

Abstract
A recent conference entitled Purines in Cell Signalling: Targets for New Drugs, held in Rockville, Maryland, in September, 1989, was one indication of the increasing interest in developing agonists and antagonists of P1-(adenosine) and P2-(ATP) purinoceptors [1] as potential therapeutic agents. Extracellular adenosine, acting at its membrane bound A1 and A2 receptors, is a ubiquitous modulator of cellular activity. The purine can arise from several sources including ATP hydrolysis by ectokinase activity in the region of the nerve terminal [2] and from S-adenosylhomocysteine [3] and ATP within the cell. Together with its more stable analogs, adenosine is a potent inhibitor of neurotransmitter release in both the central and peripheral nervous systems, and in cardiac, adipose and other tissues. Adenosine can also affect blood pressure and heart rate as well as modulate the function of the immune, inflammatory, gastrointestinal, renal and pulmonary systems, either via its effects on transmitter release or directly via receptor mechanisms altering intracellular transduction processes.
Adenosine agonists
The first reported physiological action of adenosine (1) was in regard to its ability to lower blood pressure, a finding reported by Drury and Szent-Györgyi in 1929 [4]. This seminal article stimulated interest in the potential use of adenosine as an antihypertensive agent. However, clinical evaluation of the purine in the 1930s as a hypotensive agent was ephemeral due to its poor efficacy, a result of a short half-life, and led to a degree of negativity about its therapeutic potential [5]. Interestingly, the recently approved use of adenosine as an agent for the treatment of supraventricular tachycardia [6], developed by Berne and coworkers, is successful primarily because of the short half-life of the native compound.
In the 60 years since the work of Drury and Szent-Györgyi, many other uses for adenosine agonists have been proposed (Table 1). These actions are dependent almost exclusively on the degree of selectivity of the ligands for either A1 or A2 receptor subtypes and the present knowledge related to the physiological role(s) of these receptors.
Table 1.
| Tissue/organ | Adenosine agonist | Adenosine antagonist |
|---|---|---|
| CNS | Antipsychotic [9,10], anxiolytic, sedative, analgesic, neuroprotective [12], anti-convulsant, anti-hypertensive (central mech.), anti-Parkinson | Nootropic, cognition enhancer [11] |
| Heart | Antiarrhythmic [13], congestive heart failure | Cardiotonic |
| Kidneys | Antihypertensive (by inhibition of renin release [14]) | Diuretic, kidney protective [14] |
| Vasculature | Vasodilator | — |
| Immune | — | Immunostimulant |
| Inflammatory | Anti-inflammatory | — |
| Reproductive | — | Contraceptive (lowers sperm motility)* |
| Gastrointestinal | Inhibitory to gastric acid secretion [15] | — |
| Lungs | — | Anti-asthmatic [16,†] |
Hoskins D and Vijayaraghavan S, Intracellular adenosine regulates mammalian sperm motility. Presented at Purine Nucleotides and Nucleotides in Cell Signalling: Targets for New Drugs, Bethesda, MD, Sept. 17, 1989, Abstr. D1.
Anti-asthmatic effects of theophylline are complex and appear to be related to inhibition of phosphodiesterases and other mechanisms.
Several thousand potent adenosine agonists (selected structures in Table 2) have been synthesized in industrial and other medicinal chemistry laboratories in the past 40 years [1]. Anecdotal reports of limited human trials in the early 1950s with 2-chloroadenosine (2-CADO, 3) suggested that the longer half-life of this agonist and its greater potency led to an unacceptable, near fatal drop in blood pressure that precluded further testing. In the 1970s, Boehringer-Ingelheim conducted a major program in synthesizing N6-alkyl substituted adenosine analogs [including N6-(R)-phenyliso-propyl-adenosine, R-PIA, 6] as potential hypotensive agents [17]. The vasodilatory potency of such agonists was improved by Abbott Laboratories with the synthesis of 5′-N-ethylcarboxamidoadenosine [18] (NECA, 2), which is still one of the more potent agonists at the A2 adenosine receptor subtype. Interestingly, the original patent for this compound [18] described it as a rodent poison. Takeda Pharmaceuticals discovered that arylamino substitution at the C-2 position of adenosine resulted in potent vasodilators [19], among which, CV 1808 [2-(phenyl-amino) adenosine, 4] was later found to be a weak (Ki = 100 nM) but 7-fold A2-selective adenosine agonist [20].
Table 2.
Structures of selected adenosine agonists and affinities at A1- and A2-adenosine receptors (data from references given in text)

| |||||
|---|---|---|---|---|---|
| Compound | R | R′ | R″ | KiA1 | KiA1 |
| 1 | H | H | CH2OH | 12.8 | 37 |
| 2 | H | H | CONHEt | 6.3 | 12 |
| 3 | H | Cl | CH2OH | 9.3 | 63 |
| 4 | H |

|
CH2OH | 600 | 116 |
| 5 | H |
|
CONHEt | 2600 | 15 |
| 6 |

|
H | CH2OH | 1.17 | 124 |
| 7 |

|
H | CH2OH | 6.8 | 25 |
| 8 |
|
H | CH2OH | 142 | 4.4 |
| 9 | ” | H | CONHEt | 207 | 5.6 |
| 10 | ” |

|
CH2OH | 10,300 | 340 |
| 11 |
|
H | CH2OH | 22 | 412 |
| 12 |

|
H | CH2OH | 0.3 | 1390 |
| 13 | ” | H | CH2Cl | 0.24 | 3900 |
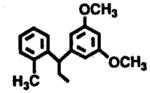
One of the most significant efforts in the medicinal chemistry of adenosine derivatives was that of Parke-Davis in the 1980s [10,20–23]. This program encompassed both agonist and antagonist pharmacophores for adenosine receptors and resulted in the identification of several classes of N6-substituted adenosine derivatives. The majority of these were A1 selective, but others including CI-936 [N6-(2,2-diphenylethyl)adenosine, 7] [10] were potent agonists at A2-adenosine receptors. Continued efforts to optimize the A2 affinity of this series of compounds resulted in a series of substituted diphenyl-ethyladenosine derivatives, from which DPMA (N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine, PD 125,944, 8) was identified as having a 32-fold selectivity for the A2 receptor, with a Ki value of 4.4 nM [21].
Interestingly, when an anilino function was incorporated into the C2-position of DPMA, such as in the case of CV-1808, with an intent to improve the A2 potency and/or selectivity, it rendered an analog (10) with significantly lower affinity at both the receptors (A1 Ki = 10,300 nM; A2 Ki = 340 nM) [22]. The decrease in the binding affinity for this analog compared to the parent compound was attributed to the steric factors involved at the C2 domain of the binding site. However, modification of the 5′-hydroxymethyl function to a carboxamidoethyl function, such as in the case of NECA, provided an analog (9) with binding affinity (A1 Ki = 207 nM; A2 Ki = 5.6 nM) similar to the parent compound [21]. These data suggest that there may exist two separate binding domains at the A2 receptor where these adenosine analogs could interact independently when substituted either in the N6-position or in the 5′-position. However, for the A1 receptor, the binding affinity seems to be dependent on the interactions at both the N6 and the 5′-domain. For example, conversion of the A1-selective agonist N6-(2-S-endo-norbonyl)adenosine (S-ENBA, 12) into a 5′-chloro derivative (13) resulted in a compound which is one of the most potent (Ki = 0.24 nM) and highly selective (16,000-fold) ligands for the adenosine A1 receptor [23], Thus, selectivity could be enhanced for the A1 receptor by simply modifying the 5′-position of the molecule. Indeed, in recent years, extensive work on structure–activity relationships has been carried out in various laboratories, which not only enhanced our understanding of the binding domain of these receptors but also has provided major insights into the key structural features required for better affinity and/ or selectivity at these receptors.
Hybrid modifications of the purine nucleoside pharmacophore in the 5′- and C2 positions by the CIBA-Geigy group led to over 200 highly A2-selective adenosine agonists, among which CGS 21680 [2-(2-p-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine, 5] was 140-fold selective for the A2-adenosine receptor subtypes with an affinity of 21 nM [24]. Similar 5 ′/N6 hybrid molecules are generally A1 selective [25].
Despite the intensive efforts on the part of many companies over the past 30 years with the attendant discovery of successive generations of potent and selective adenosine agonists, it is ironic that only adenosine itself has been approved as a therapeutic agent. Lyphomed/Medco introduced adenosine as Adenocard™ for the treatment of supraventricular tachycardia (SVT), a transitory blockade of AV node conduction leading to an attenuation of abnormal heart rhythms [6], based on work by Belardinelli, Berne, and colleagues. The crucial feature in the administration of adenosine, rather than a more stable (and potent) analog, for SVT was the very short half-life (10 sec) of the nucleoside [26]. As a result of metabolic inactivation, primarily through adenosine deaminase, the effects of intravenously-administered adenosine are limited to actions in the vascular system and, as a result, potential side-effects are avoided. Interestingly, Adenocard™ has been upgraded recently to the FDA’s coveted 1A designation for a compound fulfilling unmet medical need. Medco is also pursuing use patents for diagnostic applications of adenosine in various cardiovascular disease states, while the purine is being used for controlled hypotension in aneurysm surgery in Sweden [27] and evaluated for similar use in North America.
In contrast, the majority of potent and selective adenosine analogs have remained in the research tool category. One reason for this is the perceived potential actions of nucleosides and nucleotides as antimetabolites, which would be undesirable here. Many such compounds including the anti-AIDS drugs AZT (3-azidothymidine), ddI (2′,3′-dideoxyinosine) and ddC (2′,3′-dideoxycytosine) are effective antiviral agents and have the potential both to interfere with nucleic acid replication and to suppress the immune response. Another issue relates to the ubiquitous actions of the purine nucleoside. Because adenosine receptors, both A1 and A2, are present in nearly all tissues, it is probable that agents acting via activation or inhibition of such receptors will have a multitude of actions. Xanthine adenosine antagonists (see below) with potential as central stimulants may also function as cardiotonic agents [28]. Conversely, agonists affecting cardiovascular function will cause sedation in the CNS and also alter renal function. This liability for side-effects, perceived or real, is a major barrier to the use of synthetic adenosine agonists, which are potent and relatively metabolically stable in vivo.
For example, Parke-Davis sought to develop for human therapeutic use several promising adenosine agonists, including CI-936 (7) evaluated as an antipsychotic [9]. CI-936 exhibited antipsychotic-like activity in mice by inhibiting motor activity (ED50 = 1.3mg/kg) without producing significant ataxia (ED50 = 145mg/kg)[10]. Furthermore, in the Sidman avoidance paradigm, a test predictive of antipsychotic activity, this compound selectively blocked the response with an ED50 of 2.5 mg/kg. Extensive toxicological evaluation in mice, rats, dogs and monkeys* showed that CI-936 was well tolerated at daily doses up to 40 mg/kg in rats, 12.5 mg/kg in dogs, and 12.5 mg/kg in monkeys for up to 2 weeks. Sporadic emesis was noted at 12.5 mg/kg in dogs and at 25 mg/kg in monkeys.* Unfortunately, the lack of selectivity of CI-936 for its CNS versus cardiovascular actions and other side-effects precluded further development.
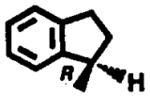
Another series of novel adenosine agonists identified at Parke-Davis, the N6-(benzocyclo-alkyl)adenosines, had excellent blood pressure lowering activity in a spontaneously hypertensive rat model.† From this series of compounds, (R)-N6-(2,3-dihydro-1H-inden-1-yl)adenosine (11) was identified as a potential antihypertensive agent. The oral doses required to lower mean arterial blood pressure by 20% (ED20) in the spontaneously hypertensive rats (SHR), renal hypertensive rats, DOCA (deoxycorticosterone) salt hypertensive rats and renal hypertensive dogs (RHD) were 1.2, 0.3, 1.3 and 1.5 mg/kg respectively [29]. At the ED20, heart rate was elevated moderately in RHD (+30 beats/min) and decreased in SHR (−40 beats/ min). A long duration of action was observed in all hypertensive models, with effects lasting beyond 10 hr [29]. However, this compound showed significant toxicity in dogs [30] including coronary arterial lesions, endothelial swelling, disruption of the internal elastic lamina, and necrosis. It is not clear whether the toxicity observed was the consequence of the pharmacological effects exerted by the compound or was due to the inherent toxicity associated with this class of chemicals.
Even agents which are selective for the periphery or more selective for a receptor subtype would suffer from the potential multiplicity of side-effects. For most of the envisioned applications of adenosine agonists (Table 1), such effects could conceivably be as serious as the conditions for which they were intended to treat. This would be unacceptable where therapeutically effective and safe drug entities are already meeting medical needs.
Such pessimism may be balanced to some extent by the fact that chronic ingestion of the adenosine antagonist, caffeine, is rarely associated with deleterious effects except in individuals who are extremely prone to tremors and convulsions. The underlying cause of the latter response is not known nor indeed is there much information available in regard to adenosine pathophysiologies in either cardiovascular or CNS disease states for which the nucleoside has been loosely targeted. An anomaly of the drug discovery process is that with few exceptions, preclinical studies are conducted using healthy tissue systems. The negativism regarding purinergic therapeutics has been discussed previously [7] in relation to the universal use of aspirin and other cyclooxygenase inhibitors such as analgesics and anti-inflammatory agents. The wisdom being applied to what is known about the action of adenosine agonists in animal models would similarly preclude using aspirin because of its wide spectrum of potential actions. Yet it is fairly obvious that because of the inflammatory process, the drug acts as a paracrine effector agent to seek out diseased tissues, leaving those in normal homeostatic balance unaffected.
Another factor relates to the fact that all currently known adenosine agonists represent modified forms of the parent. This fact tends to exacerbate the potential for actions that overlap with the endogenous effector. In other successful drug discovery programs, the breakthroughs in minimizing the therapeutic ratio for deleterious side-effects has been the identification of novel structure which in turn has usually led to the identification of receptor subtypes.
Adenosine antagonists
A similar situation exists with the xanthine adenosine antagonists. Caffeine and theophylline as the prototypic adenosine antagonists are widely ingested in foodstuffs and are probably the most avidly consumed drugs in the world. The actions of the xanthines are compounded by their activity as phosphodiesterase inhibitors. The basis for the use of theophylline in asthma has yet to be satisfactorily elucidated at the mechanistic level. Many potent and selective adenosine antagonists that have been developed (e.g. 8-substituted xanthine derivatives [1]) have yet to be introduced as therapeutic agents. Evaluation of such entities as central stimulants was disappointing [31], a fact that may be attributed to their poor bioavailability, due to hydrophobicity and highly stable crystal lattices, leading to very low (in the range of 1μM) aqueous solubility. A later generation of adenosine antagonists has now been developed including the more hydrophilic xanthine amine congener (XAC), which contains an ionizable group in the form of an amine, attached through a functionalized chain at a distance from the main pharmacophore [1].
In order for an adenosine antagonist to become a drug, it will have to be far superior to caffeine and theophylline, by being more selective and significantly less toxic. For this reason, it would be desirable to develop a selective, potent, and bioavailable non-xanthine adenosine antagonist. A number of classes of non-xanthine adenosine antagonists have been discovered [32]. Of these, CGS 15943A, 14 (Fig. 1) [33], was targeted in aerosol form as an antiasthmatic but failed in development due to skin irritation. A series of triazoloquinoxalines from Pfizer, which for a time were in clinical trials as antidepressants, were later found to include some A2-receptor selective adenosine antagonists [34], such as CP 66,713 (15).
Fig. 1.

Structures of two non-xanthine adenosine antagonists with selectivity for A2 receptors.
One promising potential site for therapeutic application of adenosine antagonists is in the kidneys. As shown in Table 3, adenosine affects nearly all aspects of renal function: renal blood flow and its distribution within the kidney [35], glomerular filtration rate [35], renin secretion [35], urine flow and sodium excretion [36], and transmitter release from the renal efferent sympathetic nerves [37]. All of these effects are mediated by adenosine receptors, since they are antagonized by alkylxanthines and/or mimicked by adenosine analogs that act as adenosine receptor agonists [37,38]. For some of these effects, the subclasses of adenosine receptors that are involved have been established, based on the order of potency of agonists. These observations, taken together with the observation that kidneys produce and release adenosine into extracellular fluids [39], suggest that variations in the concentration of endogenously-released adenosine could play important roles in renal function and/or dysfunction. Indeed, it has been postulated that adenosine is the mediator of several physiological and pathophysiological phenomena: the autoregulation of renal blood flow and glomerular filtration rate [35], the tubuloglomerular feedback response [40], the effect of macula densa cells on nearby renin-secreting juxtaglomerular cells [35,36,40], and the hemodynamic changes in acute renal failure [14]. If true, then theoretically, adenosine receptor antagonists should have equally profound and wide-ranging effects on renal function.
Table 3.
Summary of the receptor-mediated* renal effects of adenosine
| Target cells | Receptor type | Response | Comments |
|---|---|---|---|
| Vascular smooth muscle cells | |||
| Afferent arteriole | A1 | Constriction (decrease RBF† and GFR) | RBF can decrease, increase, or remain constant but GFR always decreases. There may be a redistribution of renal blood flow from outer to inner cortex. If so, urine flow and sodium excretion would decrease, since inner cortical nephrons reabsorb sodium and water very avidly. |
| A2 | Dilation (increase RBF and GFR) | ||
| Efferent arteriole | A2 | Dilation (increase RBF, decrease GFR) | |
| Juxtaglomerular cells | A1 | Inhibit renin secretion | Renin secretion can decrease, increase, or remain constant. |
| A2 | Stimulate renin secretion | ||
| Tubular epithelial cells | A1 | Decrease cyclic AMP | It is not clear which, if either, of these changes in cyclic AMP explain adenosine-induced decreases in urine flow and sodium excretion. |
| A2 | Increase cyclic AMP | ||
| Sympathetic nerve terminals | ? | Inhibit norepinephrine release | RBF would increase, renin secretion would decrease, and urine flow and sodium excretion would increase, since the nerves constrict (α-adrenergic), stimulate renin secretion (β-adrenergic), and enhance the reabsorption of sodium and water. |
Adenosine-induced increases in renal afferent (sensory) nerve activity are not receptor-mediated, since they cannot be blocked by theophylline. The receptor-mediated effects listed in the table can be blocked by theophylline and are induced by adenosine analogs as well as adenosine per se.
RBF = renal blood flow; and GFR = glomerular filtration rate.
Although the concentration of endogenously-released adenosine in renal extracellular fluids is in a range that suggests effects on renal hemodynamics [35], it is questionable whether variations in the concentration play a role in the normal physiological control of renal hemodynamics. There is evidence that adenosine mediates the tubuloglomerular feedback response [39,40], but it is very unlikely to mediate autoregulation of renal blood flow and glomerular filtration rate [14]. In fact, although the renal hemodynamic effects of theophylline are extremely variable, theophylline actually improves autoregulation in some pathological circumstances [41], and in congestive heart failure, theophylline nearly doubles glomerular filtration rate in humans [42], perhaps because of unusually high levels of endogenously released adenosine. Moreover, as outlined below, there is evidence that endogenously released adenosine explains, at least in part, the renal hemodynamic changes in some experimental animal models of acute renal failure.
It was proposed [14] that adenosine mediates the hemodynamic changes in acute renal failure, and that the changes are pathogenic in reducing glomerular filtration rate. According to this hypothesis, a state of energy deficit (impaired oxidative phosphorylation, resulting from hypoxic, ischemic, or nephrotoxic tubular cell injury) leads to decreased cellular ATP levels and increased adenosine production and release. The adenosine then acts on afferent and efferent arterioles to produce the hemodynamic changes. Several observations are consistent: (a) Renal ischemia results in decreased cellular ATP levels and increased adenosine production and release. Nephrotoxic injury of renal tubular cells also decreases cellular ATP levels, but it is not known if this accompanied by increased adenosine production and release, (b) Adenosine-induced changes in renal hemodynamics (see above) mimic the hemodynamic changes in acute renal failure [14], These include variable changes in renal blood flow and its distribution within the kidney, but consistent decreases in filtration fraction and glomerular filtration rate, (c) Pharmacological manipulation of the renal adenosine system has provided support for the hypothesis, at least in some experimental animal models, of acute renal failure. Theophylline has protective effects on renal function in an ischemic model that is produced in rats by unilateral occlusion of a renal artery for 30 or 45 min; renal plasma flow and glomerular filtration rate are higher in the previously-ischemic kidneys of rats treated with theophylline than in rats treated with the vehicle [14], during both the initiation and maintenance phases of renal failure. Moreover, the adenosine uptake blocker dipyridamole enhances the severity of failure in the initiation phase of this model [14]. Ischemia is considered to play a role in glycerol-induced myoglobinuric acute renal failure in rats, and in this model, also, theophylline has protective effects [14] that are dose-dependent and independent of any effects on sodium excretion or tubuloglomerular feedback. Bowmer et al. [43] have shown that 8-phenyltheophylline, a more potent adenosine receptor antagonist, has similar protective effects in the glycerol model, both with respect to renal function and renal morphology. Pentoxifylline [44] and theophylline [14,43–45] have protective effects in other ischemic and toxin-induced models of renal failure in rats and rabbits. On the other hand, in other models of nephrotoxic acute renal failure [46], adenosine-mediated hemodynamic changes do appear to be less important.
Exogenous adenosine produces intense antidiuretic and antinatriuretic effects in many species [14]. These effects are receptor-mediated since they are competitively antagonized by theophylline and mimicked by several adenosine analogs. It seems reasonable to assume that the well-known diuretic and natriuretic effects of methylxanthines are produced by antagonism of the effects of endogenously released adenosine. A variety of mechanisms could be involved in adenosine-induced antidiuresis and antinatriuresis. Explanations based on systemic effects (changes in cardiac output, blood pressure, neural activity, or hormone secretion) seem to be excluded by the observations that isolated perfused kidneys respond predictably to both agonists and antagonists. However, the changes in urine flow and sodium excretion could be a consequence of a change in renal hemodynamics, since adenosine may induce a vasodilation of the juxtamedullary cortex, and it is believed that juxtamedullary nephrons reabsorb filtered water and sodium more avidly than outer cortical nephrons. In addition, adenosine decreases the glomerular filtration rate and, therefore, the filtered loads of water and sodium. However, adenosine-induced percentage decreases in urine flow and sodium excretion exceed, by far, adenosine-induced percentage decreases in glomerular filtration rate [14]. Conversely, methylxanthines can produce diuresis and natriuresis in the absence of detectable increases in glomerular filtration rate [14]. Therefore, it seems reasonable to assume that adenosine-induced antidiuresis and antinatriuresis and, by inference, methylxanthine-induced diuresis and natriuresis, can be mediated by both renal hemodynamic and direct tubular mechanisms. Consistently, adenosine analogs stimulate active sodium transport in toad kidney cells [47]. Moreover, binding studies and studies of adenylate cyclase activity demonstrate the presence of both A1 and A2-adenosine receptors [48].
As with classical adenosine agonists, a multiplicity of side-effects of potent adenosine antagonists is possible. For example, 8-phenyltheophylline causes a diabetes-like condition in the rat heart, i.e. it induces total insulin refractoriness to glucose transport [49].
Non-classical approaches
The failure of existing entities and the perceived disadvantages of classical medicinal chemical approaches for adenosine receptor drugs has led a number of laboratories to explore alternate approaches, including prodrugs [50, *] and indirect adenosine agonists, i.e. uptake blockers [51]. Other laboratories have turned to applications such as the treatment of stroke [8,12], in which the potential benefit of life-saving treatment would outweigh the side-effects.
Prodrugs
A prodrug is a chemically masked derivative of an active drug, which is converted to an active form at a physiological target site [52]. The prodrug is a latent (and therefore biologically inactive or weakly active) form of a drug. The unmasking of the prodrug typically takes place through an enzymatic or a chemical cleavage step. Ideally, the free drug is present at biologically active concentrations only at the target organ or tissue. In this manner, side-effects at other sites are minimized.
A limited success was achieved when an attempt was made to separate the cardiovascular effects of CI-936 from the CNS effect by preparing a variety of prodrugs of the adenosine derivative.* Modification of the ribose hydroxyl groups in the form of more hydrophobic esters (simple acyl and various amino acid derivatives) produced compounds which were more CNS-versus cardiovascular-selective than CI-936.
To exploit the beneficial therapeutic properties of adenosine antagonists in the kidneys, a xanthine prodrug (Fig. 2) has been developed [50]. This prodrug was based on the conceptual fusion of two developments, one being the xanthine functionalized congeners, of which XAC is the prototype, and the other a kidney prodrug scheme first proposed by Orlowski, Wilk, and coworkers [53]. The prodrug scheme utilizes selective cleavage of γ-glutamyl amide derivatives by the enzyme γ-glutamyl transpeptidase (γ-GT), which is highly concentrated in kidney cells of the brush border membrane. This general prodrug scheme has been expanded to achieve a selective concentration of various drugs, including dopamine and the antibiotic sulfamethoxazole [53], in the kidney. This prodrug scheme utilizes a two-step cleavage process, in which an N-acyl-γ-glutamyl amide derivative of an amine-containing drug is cleaved first by an acylase to give the γ-glutamide and finally by γ-GT. Both enzymes are concentrated in the kidneys. Using two enzymatic cleavage steps to generate the potent drug resulted in greater specificity for the kidney than a single step dependent on γ-GT. Later work has extended use of γ-glutamyl amides of diuretics as kidney prodrugs [54].
Fig. 2.

Enzymatic transformations of xanthine prodrugs derived from XAC, 18.
XAC, like theophylline, is active as a diuretic and as a natriuretic [50] antagonizing the reduction of glomerular filtration rate induced by the adenosine analog, N6-cyclohexyladenosine (CHA). At a dose of 5mg/kg, administered in the tail vein in rats, XAC causes an increase in urine output that is comparable to that produced by theophylline. Furthermore, XAC is A1-selective (species dependent), and further manipulation of the receptor subtype selectivity is possible through the introduction of additional substituents apart from the amino group [1]. Thus, it would be desirable to achieve a selective concentration in the kidneys of a theophylline analog such as XAC, which is more potent and selective than theophylline. γ-Glutamyl-XAC, 17 (Fig. 2), and its derivatives were explored as prodrugs for cleavage in the kidneys by γ-glutamyltransferase. The basis for the expectation that γ-Glu-XAC would serve as a suitably masked prodrug (and therefore less active than the parent) is the observation that, in general, anionic xanthine derivatives, e.g. functionalized on the 8-position chain, and zwitterionic derivatives tend to be less potent as adenosine antagonists than cationic (principally amine) derivatives, such as XAC and D-Lys-XAC. This generalization was substantiated in the case of glutamyl derivatives of XAC. In binding assays [50], which for adenosine antagonists are often indicative of biological potency except in cases of insolubility, the anionic N-acetyl-γ-glutamyl-XAC (16) was much less potent than XAC. The zwitterionic γ-glutamyl-XAC was intermediate in potency as an adenosine antagonist (20-fold less potent than XAC in binding assays at rat A1-adenosine receptors). It was hoped that N-acetyl-γ-glutamyl-XAC would serve as a substrate for the two-step cleavage process. However, it was not certain that γ-glutamyl-XAC would be cleaved by γ-GT [55], although it seemed likely that N-acetyl-γ-glutamyl-XAC would be cleaved in the first step by renal acylase, known to have broad specificity. Surprisingly, N-acetyl-γ-glutamyl-XAC was not a substrate for renal acylase, and did not act as a diuretic in vivo. When N-acetyl-γ-glutamyl-XAC was injected in rats intravenously, no XAC was observed in urine or in plasma. γ-Glutamyl-XAC did act as a diuretic, and was cleaved to XAC both in vivo and in pure enzyme preparations. Since the two enzyme processes [53] could not be utilized, an additional derivative, which consisted of γ-glutamyl-γ-glutamyl-XAC (19), which would be acted upon by γ-GT in two steps, was also prepared, and found to be cleavable readily to XAC. γ-Glutamyl-γ-glutamyl-XAC was also active in vivo as a diuretic. To establish kidney selectivity of γ-glutamyl-XAC, it will be necessary to characterize the in vivo actions of this adenosine antagonist more fully, i.e. assessing for actions in the cardiovascular system. Since even the prodrug itself potentially has adenosine antagonist properties (albeit much weaker), it is possible that other biological effects will be observed. Another question which should be answered is the mechanism of entry into the cell in order to be acted upon by γ-GT, which occurs intracellularly.
Uptake blockers
The classical nucleoside transport inhibitor, dipyridamole, has been used clinically as a vasodilatory agent and to characterize the properties of adenosine uptake in a variety of mammalian tissues [3,56]. Selective inhibition of adenosine uptake would act in a manner similar to the administration of adenosine agonists although conceptually the effects of such agents would be localized to tissues where adenosine-related metabolic and paracrine activities were highest. Mioflazine, a nucleoside transport inhibitor from Janssen Pharmaceutica, has proven to be efficacious as a hypnotic in animals [51] and may have anticonvulsant activity. The structure–activity relationships of various transport inhibitors have been determined, and a newer analog R 75231 [57] (20, Fig. 3) which does not cross the blood brain barrier, shows potential for cardioprotection.
Fig. 3.

Structures of R 75231 (20), an analog of the adenosine uptake inhibitor, mioflazine; the binding enhancer PD 81,723 (21); and 5-amino-4-imidazole carboxamide (AICA) riboside (22).
Allosteric enhancers
An interesting strategy for potentially treating ischemic diseases and others that involve a “hyperproduction” of purine nucleosides is that of “allosteric” enhancers of adenosine binding [58]. As a result of a broad screening program at Warner-Lambert, a series of benzoylthiophene derivatives, including PD 81,723 (21, Fig. 3), were discovered to cause an increase in the binding of adenosine analogs selectively at A1-adenosine receptors. Moreover, the biological effects of adenosine agonists as inhibitors of cAMP production in FRTL-5 rat thyroid cells were potentiated. Such enhancers, predicated on the discovery of more potent pharmacophores, when used therapeutically, might be expected to have biological effects only in ischemic areas where the adenosine concentration was elevated, in a more selective fashion than adenosine agonists.
Adenosine potentiators
Another means of raising the extracellular levels of adenosine, so that adenosine receptors are activated without the use of synthetic agonists, is through metabolic manipulation via “potentiator” actions [59]. Pretreatment with 5-amino-4-imidazole carboxamide (AICA) riboside (22, Fig. 3) [60] causes an enhanced increase in the local adenosine concentration under conditions of myocardial ischemia in dogs. Increased blood flow, inhibition of granulocyte adherence, and less tissue injury were observed. One mechanism proposed for the cardiac protective effects of AICA riboside is an increased production of adenosine rather than inosine from ATP catabolism although the precise mechanism for this effect has yet to be determined [61].
ATP receptors as drug targets
While ATP represents a major potential source for adenosine, the nucleotide, together with ADP and AMP, can alter tissue function via specific cell surface recognition sites termed P2 (ATP) receptors, which are distinct from P1 (adenosine) receptors, except in the common structural features and the metabolic relationship of the endogenous ligands [62–64].
The concept of a role for ATP and related phosphorylated adenine nucleotides as mediators of cell-to-cell communication has a long and distinguished history via the efforts of Burnstock and his many coworkers over the past two decades [62]. ATP appears to be the main candidate as effector agent for the “non-cholinergic, non adrenergic” (NANC) neurotransmission in peripheral tissues. Four major types of P2 receptor have been identified [63].
The P2t receptor is selectively activated by 2-methylthio-ADP and is localized on platelets. It is an ADP receptor. The P2x and P2y receptors [62] are ATP receptors with α,β-methylene-ATP and 2-methylthio-ATP being selective ligands, respectively, for each receptor. The P2z receptor found in mast cells [63] is sensitive to ATP4−.
Activation of P2x receptors causes contraction of arteries, bladder and vas deferens, whereas P2y receptor activation relaxes taenia coli, trachea and vasculature, the latter effect involving nitric oxide as effector agent. Antagonists for the various P2 receptor subclasses have proven difficult to identify but β,γ-dichloromethylene-ATP is a relative potent (Ki = 21 μM) competitive antagonist [65]. The list of tisues [66] in which ATP receptors have been identified is growing rapidly, as has the availability of ATP analogs by which to study these receptors [64].
Conclusion
Commercial interest in the therapeutic potential of purinergic ligands as drug entities has continued notwithstanding serious difficulties encountered by a number of large pharmaceutical companies. Attention is now turning towards non-classical approaches to purine therapeutics such as prodrugs, allosteric binding enhancement, and modulating metabolic and uptake processes as a means of avoiding the side-effects of classical agonists and antagonists. Also gaining importance as therapeutic targets for adenosine-related drugs are acute disorders, such as stroke, in which the lack of specificity for currently available adenosine pharmacophores may be acceptable in their acute use of the context of life-threatening situations. Classical competitive pharmacophores, however, should not be overlooked given recent discoveries of novel adenosine pharmacophores from natural sources [67–69].
Footnotes
Macallum GE, Walker RM, Barsoum NJ and Smith GS, unpublished observations. Cited with permission.
Trivedi BK, Bristol JA, Blankley CJ, Hamilton JW, Patt WC, Kramer WJ, Bruns RF, Cohen DM and Ryan MJ, Synthesis of prodrugs of CI-936, an adenosine receptor agonist for CNS disorders. Abstract presented to the Division of Medicinal Chemistry at the meeting of the Third Chemical Congress of North America in Toronto, 1988, Abstr. 32.
Hamilton HW, Hawkins LD, Patt WC, Johnson SA, Trivedi BK, Heffner TG, Wiley JN and Bruns RF, Abstract presented to the Division of Medicinal Chemistry at the 196th National ACS Meeting, 1988, Abstr. 80.
References
- 1.Jacobson KA. Adenosine (P1) and ATP (P2) receptors. In: Emmett JC, editor. Comprehensive Medicinal Chemistry. Vol. 3. Pergamon Press; New York: 1990. pp. 601–642. [Google Scholar]
- 2.Pearson JD. Ectonucleotidases. Measurement of activities and use of inhibitors. Methods Pharmacol. 1985;6:83–108. [Google Scholar]
- 3.Stone TW, Lloyd HGE, Newby AC. Adenosine release. In: Williams M, editor. Adenosine and Adenosine Receptors. Humana Press; Clifton, NJ: 1990. pp. 173–223. [Google Scholar]
- 4.Drury A, Szent-Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol (Lond) 1929;68:213–237. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Honey RM, Ritchie WT, Thompson WAR. The action of adenosine upon the human heart. Q J Med. 1930;23:485–490. [Google Scholar]
- 6.Medco’s 1st approval—Adenocard. Scrip No. 1989;1464:22. [Google Scholar]
- 7.Williams M. Adenosine receptors as drug targets: Fulfilling the promise? In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 174–183. [Google Scholar]
- 8.Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiol Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- 9.Heffner TG, Wiley JN, Williams AE, Bruns RF, Cougenhour LJ, Downs DA. Comparison of the behavioral effects of adenosine agonists and dopamine antagonists in mice. Psychopharmacology. 1989;98:31–38. doi: 10.1007/BF00442002. [DOI] [PubMed] [Google Scholar]
- 10.Bridges AJ, Moos WH, Szotek DL, Trivedi BK, Bristol JA, Heffner TG, Bruns RF, Downs DA. N6-(2,2-Diphenylethyl)adenosine, a novel adenosine receptor agonist with antipsychotic-like activity. J Med Chem. 1987;30:1709–1711. doi: 10.1021/jm00393a003. [DOI] [PubMed] [Google Scholar]
- 11.Schingnitz G, Küfner-Mühl U, Ensinger H, Lehr E, Kuhn FJ. Selective A1-antagonists for treatment of cognitive deficits. Nucleosides Nucleotides. in press. [Google Scholar]
- 12.Marangos PJ, Von Lubitz D, Miller LP, Daval JL, Deckert J. The central adenosine system as a therapeutic target in stroke. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 100–107. [Google Scholar]
- 13.Belardinelli L, Pelleg A. Rationale for the use of adenosine in the diagnosis and treatment of cardiac arrhythmias. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 95–99. [Google Scholar]
- 14.Churchill PC, Bidani AK. Adenosine and renal function. In: Williams M, editor. Adenosine and Adenosine Receptors. Humana Press; Clifton, NJ: 1990. pp. 335–380. [Google Scholar]
- 15.Gerber JG, Payne NA. The role of adenosine on the gastric acid secretory response. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 213–219. [Google Scholar]
- 16.Howell RE, Gaudette RF, Raynor MC, Warner AT, Museham WT, Schenden JA, Perumattam J, Hiner RN, Kinnier WJ. Mechanisms of xanthine actions in models of asthma. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 370–375. [Google Scholar]
- 17.Daly JW. Adenosine receptors: Targets for future drugs. J Med Chem. 1982;25:197–207. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]
- 18.Stein HH, Prasad RN, Tietje KR. Adenosine-5′-carboxamides for controlling undesired animals. 4,167,565. US Patent. 1979
- 19.Kawazoe K, Matsumato M, Tanabe S, Fujiwara M, Yanagimoto M, Hirata M, Kakiuchi K. Coronary and cardiohemodynamic effects of 2-phenyl-aminoadenosine (CV1808) in anaesthetized dogs and cats. Arzneimittelforschung. 1980;30:1083–1087. [PubMed] [Google Scholar]
- 20.Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- 21.Bridges AJ, Bruns RF, Ortwine DF, Priebe SR, Szotek DL, Trivedi BK. N6-[2-(3,5-Dimethoxyphenyl)-2-(2-methylphenyl)ethyl]adenosine and its uronamide derivatives. Novel adenosine agonists with both high affinity and high selectivity for the adenosine A2 receptor. J Med Chem. 1988;31:1282–1285. doi: 10.1021/jm00402a004. [DOI] [PubMed] [Google Scholar]
- 22.Trivedi BK, Bruns RF. C2, N6-Disubstituted adenosines: Synthesis and structure–activity relationships. J Med Chem. 1989;32:1667–1673. doi: 10.1021/jm00128a002. [DOI] [PubMed] [Google Scholar]
- 23.Trivedi BK, Bridges AJ, Patt WC, Priebe SR, Bruns RF. N6-Bicycloalkyladenosines with unusually high potency and selectivity for the adenosine A1 receptor. J Med Chem. 1989;32:8–11. doi: 10.1021/jm00121a002. [DOI] [PubMed] [Google Scholar]
- 24.Hutchison AJ, Williams M, deJesus R, Oei HH, Ghai GR, Webb RL, Zoganas HC, Stone GA, Jarvis MF. 2-Arylalkylamino-adenosine 5′-uronamides: A new class of highly selective adenosine A2 receptor agonists. J Med Chem. 1990;33:1919–1923. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]
- 25.Olsson RA, Kusachi S, Thompson RD, Ukena D, Padgett W, Daly JW. N6-Substituted N-alkyladenosine-5′-uronamides: Bifunctional ligands having recognition groups for A1 and A2 adenosine receptors. J Med Chem. 1986;29:1683–1689. doi: 10.1021/jm00159a020. [DOI] [PubMed] [Google Scholar]
- 26.Klabunde RE. Dipyridamole inhibition of adenosine metabolism. Eur J Pharmacol. 1983;93:21–26. doi: 10.1016/0014-2999(83)90026-2. [DOI] [PubMed] [Google Scholar]
- 27.Sollevi A. Cardiovascular effects of adenosine in man: Possible clinical implications. Prog Neurobiol. 1986;27:319–349. doi: 10.1016/0301-0082(86)90005-5. [DOI] [PubMed] [Google Scholar]
- 28.Fredholm BB, Jacobson KA, Jonzon B, Kirk KL, Li YO, Daly JW. Evidence that a novel 8-phenyl-substituted xanthine derivative is a cardioselective adenosine receptor antagonist in vivo. J Cardiovasc Pharmacol. 1987;9:396–400. doi: 10.1097/00005344-198704000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor DG, Bruns RF, Bjork FA, Cohen DM, Singer RM, Olszewski BJ, Ryan MJ, Trivedi BK, Kaplan HR. Antihypertensive profile of an adenosine analog PD 117,519. FASEB J. 1988;2:1799. [Google Scholar]
- 30.Griffin HE, Metz AL. Cardiac arrhythmias and coronary arteriopathy induced by an adenosine agonist-antihypertensive in beagle dogs. J Mol Cell Cardiol. 1989;21(Suppl II):S.45. [Google Scholar]
- 31.Snyder SH, Katims JJ, Annau Z, Bruns RF, Daly JW. Adenosine receptors and the actions of methylxanthines. Proc Natl Acad Sci USA. 1981;78:3260–3264. doi: 10.1073/pnas.78.5.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daly JW, Hong O, Padgett WL, Shamim MT, Jacobson KA, Ukena D. Non-xanthine heterocycles: Activity as antagonists of A1- and A2-adenosine receptors. Biochem Pharmacol. 1988;37:655–664. doi: 10.1016/0006-2952(88)90139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Francis JE, Cash WD, Psychoyos S, Ghai G, Wenk P, Friedmann RC, Atkins C, Warren V, Furness P, Hyun JL, Stone GA, Desai M, Williams M. Structure activity profile of a series of novel triazoloquinazoline adenosine agonists. J Med Chem. 1988;31:1014–1020. doi: 10.1021/jm00400a022. [DOI] [PubMed] [Google Scholar]
- 34.Sarges R, Howard HR, Browne RG, Koe BK. 4-Amino -[1,2,4]triazolo[4,3-a]quinoxalines — Highly selective adenosine and potential antidepressants. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 417–418. [Google Scholar]
- 35.Spielman WS, Thompson CI. A proposed role for adenosine in the regulation of renal hemodynamics and renin release. Am J Physiol. 1982;242:F423–F435. doi: 10.1152/ajprenal.1982.242.5.F423. [DOI] [PubMed] [Google Scholar]
- 36.Tagawa H, Vander AJ. Effects of adenosine compounds on renal function and renin secretion in dogs. Circ Res. 1970;26:327–338. doi: 10.1161/01.res.26.3.327. [DOI] [PubMed] [Google Scholar]
- 37.Hedqvist P, Fredholm BB, Olundh S. Antagonistic effects of theophylline and adenosine on adrenergic neuroeffector transmission in the rabbit kidney. Circ Res. 1978;43:592–598. doi: 10.1161/01.res.43.4.592. [DOI] [PubMed] [Google Scholar]
- 38.Murray RD, Churchill PC. The concentration-dependency of the renal vascular and renin secretory responses to adenosine receptor agonists. J Pharmacol Exp Ther. 1985;232:189–193. [PubMed] [Google Scholar]
- 39.Osswald H, Schmitz H-J, Kemper R. Tissue content of adenosine, inosine and hypoxanthine in the rat kidney after ischemia and postischemic recirculation. Pflugers Arch. 1977;371:45–49. doi: 10.1007/BF00580771. [DOI] [PubMed] [Google Scholar]
- 40.Osswald H, Nabakowski G, Hermes H. Adenosine as a possible mediator of metabolic control of glomerular filtration rate. Int J Biochem. 1980;12:263–267. doi: 10.1016/0020-711x(80)90082-8. [DOI] [PubMed] [Google Scholar]
- 41.Osborn JE, Hoversten LG, DiBona GF. Impaired blood flow autoregulation in nonfiltering kidneys: Effects of theophylline administration. Proc Soc Exp Biol Med. 1983;174:328–335. doi: 10.3181/00379727-174-41744. [DOI] [PubMed] [Google Scholar]
- 42.Davis JO, Shock NW. The effect of theophylline ethylene diamine on renal function in control subjects and in patients with congestive heart failure. J Clin Invest. 1949;28:1459–1468. doi: 10.1172/JCI102211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowmer CJ, Collis MG, Yates MS. Effect of the adenosine antagonist 8-phenyltheophylline on glycerol-induced acute renal failure in the rat. Br J Pharmacol. 1986;88:205–212. doi: 10.1111/j.1476-5381.1986.tb09488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellermann J, Grunder W, Keller T. Effect of pentoxifylline on the ischemic rat kidney monitored by 31P NMR spectroscopy in vivo. Biomed Biochim Acta. 1988;47:515–521. [PubMed] [Google Scholar]
- 45.Gouyon J-B, Guignard J-P. Functional renal insufficiency: Role of adenosine. Biol Neonate. 1988;53:237–242. doi: 10.1159/000242796. [DOI] [PubMed] [Google Scholar]
- 46.Rossi N, Ellis V, Kontry T, Gunther S, Churchill P, Bidani A. The role of adenosine in HgCl2-induced acute renal failure in rats. Am J Physiol. 1989;258:F1554–F1560. doi: 10.1152/ajprenal.1990.258.6.F1554. [DOI] [PubMed] [Google Scholar]
- 47.Lang MA, Preston AS, Handler JS, Forrest JN., Jr Adenosine stimulates sodium transport in kidney A6 epithelia in culture. Am J Physiol. 1985;249:C330–C336. doi: 10.1152/ajpcell.1985.249.3.C330. [DOI] [PubMed] [Google Scholar]
- 48.Spielman WS, Arend LJ, Klotz K-N, Schwabe U. Adenosine receptors and signaling in the kidneys. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 220–225. [Google Scholar]
- 49.Law WR, McLane MP, Raymond RM. Adenosine is required for myocardial insulin responsiveness in vivo. Diabetes. 1988;37:842–845. doi: 10.2337/diab.37.6.842. [DOI] [PubMed] [Google Scholar]
- 50.Barone S, Churchill PC, Jacobson KA. Adenosine receptor prodrugs: Towards kidney-selective dial-kylxanthines. J Pharmacol Exp Ther. 1989;250:79–85. [PMC free article] [PubMed] [Google Scholar]
- 51.Wauquier A, Van Belle H, Van der Broeck WAE, Janssen PAJ. Sleep improvement in dogs after oral administration in mioflazine, a nucleoside transport inhibitor. Psychopharmacology. 1987;91:434–440. doi: 10.1007/BF00216007. [DOI] [PubMed] [Google Scholar]
- 52.Bundgaard H. Design of prodrugs: Bioreversible derivatives for various functional groups and chemical entities. In: Bundgaard H, editor. Design of Prodrugs. Elsevier; Amsterdam: 1985. pp. 1–92. [Google Scholar]
- 53.Orlowski M, Mizoguchi H, Wilk S. N-Acyl-γ-glutamyl derivatives of sulfamethoxazole as models of kidney-selective prodrugs. J Prodrug Exp Ther. 1980;212:167–172. [PubMed] [Google Scholar]
- 54.Hoffbauer KG, Sonnenburg C, Stalder R, Criscione L, Kraetz J, Fuhrer W, Habicht E. GP 22979A, a renal vasodilator with natriuretic properties. J Pharmacol Exp Ther. 1985;232:838–844. [PubMed] [Google Scholar]
- 55.Magnan SDJ, Shirota FN, Nagasawa HT. Drug latentiation by γ-glutamyl transpeptidase. J Med Chem. 1982;25:1018–1021. doi: 10.1021/jm00351a003. [DOI] [PubMed] [Google Scholar]
- 56.Marangos PJ, Deckert J, Bisserbe J-C. Central sites of adenosine action and their interaction with various drugs. In: Gerlach E, Becker BF, editors. Topics and Perspectives in Adenosine Research. Springer; Berlin: 1987. pp. 74–88. [Google Scholar]
- 57.van Belle H. Comparative pharmacology of nucleoside transport inhibitors. Nucleosides Nucleotides. in press. [Google Scholar]
- 58.Bruns RF, Fergus JH. Allosteric enhancers of adenosine A1 receptor binding and function. In: Ribeiro JA, editor. Adenosine Receptors in the Nervous System. Taylor & Francis; London: 1989. pp. 53–60. [Google Scholar]
- 59.Engler R. Consequences of activation and adenosine-mediated inhibition of granulocytes during myocardial ischemica. Fed Proc. 1987;46:2407. [PubMed] [Google Scholar]
- 60.Gruber HA, Hoffer ME, McAllister DR, Laikind PK, Lane TA, Schmid-Schoenbein GW, Engler RL. Increased adenosine concentration in blood from ischemic myocardium by AICA riboside. Effects on flow, granulocytes, and injury. Circulation. 1989;80:1400–1411. doi: 10.1161/01.cir.80.5.1400. [DOI] [PubMed] [Google Scholar]
- 61.Mullane KM, Williams M. Adenosine and cardiovascular function. In: Williams W, editor. Adenosine and Adenosine Receptors. Humana Press; Clifton, NJ: 1990. pp. 289–319. [Google Scholar]
- 62.Burnstock G, Kennedy C. Is there a basis for distinguishing two types of P2-purinoceptors? Gen Pharmacol. 1985;16:443. doi: 10.1016/0306-3623(85)90001-1. [DOI] [PubMed] [Google Scholar]
- 63.Gordon JL. Extracellular ATP: Effects, sources, and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams M, Cusack NJ. Neuromodulatory roles of purine nucleosides and nucleotides: Their receptors and ligands. Neurotransmissions. 1990;6:1–6. [Google Scholar]
- 65.Krishtal OA, Marchenko SM, Obukhov AG, Volkova TM. Receptors for rat sensory neurons: The structure–function relationships for ligands. Br J Pharmacol. 1988;95:1057–1062. doi: 10.1111/j.1476-5381.1988.tb11739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burnstock G. Classification and characterization of purinoceptors. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling: Targets for New Drugs. Springer; New York: 1990. pp. 241–253. [Google Scholar]
- 67.Davies LP, Jamieson DD, Baird-Cambert JA, Kazlauskas R. Halogenated pyrrolopyrimidine analogues of adenosine from marine organisms: Pharmacological activities and potent inhibition of adenosine kinase. Biochem Pharmacol. 1984;33:347–355. doi: 10.1016/0006-2952(84)90225-9. [DOI] [PubMed] [Google Scholar]
- 68.Balduini W, Cattabeni F. Displacement of [3H]N6-cyclohexyladenosine binding to rat cerebral cortical membranes by a hydroalcoholic extract of Valeriana officinalis. Med Sci Res. 1989;17:639–640. [Google Scholar]
- 69.Araki H, Karasawa Y, Kawashima K, Hayashi M, Aihara H, Jun-Hua H. The adenosine analogue and cerebral protecting agent AMG-1 has no effect on delayed neuronal death following ischemia. Methods Find Exp Clin Pharmacol. 1989;11:731–736. [PubMed] [Google Scholar]