction in the cortical area in the Lis1+/ko;Dcxko/Y male double mutant. Scale bar is 100 μm. The confidence interval indicates the SD. Two-tailed t test distribution was used to calculate p values.
We examined all genotypes in males prenatally at two developmental stages (P0 and E13.5). At P0 there was an increasing severity of the hippocampal disorganization across the mutants. In agreement with previous reports (Corbo et al., 2002), the hippocampal pyramidal layer of Dcxko/Y males displayed moderate splitting of the CA3 region, while in Lis1+/ko males the splitting extended into the CA1 region (Fig. 1C). The hippocampus of Lis1+/ko;Dcxko/Y males was much smaller with severe disintegration and layering of the pyramidal layer. At E13.5 major structural abnormalities were not detected, but there was a reduction in the cortical thickness of Lis1+/ko and Lis1+/ko;Dcxko/Y brains. There was no significant difference between the Dcxko/Y male single mutant compared with WT, but there were 9–13% (p < 0.001) and 15% (p < 0.001) differences in the cortical thickness in Lis1+/ko;Dcxko/Y males compared with the single mutants and WT males respectively (Fig. 1C). There was also a slight significant difference between the Lis1+/ko and the Dcxko/Y single mutants (p < 0.01). The overall severe phenotype of Lis1+/ko;Dcxko/Y and the Lis1+/ko;Dcx+/ko mice compared with the single mutants strongly suggested a synergistic effect of the two genes during brain development.
Cortical neuronal migration defects in Dcxko/Y and Lis1+/ko;Dcxko/Y mutants
We next investigated neuronal migration ex-vivo using organotypic slice cultures from E14.5 male WT, Dcxko/Y or Lis1+/ko single mutants and Lis1+/ko;Dcxko/Y double mutant brains (Fig. 2A). Most cells in WT slices migrated away from the starting position (dotted lines) and only a few were stationary (Fig. 2A, blue outlines). In the Lis1+/ko or Dcxko/Y single mutants and Lis1+/ko;Dcxko/Y double mutant male slices there was a decreasing proportion of moving cells (Fig. 2A; supplemental Movies 1–4, available at www.jneurosci.org as supplemental material). In WT slices 73% of the cells moved away from the starting position. In the Dcxko/Y and Lis1+/ko single mutant slices only 37% and 18% of cells moved, respectively, while cells in Lis1+/ko;Dcxko/Y slices displayed a very severe phenotype with very few moving cells (Fig. 2B). The migration of moving cells was tracked at each time point across the cortex independently of the direction of the movement. WT cells migrated with a speed of 55 ± 2.35 μm/h, while Dcxko/Y and Lis1+/ko displayed slower migration speeds of 38 ± 2.42 and 36 ± 4.29 μm/h respectively (Fig. 2C). Lis1+/ko;Dcxko/Y double mutant cells were mostly stationary and did not move from their initial position, displaying a rounded shape without forming processes (Fig. 2C; supplemental Movies 1–4, available at www.jneurosci.org as supplemental material). When migration velocities for moving and nonmoving cells were combined the difference in the average velocity across the four genotypes was markedly different (Fig. 2D). Of note, the speed of moving cells in the Dcxko/Y and Lis1+/ko single mutants were approximately the same, suggesting that a major difference in the cortical outcomes of these mutants may be due to the proportion of moving cells. We found similar differences for the average velocity, maximal velocity, maximal acceleration and total distance traveled when analyzing migrating cells that were migrating in radial, retrograde and tangential directions (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). There was also no evidence for a bimodal distribution of migrating cells in female Dcx+/ko cortical slices (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) as would be expected based on the phenotype of heterozygous mutations in females, although this measurement may not be sensitive enough to detect such differences.
Figure 2.

Neuronal migration is slower in Dcxko/Y and severely impaired in Lis1+/ko;Dcxko/Y double mutant. Analysis of neuronal migration using organotypic slice cultures. A, Single representative images from time lapse videomicroscopy (supplemental Movies 1–4, available at www.jneurosci.org as supplemental material) demonstrate the gradual decrease of moving cells across the genotypes. The top panel shows the starting position of moving cells with the direction of movement (outlines with arrow). Nonmoving cells (blue outline) remain at approximately the same position until the end of the movie. The bottom panel shows the end position of moving cells and the trajectory they followed (dotted lines). B–D, Quantification of neuronal migration across the genotypes. B, The percentage of moving and stationary cells within three arbitrary areas from each time-lapse video-microscopy movie at the level of the IZ is shown. Cells with an appreciable movement from the starting position were considered to be migrating and were included in the tracking analysis. C, Each movie was calibrated and analyzed to determine the velocity of each single neuron. The average velocity values displayed are independent from the movement direction (for direction-dependent values, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). D, Velocity values were corrected by combining data from moving and nonmoving cells. Error bars indicate SD; *p < 0.05, ***p < 0.0001 by two-tailed t test.
Cortical migration of Dcx knock-out cells is multidirectional with increased direction changes and branching
The vast majority of WT neurons migrated exclusively in radial, retrograde and tangential directions Surprisingly, we noticed that many cells in both Dcx+/ko and Dcxko/Y genotypes displayed from a few to several changes of direction during their migration (Fig. 3A,B). These cells moved with unpredictable patterns, such as L-shape, C-shape, S-shape, or zig-zag patterns (Fig. 3A,B; supplemental Movie 5, available at www.jneurosci.org as supplemental material) or even more complex patterns (Fig. 3A, pink track in Dcxko/Y slice, see enlarged inset). Multidirectional movements were at least three times more frequent in Dcx+/ko and Dcxko/Y neural progenitor cells than in WT or Lis1+/ko progenitor cells (Fig. 3C). Most of these multidirectional-migrating cells (70% of WT and 60% of Dcx+/ko cells) changed direction once, and only a small proportion changed direction 2, 3 or 4 times. In contrast, the majority of Dcxko/Y cells changed direction at least twice, and a number of cells changed direction 3 or 4 times with a few cells changing direction 7 or 8 times (Fig. 3D). Such high numbers of directional changes were never seen in WT or Dcx+/ko mutant. This peculiar migration phenotype was accompanied by abnormal morphology of the Dcxko/Y cells, with these migrating neuronal precursors displaying a typical neurite swelling detached from the cell body and an increased number of branches (Fig. 3E,F), as previously described in similar studies (Kappeler et al., 2006; Koizumi et al., 2006a).
Figure 3.
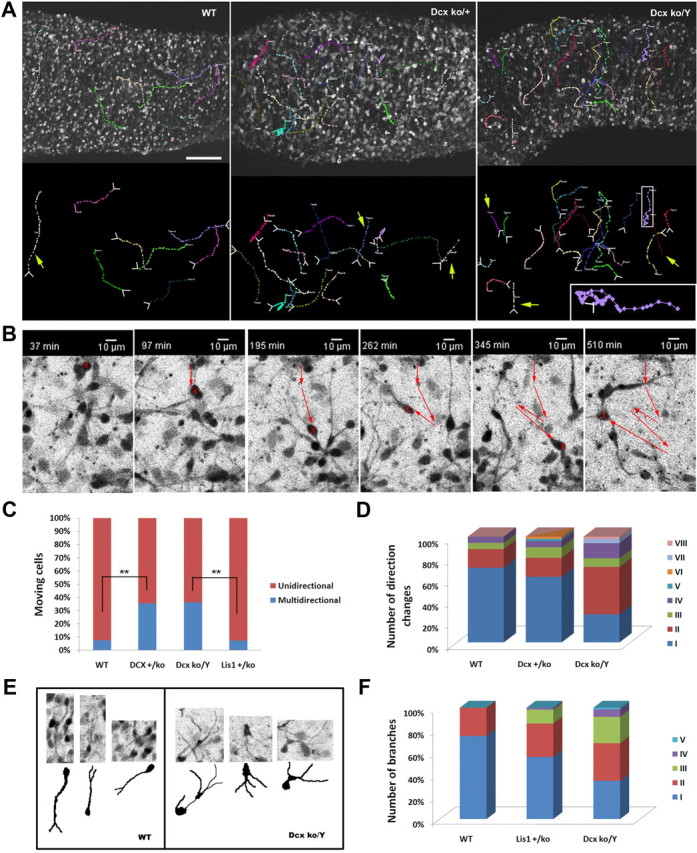
Neuronal migration of Dcx knock-out cortical neurons is multidirectional. Neural precursor cells from Dcx mutant brains displayed a multidirectional phenotype with increased branching and morphological abnormalities. A, Dcx heterozygous and hemizygous mutations cause neurons to migrate with several changes of direction. The starting position and the direction of the movements are indicated by white arrows at the tracking lines. Yellow arrows indicate unidirectional movements. Scale bar, 100 μm. B, Dcx knock-out cell migrated with a zig-zag movement and displayed increased branching. C, Quantification of the number of cells with multidirectional movements. In the Dcx mutants, the proportion of cells with this atypical pattern is at least three times more frequent than WT and Lis1+/ko cells. D, Quantification of the number of direction changes in the multidirectional-migrating cells. The number of changes of direction is higher in the Dcxko/Y mutants than in the Dcx+/ko mutants. E, F, Dcxko/Y migrating neurons displayed increased branching and morphological abnormalities with a swelling protruding or detached from the cell body. **p < 0.001 by two-tailed t test.
Cortical layering defects in the male Lis1+/ko;Dcxko/Y mutant
To determine whether these neuronal migration defects were associated with anatomical disorganization in the mutants, we examined laminar organization using layer-specific markers on P0 cortical sections. Cux1 (layers II–III) and Foxp1 (layers III–V) were similar to WT in Dcxko/Y and Lis1+/ko single mutants, but a dramatic reduction of Cux1 staining was evident in Lis1+/ko;Dcxko/Y double mutants with very few or no cells present in the outermost layers (Fig. 4, yellow arrowheads). Foxp1-positive cells were mildly abnormal in the Dcxko/Y and Lis1+/ko single mutants, but in the Lis1+/ko;Dcxko/Y double mutant, Foxp1-positive cells were shifted toward the surface. Foxp2 cells (layers V–VI) were in similar places in the lower layers of the cortex in WT and single mutants although there was some reduction of these cells in the Lis1+/ko mutant. In contrast, in the Lis1+/ko;Dcxko/Y double mutant, Foxp2-positive cells were near the pial surface and substantially overlapped with Foxp1-positive cells (Fig. 4, yellow arrows). It appears that Foxp1 and Foxp2-positive cells are mixed, similar to the mixing of cortical layers that we previously showed in Lis1hc/ko brains (Hirotsune et al., 1998; Gambello et al., 2003).
Figure 4.
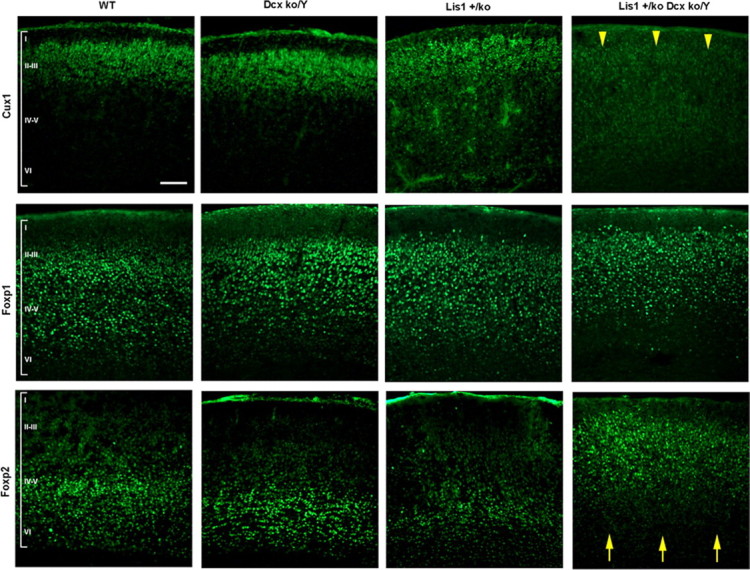
Cortical organization is severely abnormal in the Lis1+/ko;Dcxko/Y male mutants. Cortical layer-specific immunostaining in each of the genotypes, using Cux1 (layers II–III), Foxp1 (layers III–V) and Foxp2 (layer V–VI) antibodies. Major abnormalities are evident in the Lis1+/ko;Dcxko/Y double mutant with absence of Cux1-positive cells, misplacement of Foxp2-positive cells (yellow arrowheads and arrows). Milder abnormalities are present in the Lis1+/ko mutant with reduction in the number of positive cells. Later born neurons (Cux1 positive) are reduced or absent in the Lis1+/ko;Dcxko/Y double mutant suggesting that there is a neurogenesis defect in addition to defects in neuronal migration. Scale bar, 100 μm.
Lis1 and Dcx control apical spindle orientation in radial glial progenitor cells
We recently demonstrated that Lis1 controls neuroepithelial expansion and/or radial glial neurogenesis by promoting the capture of microtubules at the cell cortex and controlling the mitotic spindle orientation of neuronal progenitors at the ventricular surface (Yingling et al., 2008). We tested whether haploinsufficiency for Lis1 combined with the absence of Dcx would lead to a more severe spindle orientation phenotype at E9.5 and E14.5 (Fig. 5A). During neuroepithelial expansion (E9.5), when Dcx is not expressed there were no significant changes in the spindle orientation for Dcxko/Y and Lis1+/ko;Dcxko/Y male mutants compared with WT and Lis1+/ko respectively, although as expected Lis1 haploinsufficiency resulted in a more randomized spindle orientation compared with WT (Fig. 5B). During radial glial neurogenesis (E14.5) we surprisingly found that the average angle of spindle orientation in the Dcxko/Y brains was randomized to a similar degree as Lis1+/ko brains (Fig. 5B), and the Lis1+/ko;Dcxko/Y double mutant brains displayed more randomized spindle orientation compared with the single mutants. We found dividing cells at E14.5 that coexpressed DCX in the SVZ/IZ (Fig. 5C) (Brown et al., 2003). Low levels of DCX have been previously reported in cells isolated from the VZ/SVZ of P2 mouse brains, while at E14 20% of same population coexpressed the precursor marker nestin (Walker et al., 2007). Apical-basal polarity was preserved in all mutants, indicated by the correct localization of atypical PKC (Fig. 5A), β-catenin (data not shown) and the presence of the cadherin hole at the apical membrane (data not shown; Kosodo et al., 2004). Although aPKC was normally localized in the mutants, analysis of the projected Z-stack images revealed a broader expression pattern with a thickened distribution at the ventricular surface, and the distribution of the centrosomes was less organized (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Nestin immunostaining displayed a mild reduction of labeled fibers at the VZ/SVZ in the single Lis1+/ko mutants and a normal pattern in Dcxko/Y mutants. Nestin staining in the Lis1+/ko;Dcxko/Y double mutant was significantly reduced at the VZ/SVZ with less organized and more fragmented fibers (Fig. 5D). Thus, the abnormal distribution of atypical PKC likely resulted from a general cellular disorganization.
Lis1 and Dcx are required to maintain the progenitor pool during neurogenesis
During radial glial neurogenesis, the positioning of the spindle determines the fate of the two dividing daughter cells. A higher number of asymmetric divisions during neurogenesis should commit more cells to the neuronal lineage and lead to a depletion of the progenitor cells. We investigated both cell proliferation and cell-cycle exit in all genotypes by injecting BrdU 30 min before collection of the brains and performing double staining with BrdU and Ki-67 antibodies. In WT brains, 41% of cells were BrdU-positive, while in mutants this was reduced to 39% and 33.7% in the Dcxko/Y and Lis1+/ko single genotypes respectively and 27.2%in the Lis1+/ko;Dcxko/Y double mutant brains (Fig. 6A,B). In WT brains, 17% of dividing cells exited the cell-cycle, while in Dcxko/Y and Lis1+/ko single mutants, 21.4% and 26.8% of dividing cells exited the cell-cycle respectively, and in Lis1+/ko;Dcxko/Y double mutants, 44.5% of dividing cells exited the cell-cycle (Fig. 6A,C). We further investigated whether cell cycle exit was increased in intermediate and/or radial glial progenitors using Tbr2 as third marker. In WT, Dcxko/Y and Lis1+/ko single mutants, cell cycle exit occurred exclusively in the radial glial population, while in the Lis1+/ko;Dcxko/Y double mutants both populations were affected, although with different degree of severity (Fig. 6A,D). We quantified the number of ventricular and abventricular mitoses by staining with phospho-histone H3 antibody and found that the double mutant Lis1+/ko;Dcxko/Y brains had a higher number of abventricular mitoses compared with the other genotypes (Fig. 6A,E). Staining with the neuronal marker TuJ confirmed the presence of more postmitotic neurons in the Lis1+/ko;Dcxko/Y genotype compared with WT, while there were no major differences in the single mutants (Fig. 6A). We determined whether the increase in cell-cycle exit was accompanied by an increase in apoptosis. Although more apoptotic cells were found in the Lis1+/ko single mutants compared with WT or Dcxko/Y single mutants (0.81% ± 0.3 compared with 0.22% ± 0.15 or 0.32% ± 0.11 respectively), no significant differences were seen in the Lis1+/ko;Dcxko/Y double mutants compared with the Lis1+/ko single mutants (0.8% ± 0.25). These data suggest that there was depletion of the progenitor pool during neurogenesis in the Lis1+/ko;Dcxko/Y double mutant, perhaps due to more randomized mitotic divisions of progenitor cells at the VZ.
Figure 6.
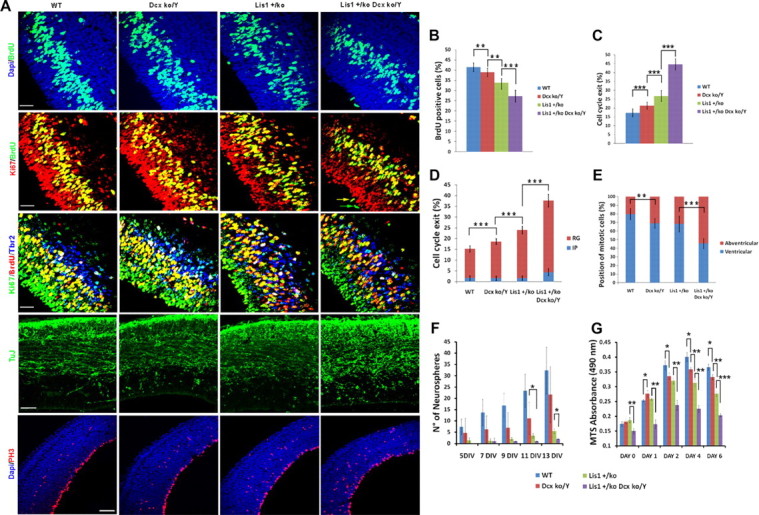
In vivo and in vitro proliferation is defective in Lis1+/ko;Dcxko/Y male mutants with depletion of the progenitor pool during development. In vivo and in vitro analysis of proliferation in each of the genotypes at E14.5. A, BrdU immunostaining (top; green) demonstrates a reduction in number of proliferating cells, especially in the Lis1+/ko;Dcxko/Y double mutant. Double with BrdU/Ki-67 antibodies (second row) and triple immunostaining with BrdU/Ki-67/Tbr2 (third row) antibodies was used to measure overall cell cycle exit and cell cycle exit in intermediate progenitor and radial glial cells. The two arrows in the Lis1+/ko;Dcxko/Y double mutant indicate the different fates of the two dividing daughter cells. TuJ immunostaining is stronger in the Lis1+/ko;Dcxko/Y double mutants, consistent with the increase number of neurogenic divisions (fourth panel from top). PH3 immunostaining demonstrates an increasing proportion of mitotic cells dividing away from the ventricular surface in each of the mutants (fifth panel from top). Scale bars: 30 μm for BrdU and cell cycle exit, 50 μm for TuJ staining, 100 μm for PH3 staining. B, Quantification of the BrdU incorporation. C, Quantification of total cell cycle exit using BrdU/Ki-67 double staining. D, Proportion of cells exiting the cell cycle in the intermediate progenitors (IP) and radial glial (RG) cells using BrdU/Ki-67/Tbr2 triple staining. E, Quantification of PH3-positive cells: the proportion of abventricular mitoses increases in each of the mutant genotypes, with the highest proportion present in the Lis1+/ko;Dcxko/Y double mutants. F, Quantification of the number of neurospheres generated per well during 13 d of neurosphere culture. G, Quantification of the MTS absorption during 6 d of neurosphere culture. Error bars indicate SD; *p < 0.05, **p < 0.001, ***p < 0.0001 by two-tailed t test.
Lis1 and Dcx mutant precursor cells display proliferation defects in vitro
We tested whether there were proliferation defects in these mutants in vitro using neurosphere cultures. We counted the number of neurospheres generated at 5 DIV after establishment of primary cortical cultures up to 13 DIV. We collected data using 9, 5, 3, and 2 plate-replicates for WT, Dcxko/Y, Lis1+/ko and Lis1+/ko;Dcxko/Y cultures, respectively (Fig. 6F). Although there were differences in the number of neurospheres at each time point, the only significant differences were at 11 and 13 DIV between Dcxko/Y and Lis1+/ko;Dcxko/Y and at 13 DIV between Lis1+/ko and Lis1+/ko;Dcxko/Y probably due to the intraindividual variability among replicates and the inability of Lis1+/ko and Lis1+/ko;Dcxko/Y cells to reproducibly generate neurospheres in suspension. Therefore we used a second method to assess proliferation and viability based on the absorption of the soluble version of MTT (MTS). Trypsinized P0 neurospheres were plated as single cells and cultured for 6 d under the same conditions used to grow neurospheres and proliferation was measured using an automated plate reader (day 0 was considered 12 h after plating). The single Dcxko/Y and Lis1+/ko mutants displayed a moderate but significant decrease in proliferation compared with WT starting at 2 DIV (Fig. 6G), while cell viability of the Lis1+/ko;Dcxko/Y double mutant instead was reduced at day 0 and proliferation of these cells severely was reduced starting at 1 DIV. In summary, this in vivo and in vitro data strongly suggest a critical role for Dcx in proliferation, resulting in depletion of the progenitor pool that may be related to the differences seen in the positioning of the mitotic spindle of radial precursor cells at the VZ.
Discussion
We initiated this study to investigate the genetic interaction between Lis1 and Dcx in vivo, and we demonstrated here that the genetic interaction of the two genes results in more severe defects in both neuronal migration and the proliferation of neuronal precursor during brain development. Moreover these findings led us to examine the Dcx single mutant, where we found that neuronal migration and proliferation are also defective, although cortical organization in the Dcx knock-out mouse is fairly well preserved at the histological level (Corbo et al., 2002; Kappeler et al., 2006). These studies demonstrate an essential role for Dcx in neuronal migration and, surprisingly, during neurogenesis.
Cortical and hippocampal development is severely disrupted in Lis1+/ko;Dcxko/Y mutant brains
Based on the neonatal lethality of Dcxko/Y males, it is not surprising that Lis1+/ko;Dcxko/Y double mutant also died shortly after birth. Therefore, we analyzed the morphology of adult Lis1+/ko;Dcx+/ko female mutant brains, which displayed severe disruption of the hippocampus, enlargement of the ventricles and loss of other brain areas compared with the milder disorganization of the hippocampus of the Lis1+/ko and Dcxko/Y mice. During embryogenesis, we confirmed the severe disorganization of the hippocampus in the Lis1+/ko;Dcxko/Y male mutants. At E13.5 we detected a reduction in the cortical area in the double male mutant compared with the WT and single mutant genotypes. Subsequent analysis of the P0 brains with layer-specific markers suggested that both neurogenesis and neuronal migration defects affected the normal development of the Lis1+/ko;Dcxko/Y male cortex. Since neurogenesis was found to be severely defective in the double mutant with a conspicuous increase of cells exiting cell cycle, this shift toward more neurogenic divisions may trigger a depletion of the progenitor pool and may explain the reduced size the cortex during early stages and the mislocalization or absence of neurons in the most outer cortical layers at later times.
Neuronal migration is severely impaired in Lis1+/ko;Dcxko/Y mutant brains
As previously reported, heterozygous mutation of Lis1 in mice is sufficient to affect neuronal migration (Hirotsune et al., 1998; Gambello et al., 2003). However, the inactivation of the Dcx gene in males did not lead to any major cortical phenotype, although these mice did display moderate defects of the hippocampus (Corbo et al., 2002). Previous studies suggested that there is a cross-talk between the two proteins regarding the binding and stabilization of microtubules in the developing cortex (Caspi et al., 2000). In addition, the migration defects of Lis1+/ko neuronal cells were rescued by Dcx overexpression (Tanaka et al., 2004). In contrast, RNA interference of Dcx in migrating neurons resulted in migration defects, but neither additive or synergistic effects on migration were found when knockdown of both Lis1 and Dcx genes were combined (Bai et al., 2003).
Here we demonstrated that the concomitant genetic deficiency of the Lis1 and Dcx gene products resulted in the inability of most of the neurons to migrate in the cortex and the migration ability of the few remaining neurons was severely reduced. Moreover, we detected a similar decreased average velocity in Lis1 heterozygotes and Dcxko/Y males, supporting the notion that both genes are essential for neuronal migration. The maximal velocity and acceleration of neurons were not severely impaired in the single mutants compared with the WT neurons, suggesting that some cells maintain the mechanical ability to effect short efforts in migration, which likely depends on the availability of the proteins responsible to promote nuclear translocation. The concomitant reduction of the two proteins resulted in a dramatic loss of all aspects of migration, including maximal velocity and acceleration.
Dcx is essential for neuronal migration in the murine neocortex
In our initial description of the Dcx knock-out mouse, we found no major abnormalities in cortical lamination, although migration of cortical neurons was not directly investigated (Corbo et al., 2002). Radial migration was found to be affected in rat neocortex after Dcx RNA interference (Bai et al., 2003), and knockdown of Dcx significantly slowed the migration of rat interneurons from the ganglionic eminence to the cerebral cortex (Friocourt et al., 2007).
Our observations shed some light on the unexplained discrepancy between the phenotypes of the human patients and the normal cortex so far reported for Dcx knock-out mice, suggesting that the difference in phenotype may be due to the way the tissue develops and perhaps the extended migratory path needed in human cortex to reach the cortical plate rather than a change in Dcx requirement or redundancy.
We detected an overall reduced velocity of migration and an atypical migration behavior consisting of frequent changes of direction in the cortex. Defective neuronal migration in the Dcx mouse knock-outs have been reported only for tangentially migrating interneurons derived from the medial ganglionic eminence or in the RMS (Kappeler et al., 2006; Koizumi et al., 2006a). In these studies, the migration of Dcx mutant neurons was disorganized with reduced long-distance movements of the nucleus, and migration was characterized by long stuttering-like pauses resulting in a delayed migration that was independent of direction or responsiveness to Slit chemorepulsion. In both studies, Dcx mutant neurons displayed atypical multipolar morphology with increased number of branches. Our studies definitely demonstrate that Dcx is essential for radial, tangential and VZ-directed migration in the cortex.
DCX is on the X chromosome in humans and mice. Heterogyzote females with DCX mutations display a “double cortex” phenotype due to random X inactivation. Neurons with inactivation of the X chromosome with the normal DCX allele are completely missing DCX protein, and remain as a band of heterotopic neurons, while neurons that inactivate the X chromosome with the mutated DCX gene have normal levels of DCX protein and migrate normally (des Portes et al., 1998; Gleeson et al., 1998; Kato and Dobyns, 2003). In rodents, genetic studies have demonstrated a cell-autonomous mechanism for Dcx effects on neuronal migration (Koizumi et al., 2006a; Kerjan et al., 2009), while RNA interference studies have shown that partial non-cell-autonomous effects may arise likely due to the cooperative property of radially migrating cells in the neocortex (Bai et al., 2003; Ramos et al., 2006;). In our study the reduced velocity of Dcx deficient cells did not demonstrate the clear cell-autonomous mechanism for migration in female heterozygotes, since the distribution of both WT and Dcx-deficient populations in the Dcx+/ko female genotype was not bimodal, but rather uniform and in between the WT and Dcxko/Y speed of migration. Similarly the quantification of the multidirectional phenotype and the number of direction changes of the Dcx+/ko and Dcxko/Y genotypes may be interpreted as the effects of both cell- and non-cell-autonomous mechanisms. These findings are in agreement with Bai et al. (2003) that demonstrated dual effects on radial migration of cortical rat neurons after inactivation of the DCX protein function. However, an alternative possibility is that there was not an adequate differential velocity between WT and Dcx mutant cells to observe a bimodal distribution, as would be expected based on the phenotype in human female heterozygous (Matsumoto et al., 2001). We have previously shown cell-autonomous effects for Lis1 in neuronal migration defects in vitro (Hirotsune et al., 1998; Gambello et al., 2003). Subsequently, using embryonic brain slice transplant cultures of tangentially migrating interneurons, mixing wild type and mutant substrates and migrating cells, we clearly demonstrated that the main effects of Lis1 during tangential migration are cell autonomous, although small non-cell-autonomous effects were found to be due to alterations of the migration substrate (McManus et al., 2004). Thus, it is likely that there are major cell-autonomous and some additional non-cell-autonomous effects when either Lis1 or Dcx is mutated.
Dcx is essential for neurogenesis
A role for Dcx in neurogenesis and proliferation has not previously been reported, and DCX is commonly used as a marker for postmitotic neurons (des Portes et al., 1998; Gleeson et al., 1999). Here, we provide several lines of evidence that Dcx does in fact play a role in neurogenesis and proliferation of neuronal progenitors. First, the average spindle orientation of Dcxko/Y and Lis1+/ko;Dcxko/Y radial glial progenitors was shifted toward more asymmetric divisions compared with WT and Lis1+/ko progenitors, respectively. Second, this change correlated with moderate but significant differences in the number of cells positive for BrdU and in cell cycle exit. Third, cell cycle exit occurred significantly in the radial glial population in both single and double mutants, with Lis1+/ko and Dcxko/Y single mutants showing no change in the intermediate progenitor population; this strongly correlates with the phenotypic differences in the spindle orientation. Fourth, there was an increase in abventricular mitoses in Dcxko/Y and Lis1+/ko;Dcxko/Y embryos compared with WT and Lis1+/ko or Dcxko/Y embryos, respectively. Finally, in vitro studies using embryonic cortical cultures directly support a role for Dcx in proliferation. Importantly, there was no effect of Dcx mutation on spindle orientation in neuroepithelial stem cells at E9.5, when Dcx is not expressed. Additionally, the differences found in the number of BrdU-positive cells, the excessive percentage of cells exiting the cell cycle and the reduction in number of later born Cux1-positive neurons correlated with the change in the average spindle orientation of the Lis1+/ko;Dcxko/Y mutant. Together, these data indicated that, like Lis1, Dcx plays a crucial role in neuronal migration and a significant role in maintaining the correct balance between neurogenic and stem cell-like mitotic divisions. Loss of Dcx eventually contributes to the depletion of the progenitor pool during cortical development.
Supporting a role for Dcx in proliferation, a subpopulation of cells expressing Dcx at low levels was detected that proliferates in a stem cell-like manner both at E14.5 and P2 (Walker et al., 2007). A role for Dcx in cell proliferation might be expected based on the study of mutation of its ortholog zyg-8 in C. elegans that demonstrated defective mitotic spindle positioning and microtubule assembly (Gönczy et al., 2001). Other Dcx homologues are clearly involved in neurogenesis; Dclk is highly expressed in regions of active neurogenesis in the cortex and regulate the formation of bipolar mitotic spindles and the proper transition from prometaphase to metaphase during mitosis (Shu et al., 2006). Dcl, a splice variant of Dclk, is expressed in radial glial and neural progenitors and regulates mitotic spindle stability, precursor proliferation and integrity of the radial fiber network (Vreugdenhil et al., 2007). As we demonstrate, Dcx itself plays a significant role in neurogenesis as well.
Conclusion
Our study provides definitive evidence, from both single and double mutants, that Dcx plays important roles in neuronal migration and neurogenesis (Fig. 7), and strongly suggests that functional relationships of these two proteins are critically important in regulating specific cellular functions required for migration and neurogenesis. DCX binds ubiquitously to microtubules in non-neuronal cells and its activity is essential for their bundling and stabilization (Gleeson et al., 1999). Perhaps this function is impaired in Dcx mutants resulting in abnormal branching and change of direction during migration. We have recently uncovered an important role of Lis1 in mitotic divisions of neuroepithelial and radial glial precursor cells at the VZ (Yingling et al., 2008), required for proper attachment of microtubules at the cell cortex. Perhaps DCX plays an important role with LIS1 in cortical dynein capture, although the precise mechanisms have yet to be defined. These findings suggest that the molecular interaction of the LIS1 and DCX is important in both in neuronal migration and neurogenesis. In addition, there is a cortical role of DCX in nuclear translocation and positioning of the mitotic spindle in radial glial mitotic division.
Figure 7.

Summary of neuronal migration and neurogenesis defects found in single Lis1+/ko and Dcxko/Y mutants and double Lis1+/ko;Dcxko/Y mutants. In the WT, neuroepithelial stem cells (NESCs, E9.5 stage) actively proliferate in a stem cell like manner with the spindle orientation mostly perpendicular to the surface. At later stages (E14.5), radial glial progenitors (RGPCs) divide both symmetrically and asymmetrically to generate postmitotic neurons that migrate in an inside-out manner to organize the cortical plate into six layers. Loss of Dcx does not affect spindle positioning during neuroepithelial expansion but does have effects on radial glial progenitor cells, randomizing spindle orientation similar to Lis1 heterozygotes. Although the final cortical organization is preserved in Dcx male mutants, neuronal migration is slower with a multidirectional pattern of migration. Lis1 haploinsufficiency, as previously described (Yingling et al., 2008) results in spindle orientation defects both during neuroepithelial expansion and radial glial mitotic divisions. Cortical organization is mostly preserved with a mild disorganization and a reduction of the number of neurons in the most outer and inner layers of the cortex. In contrast the Lis1+/ko;Dcxko/Y double mutants displayed a more severe randomization of the spindle during radial glial mitotic divisions with abventricular divisions leading to increased cell-cycle exit and depletion of the progenitor pool at the ventricular surface. Neuronal migration and proliferation defects lead to a severe disorganization of the cortex with misplaced neurons and the lack of Cux1-positive cells. In contrast, during neuroepithelial cell divisions the spindle orientation is not significantly different from the Lis1+/ko brains.
Footnotes
This work was supported in part by the National Institutes of Health (Grants NS041310 and HD047380 to A.W.-B.). We thank Lana Bogdanova and Donna Holland for animal handling, and Jeehee Hong and Patricia LaPorte for technical assistance. We especially thank Brendan Brinkman for technical expertise and support at the University of California, San Diego, Neuroscience Microscopy Shared Facility (P30 NS047101).
References
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bantubungi K, Blum D, Cuvelier L, Wislet-Gendebien S, Rogister B, Brouillet E, Schiffmann SN. Stem cell factor and mesenchymal and neural stem cell transplantation in a rat model of Huntington's disease. Mol Cell Neurosci. 2008;37:454–470. doi: 10.1016/j.mcn.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Caspi M, Atlas R, Kantor A, Sapir T, Reiner O. Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum Mol Genet. 2000;9:2205–2213. doi: 10.1093/oxfordjournals.hmg.a018911. [DOI] [PubMed] [Google Scholar]
- Corbo JC, Deuel TA, Long JM, LaPorte P, Tsai E, Wynshaw-Boris A, Walsh CA. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci. 2002;22:7548–7557. doi: 10.1523/JNEUROSCI.22-17-07548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrié A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838–2842. doi: 10.1001/jama.270.23.2838. [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Liu JS, Antypa M, Rakic S, Walsh CA, Parnavelas JG. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J Neurosci. 2007;27:3875–3883. doi: 10.1523/JNEUROSCI.4530-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambello MJ, Darling DL, Yingling J, Tanaka T, Gleeson JG, Wynshaw-Boris A. Multiple dose-dependent effects of Lis1 on cerebral cortical development. J Neurosci. 2003;23:1719–1729. doi: 10.1523/JNEUROSCI.23-05-01719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Gönczy P, Bellanger JM, Kirkham M, Pozniakowski A, Baumer K, Phillips JB, Hyman AA. zyg-8, a gene required for spindle positioning in C. elegans, encodes a doublecortin-related kinase that promotes microtubule assembly. Dev Cell. 2001;1:363–375. doi: 10.1016/s1534-5807(01)00046-6. [DOI] [PubMed] [Google Scholar]
- Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008;31:154–162. doi: 10.1016/j.tins.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3:342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- Ishii Y, Matsumoto Y, Watanabe R, Elmi M, Fujimori T, Nissen J, Cao Y, Nabeshima Y, Sasahara M, Funa K. Characterization of neuroprogenitor cells expressing the PDGF beta-receptor within the subventricular zone of postnatal mice. Mol Cell Neurosci. 2008;37:507–518. doi: 10.1016/j.mcn.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kappeler C, Saillour Y, Baudoin JP, Tuy FP, Alvarez C, Houbron C, Gaspar P, Hamard G, Chelly J, Métin C, Francis F. Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet. 2006;15:1387–1400. doi: 10.1093/hmg/ddl062. [DOI] [PubMed] [Google Scholar]
- Kato M, Dobyns WB. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12:R89–R96. doi: 10.1093/hmg/ddg086. (Spec No 1) [DOI] [PubMed] [Google Scholar]
- Kerjan G, Koizumi H, Han EB, Dubé CM, Djakovic SN, Patrick GN, Baram TZ, Heinemann SF, Gleeson JG. Mice lacking doublecortin and doublecortin-like kinase 2 display altered hippocampal neuronal maturation and spontaneous seizures. Proc Natl Acad Sci U S A. 2009;106:6766–6771. doi: 10.1073/pnas.0812687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Gleeson JG. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci. 2006a;9:779–786. doi: 10.1038/nn1704. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006b;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Kosodo Y, Röper K, Haubensak W, Marzesco AM, Corbeil D, Huttner WB. Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 2004;23:2314–2324. doi: 10.1038/sj.emboj.7600223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Learish RD, Bruss MD, Haak-Frendscho M. Inhibition of mitogen-activated protein kinase kinase blocks proliferation of neural progenitor cells. Brain Res Dev Brain Res. 2000;122:97–109. doi: 10.1016/s0165-3806(00)00064-x. [DOI] [PubMed] [Google Scholar]
- Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- Matsumoto N, Leventer RJ, Kuc JA, Mewborn SK, Dudlicek LL, Ramocki MB, Pilz DT, Mills PL, Das S, Ross ME, Ledbetter DH, Dobyns WB. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur J Hum Genet. 2001;9:5–12. doi: 10.1038/sj.ejhg.5200548. [DOI] [PubMed] [Google Scholar]
- McManus MF, Nasrallah IM, Pancoast MM, Wynshaw-Boris A, Golden JA. Lis1 is necessary for normal non-radial migration of inhibitory interneurons. Am J Pathol. 2004;165:775–784. doi: 10.1016/S0002-9440(10)63340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb Cortex. 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12:4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietze RL, Reynolds BA. Neural stem cell isolation and characterization. Methods Enzymol. 2006;419:3–23. doi: 10.1016/S0076-6879(06)19001-1. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Shu T, Tseng HC, Sapir T, Stern P, Zhou Y, Sanada K, Fischer A, Coquelle FM, Reiner O, Tsai LH. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. 2006;49:25–39. doi: 10.1016/j.neuron.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14–3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Tsai JW. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 2006;20:1384–1393. doi: 10.1101/gad.1417206. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil E, Kolk SM, Boekhoorn K, Fitzsimons CP, Schaaf M, Schouten T, Sarabdjitsingh A, Sibug R, Lucassen PJ. Doublecortin-like, a microtubule-associated protein expressed in radial glia, is crucial for neuronal precursor division and radial process stability. Eur J Neurosci. 2007;25:635–648. doi: 10.1111/j.1460-9568.2007.05318.x. [DOI] [PubMed] [Google Scholar]
- Walker TL, Yasuda T, Adams DJ, Bartlett PF. The doublecortin-expressing population in the developing and adult brain contains multipotential precursors in addition to neuronal-lineage cells. J Neurosci. 2007;27:3734–3742. doi: 10.1523/JNEUROSCI.5060-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynshaw-Boris A. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72:296–304. doi: 10.1111/j.1399-0004.2007.00888.x. [DOI] [PubMed] [Google Scholar]
- Yingling J, Youn YH, Darling D, Toyo-Oka K, Pramparo T, Hirotsune S, Wynshaw-Boris A. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;132:474–486. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn YH, Pramparo T, Hirotsune S, Wynshaw-Boris A. Distinct dose-dependent cortical neuronal migration and neurite extension defects in Lis1 and Ndel1 mutant mice. J Neurosci. 2009;29:15520–15530. doi: 10.1523/JNEUROSCI.4630-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]