

Abstract
Medicinal plants have long been an excellent source of pharmaceutical agents. Accordingly, the long term objectives of the author's research program are to discover and design new chemotherapeutic agents based on plant-derived compound leads by using a medicinal chemistry approach, which is a combination of chemistry and biology. Different examples of promising bioactive natural products and their synthetic analogs, including sesquiterpene lactones, quassinoids, naphthoquinones, phenylquinolones, dithiophenediones, neo-tanshinlactone, tylophorine, suksdorfin, DCK, and DCP, will be presented with respect to their discovery and preclinical development as potential clinical trial candidates. Research approaches include bioactivity- or mechanism of action-directed isolation and characterization of active compounds, rational drug design-based modification and analog synthesis, as well as structure-activity relationship and mechanism of action studies. Current clinical trials agents discovered by the Natural Products Research Laboratories, University of North Carolina, include bevirimat (dimethyl succinyl betulinic acid), which is now in Phase IIb trials for treating AIDS. Bevirimat is also the first in a new class of HIV drug candidates called “maturation inhibitors”. In addition, an etoposide analog, GL-331, progressed to anticancer Phase II clinical trials, and the curcumin analog JC-9 is in Phase II clinical trials for treating acne and in development for trials against prostate cancer. The discovery and development of these clinical trials candidates will also be discussed.
Introduction
In the Natural Products Research Laboratories (NRPL), our objectives are to discover and develop bioactive natural products and their analogs as clinical trials candidates. The three approaches used to achieve these objectives are (1) bioactivity- or mechanism of action-directed isolation and characterization of active compounds, (2) rational drug design-based modification and analog synthesis, and (3) mechanism of action (MOA) studies. The scientific disciplines covered include natural products chemistry, molecular biology and biochemistry, and pharmacology, to discover promising new leads based on bioactivity- or mechanism of action-directed approaches; medicinal chemistry and synthetic organic chemistry to achieve new leads optimization based on modern medicinal chemistry approaches; and analytical chemistry to apply state-of-the-art analytical instrumental chromatography technologies to support the above two tasks. MOA and in vivo evaluation studies are supported by collaborations with more than 60 active established researchers worldwide to enhance the programs of the NPRL. Current research programs in the NPRL include the investigation of (1) novel plant cytotoxic antitumor and anti-HIV principles and synthetic analogs as antitumor and anti-AIDS agents and (2) other chemotherapeutic agents, such as antimalarial, antifungal, antiviral and anti-inflammatory agents, as well as (3) traditional Chinese medicines (TCM), targeting to their active principles, fractions and prescriptions.
General Concepts on Drug Discovery and Development
Drug discovery can build on several sources; however, my laboratories focus on bioactivity-directed isolation and characterization of lead natural product principles from single medicinal herbs and formulations. As shown in Figure 1, the subsequent preclinical optimization of a lead compound is an cyclical process of obtaining bioassay screening results, analyzing activity data, designing new target compounds, and synthesizing new analogs.1 In this iterative process, I feel that chemistry and biology are complementary and co-dependent areas of science, similar to the Chinese concepts of Yin and Yang – one is not present or sufficient without the other (Figure 2). The discovery of new bioactive compounds depends on valid biological assays, while new chemistry can make the discovery of new biological targets possible. I feel that medicinal chemistry combines techniques from chemistry and from biology to facilitate new drug discovery. By using these concepts and techniques, my NPRL has been able to discover more than 3,000 bioactive natural products and their synthetic derivatives/analogs since 1971, as briefly summarized below.
Figure 1.

Flowchart for drug discovery and development of natural products-derived chemotherapeutic agents. Qian, K.; Nitz, T. J.; Yu, D.; Allaway, G. P.; Morris-Natschke, S. L.; Lee, K. H. In Natural Product Chemistry for Drug Discovery; Buss, A. D., Butler, M. S., Eds., RSC Publishing: Cambridge, UK, 2010; p 376. Reproduced by permission of The Royal Society of Chemistry.
Figure 2.

Complementarity of chemistry and biology: medicinal chemistry is an art of combining chemistry and biology for drug discovery
Antitumor Agents
Sesquiterpene Lactones
Helanalin and Its Analogs. In the mid-1970s, three sesquiterpene lactones, molephantin (1),2 molephantinin (2),3 and phantomolin (3) (Figure 3)4 were discovered as new cytotoxic principles from Elephantopus mollis. The latter two compounds showed similar T/C values (397% and 378%, respectively) against Walker 256 carcinoma (W-256) in rats (dose = 2.5 mg/kg). Microlenin (4), a structurally related dimeric sesquiterpene lactone from Helenium microcephalum, had a T/C value of 173% against W-256 (dose = 2.5 mg/kg),5 while its monomer, helenalin (5), had a T/C value of 316% under the same conditions (Figure 4). Helenalin contains both an α-methylene-γ-lactone moiety, and an α,β-unsaturated ketone; therefore, studies were performed to determine the relative contributions of the two O=C-C=CH2 systems. The rank order of potency against Hep-2 human epidermoid laryngeal carcinoma was 5 (ED50 0.08 μg/mL) > 11,13-dihydrohelenalin (6) (ED50 0.80 μg/mL) > 2,3-dihydrohelenalin (7) (ED50 3.84 μg/mL) > 2,3,11,13-tetrahydrohelenalin (8) (inactive) (Figure 5).6 Thus, reduction of the double bond of the α,β-unsaturated ketone had a greater effect on potency than reduction of the α-methylene-γ-lactone. In mechanistic studies, the cytotoxicity of helenalin and structurally related sequiterpene lactones was linked to a Michael-type addition reaction of the O=C-C=CH2 systems in the molecule with sulfhydryl groups of reduced glutathione and l-cysteine.7,8 Finally, bis(helenalinyl) esters (two helenalin molecules connected through their hydroxy group by a diester linkage) were generally more potent and less toxic than the parent alcohol.9 At 8 mg/kg, 5 had a T/C of 162% against P388 leukemia in mice, while bis(helenalinyl)glutarate (9, Figure 6) had a T/C of 195%.9
Figure 3.

Structures of cytotoxic natural sesquiterpene lactones from Elephantopus mollis
Figure 4.

Structures of cytotoxic natural pseudoguaianolides from Helenium microcephalum
Figure 5.

Rank order of cytotoxic potency of helenalin analogs with varying degrees of molecular unsaturation
Figure 6.

Structure of bis(helenalinyl)glutarate
Quassinoids: Brusatol and Its Analogs
The fruits of Brucea javanica (Chinese medicine “Ya-Tan-Tzu”) yielded many new quassinoids, including several compounds with significant cytotoxicity against various cancers, such as bruceosides A-F (10–15) and brusatol (16) (Figure 7).10-12 Bruceantin (17), which has a terminal isopropyl rather than methyl group in the C-15 ester side chain compared with 16 (Figure 7), was previously isolated from B. antidysenterica by Kupchan et al. by bioactivity-guided fractionation.13 Our laboratories first reported two synthetic methods for the conversion of 10 into 17, which was in anticancer clinical trials.14 Connecting two molecules of 16 or 17 at the C-3 hydroxy group through malonate, glutarate, adipate, and sebacate esters gave bis-esters (18–23, Figure 8) that were as active or more active than the parent alcohols against P-388 leukemia.15 In addition to C-3 esterification, other structural features essential for enhanced cytotoxic activity include free hydroxy groups at C-11 and -12, an enone double bond in ring A, and an unsaturated ester at C-15.16,17 The identity of the C-15 ester side chain can significantly affect cytotoxicity, and oxidation of the C-15 side chain has been postulated to cause deactivation of 16- or 17-related quassinoids. Therefore, trifluoromethyl groups were incorporated into the side chain at this position, as well as in the C-3 ester side chain. The most potent analog was 15-[3'-trifluoromethyl)-butanoyl]-bruceolide (24, Figure 8), which had similar potency and log GI50 values (-7.0 – -8.7) compared with 17 against a human cancer cell line panel.18
Figure 7.

Structures of cytotoxic natural quassinoids from Brucea species
Figure 8.

Structures of cytotoxic synthetic quassinoids
Phenylquinolones and Naphthyridinones: NSC-656158 and Its Analogs
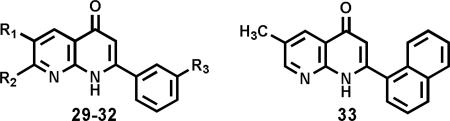
The natural flavonoids, tricin (25) and kaempferol-3-O-β-d-glucopyranoside (26), and the lignan (+)-nortrachelogenin (27) were isolated by bioactivity-guided fractionation as antileukemic principles from Wikstroemia indica (Figure 9).19 2-Phenyl-4-quinolones are structurally related compounds with nitrogen rather than oxygen in the heterocyclic ring. Synthetic modification led to several fluorinated 2-phenyl-4-quionolones as potent antimitotic antitumor agents. NSC-656158 (28, Figure 10) showed potent cytotoxicity against a human tumor cell line (HTCL) panel (average log GI50 -6.47), inhibited tubulin polymerization (IC50 0.85 μM), and exhibited good in vivo antitumor activity (130% prolonged life span of mice bearing OVCAR-3 xenografts).20 An analog (29, Figure 10) substituted with a pyrollidine ring rather than methylenedioxy group was even more potent with an average in vitro log GI50 of -7.65 and tubulin inhibitory IC50 of 0.46 μM.20 Mechanistically, 28 inhibits hepatocyte growth factor-induced invasion of SK-Hep-1 human hepatocellular carcinoma cells by suppressing matrix metalloproteinase-9 expression at the micromolar range. Therefore, 28 is a potential therapeutic agent against tumor invasion.21 2-Aryl-1,8-naphthyridin-4(1H)-ones were also synthesized as antimitotic antitumor agents. 3'-Methoxy and halogenated-2-phenyl compounds (29–32), as well as 2-(α-naphthyl) (33) but not 2-(β-naphthyl) compounds, were highly active in both antitumor and antitubulin assays, with average log GI50 values comparable to those of positive controls colchicine and podophyllotoxin (Table 1).22 The identities of the halogens on the 2-phenyl ring and of substituents on the pyridine ring of the naphythyridinone system also influenced the tumor cell line selectivity at the total growth inhibition level.22
Figure 9.

Structures of antileukemic natural flavonoids and lignan from Wikstroemia indica
Figure 10.

Structures of cytotoxic synthetic fluorinated 2-phenyl-4-quinolones
Table 1.
Antimitotic and Cytotoxic Activity of Synthetic Naphthyridinones
 | ||||||
|---|---|---|---|---|---|---|
| cmpd | R1 | R2 | R3 | ITPa IC50 (μM) | ICBb % inhibition | cytotoxicity logGI50 |
|
29
|
H |
Me |
OMe |
0.75 |
29 |
-7.24 |
|
30
|
Me |
H |
F |
0.63 |
43 |
-7.30 |
|
31
|
H |
Me |
F |
0.53 |
41 |
-7.37 |
|
32
|
Me |
H |
Cl |
0.72 |
33 |
-6.57 |
|
33
|
See structure above |
0.55 |
46 |
-7.72 |
||
| positive control |
|
|
|
|
|
|
| colchicine |
|
|
|
0.80 |
-- |
-7.24 |
| podophyllotoxin | 0.46 | 76 | -7.54 | |||
ITP = inhibition of tubulin polymerization.
ICB = inhibition of colchicine binding
Naphthoquinones and Dithiophenedione Analogs
Psychorubrin (34, Figure 11) from Psychotria rubra is a cytotoxic natural product with a naphthoquinone skeleton. Related compounds with furanonaphthoquinone (35) and naphthothiophenedione (36–38) skeletons (Figure 11) also show potent cytotoxicity.23 Continued work led to a series of potent 2- and 3-methyl-4,8-dihydrobenzo[1,2-b:5.4-b']dithiophene-4,8-diones (39–46, Figure 12). Several compounds showed high cytotoxic activity against melanoma, non-small cell lung cancer, and breast cancer cell lines. 2-Hydroxymethyl-4,8-dihydrobenzo[1,2-b:5.4-b']dithiophene-4,8-dione (42) showed the highest activity against melanoma (mean log GI50 = -7.74) and the highest overall potency (mean log GI50 = -6.99) against the NCI HTCL panel.24 Dithiophene compounds were also found to significantly enhance retinoic acid-induced differentiation in leukemia cell lines. N-(2-Dimethylaminoethyl)-4,8-dihydrobenzo[1,2-b:5.4-b']dithiophene-4,8-dione-2-carboxamide (47, Figure 12) showed the greatest effect, inducing nearly complete differentiation at 0.02 μM.25
Figure 11.

Structures of cytotoxic psychorubrin and other quinone analogs
Figure 12.

Structures of cytotoxic dithiophenedione analogs
Thiocolchicone Analogs
Both colchicine (48) and thiocolchicine (49) are potent antimitotic agents, inhibiting tubulin polymerization with IC50 values of 1.5 and 0.65 μM, respectively.26 Thiocolchicone (50–54) derivatives, which have an oxygen moiety rather than a acetamido group at C-7, showed comparable or greater activity in tubulin polymerization, colchicine-binding, and cytotoxicity assays (Figure 13).27 Allocolchinoids (55–57), with a six-membered rather than seven-membered C-ring, also showed significant antitubulin effects and cytotoxicity, even against three drug-resistant KB cell lines compared with the parental KB cell line (Table 2).28
Figure 13.

Structures and antimitotic activity of colchicine, thiocolchicine, and thiocolchicone analogs
Table 2.
Cytotoxicity of Allocolchicinoids in Drug-sensitive and Drug-resistant KB Cell Lines
 | |||||
|---|---|---|---|---|---|
| cmpd | cytotoxicity EC50 (μg/mL) |
ITP IC50 (μM±SD) | |||
| KBa | KB-7db | KB-VCRc | KB-CPTd | ||
|
55
|
0.057 |
0.025 |
>0.063 (42-69) |
0.052 |
1.0±0.1 |
|
56
|
0.029 |
0.024 |
0.016 |
0.042 |
1.0±0.03 |
| 57 | 0.020 | 0.013 | 0.013 | 0.022 | 2.0±0.2 |
KB, epidermoid carcinoma of the nasopharynx.
KB-7d, KB cells with multidrug-resistant protein.
KB-VCR, KB cells with over-expression of P-glycoprotein.
KB-CPT, KB cells with reduced level of topoisomerase.
Epipodophyllotoxins: GL331 and Its Analogs
Podophyllotoxin (58) is a natural lignan found in Podophyllum peltatum (or Mayapple) and related species. It inhibits mitosis by preventing polymerization of microtubules into tubulin. In contrast, etoposide (59) and teniposide (60), two semi-synthetic glycosidic 4'-demethylepipodophyllotoxin derivatives (Figure 14),29 do not affect tubulin, but instead act by inhibiting DNA topoisomerase II (topo II).30 Although they are used clinically against various cancers, both compounds are poorly bioavailable, can cause myelosuppression, and suffer from drug resistance.30 As a possible solution, 4-alkyl-,31 benzyl-,32 and aryl-32 amino analogs of 4'-demethyl-epipodophyllotoxin were synthesized. Many compounds exhibited more potent DNA topo II inhibition and a greater percentage of protein-linked DNA breakage compared with etoposide.32 When compared with etoposide, several arylamino compounds (61–66) were as cytotoxic against KB cells and more active against three KB cell lines (KB1C, KB7D, KB50) showing resistance to etoposide (Table 3).32,33 GL331 [4'-O-demethyl-4β-(4”-nitroanilino)-4-deoxypodophyllotoxin] (63) emerged as the lead compound from these studies. Compound 63 (NSC-628679) was tested in Phase I anticancer clinical trials at M.D. Anderson Cancer Center, after favorable toxicological evaluation by Genelabs Technologies, Inc. (Redwood City, CA). It showed markedly favorable results in these trials, with primary indications against colon and small cell lung cancers, and progressed to Phase II clinical trials. Advantages of 63 over etoposide are easier manufacture, greater activity, particularly against drug-resistant cancer cell lines, and possibly a superior safety profile.34 A Comparative Field Analysis (CoMFA) computer model was generated using 102 epipodophyllotoxin, which showed that active compounds should have a positively charged functional group near the DNA minor groove.35 Subsequent new analogs are shown in Figure 15. Adding bulky tails at the para position of the 4'-aniline resulted in improved activity profiles. Analog 67 with a pyrollecarboxamidine moiety displayed increased cytotoxicity (ED50 = 0.04 and 0.2 μM) compared with etoposide (ED50 = 0.2 and 25 μM) against both KB and KB-7d cells.36 Similarly, compound 68 with an amino acid (benzyl L-alanyl-N-carbonyl) incorporated at this same position showed even better activity (ED50 KB = 0.5 μg/mL, KB-7d = 0.25 μg/mL) than 63 (ED50 = 0.2 μg/ml, 2 μg/mL) against drug resistant cell lines.37 Finally, the 4'-hydroxy group of 4β-para-substituted arylamino-epipodophyllotoxin analogs was esterified with N,N-dimethylglycine (69) to enhance drug resistance and water solubility simultaneously. Cytotoxicity and drug resistance profiles of most analogs were similar to those of 63, while esterification caused some decrease in protein-linked DNA breaks and, thus, perhaps interaction with DNA.38
Figure 14.

Structures of podophyllotoxin, etoposide, and teniposide
Table 3.
Cytotoxicity of Arylamino Analogs of Etoposide Against KB and Drug-resistant Sublines
 | |||||
|---|---|---|---|---|---|
| cmpd | R | IC50 (μM) |
|||
| KB | KB1C | KB7D | KB50 | ||
|
61
|
NH2 HCl |
0.59 |
3.5 |
7.6 |
2.0 |
|
62
|
CN |
0.61 |
2.7 |
5.0 |
4.0 |
|
63
|
NO2
|
0.49
|
6.1
|
7.7
|
3.0
|
|
64
|
F |
0.67 |
4.0 |
8.3 |
7.2 |
|
65
|
CO2CH3 |
0.84 |
2.5 |
7.0 |
3.3 |
|
66
|
-OCH2CH2O- |
0.68 |
0.5 |
1.0 |
1.6 |
| positive control |
|
|
|
|
|
| etoposide | 0.60 | 34.8 | 77.5 | 28.7 | |
Figure 15.

Structures of novel 4β-arylamino etoposide analogs
Curcumins: JC-9 (ASC-J9) and Its Analogs
The diarylheptanoid curcumin (70, Figure 16) is found in Curcuma longa and other related species. The rhizomes of these plants are used as both spices (turmeric) and medicines, particularly for hepatic disorders. Curcumin and other polyphenolic curcuminoids give turmeric its yellow color and stimulate bile secretion in the treatment of hepatitis. Curcumin itself shows moderate inhibitory activity against prostate cancer cell lines, and synthetic modifications yielded two lead compounds, JC-9 (71) and LL-80 (72), with increased activity against PC-3 (IC50 = 1.1 and 1.0 μM, respectively) and LNCaP (IC50 = 1.3 and 0.2 μM, respectively) cell lines.39,40 Compound 71 (also known as ASC-J9) was licensed by Androscience Corp. (San Diego, CA), and has succeeded in Phase II clinical trials against acne. Anti-prostate clinical trials with 71 are being planned. Mechanistically, 71 enhances androgen receptor degradation.41-43
Figure 16.

Structures of cytotoxic curcuminoids
Neo-tanshinlactone Analogs
The rhizomes of Salvia miltiorrhiza are well known in TCM as “Tanshen” and used mainly to treat coronary disorders, such an angina pectoris. Among its lipophilic bioactive constituents are the tanshinones, including tanshinone I (73) and IIA (74) (Figure 17). The water-soluble sulfonate (75) of the latter compound may act as a calcium antagonist and anti-calmodulin drug similar to the clinically used verapamil. Another related constituent of “Tanshen” is neo-tanshinlactone (76), which has a lactone rather than o-quinone ring-C (Figure 17).44 This compound showed selective cytotoxicity against two estrogen receptor-positive (ER+) breast cancer cell lines, MCF-7 and ZR-75-1, and was ten-fold more potent than tamoxifen.45 In initial SAR studies, 4-ethyl-neo-tanshinlactone (77) was even more potent (ED50 = 0.45 and 0.18 μg/mL) than neo-tanshinlactone against these two cell lines, and, in addition, potently inhibited (ED50 = 0.1 μg/mL) the SK-BR-3 breast cancer cell, which is estrogen receptor negative (ER-), but over-expresses HER2 (HER2+).46 In SAR analog studies, the aromatic rings A and D were found to be important for anti-breast cancer activity. In addition, certain ring C-opened analogs (80, 82) retained activity and had increased selectivity towards specific breast cancer subtypes (Table 4).47,48 The lead compound 76 was also tested in vivo against cancer cell xenografts in mice. At 10 mg/kg, it remarkably delayed tumor growth compared to control, and thus, showed significant and selective antitumor activity against the human ZR-75-1 breast ductal carcinoma xenograft.49
Figure 17.

Structures of cytotoxic tanshinones and neo-tanshinlactones
Table 4.
Cytotoxicity of Neo-tanshinlactone Analogs Against Four Breast Cancer Cell Lines (in μg/mL)
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| cmpd | type | R | R′ | R″ | MCF-7 (ER+) | SK-BR-3 (HER2+) | ZR-75-1 (ER+,HER2+) | MDA MB-231 (ER-) |
| 77 | A | Et | -- | -- | 0.2 | 0.1 | 0.1 | >10 |
| 78 | A | Pr | -- | -- | 1.2 | 0.1 | 0.3 | >10 |
| 79 | A | OMe | -- | -- | 2.3 | 0.2 | 0.1 | 6.4 |
| 80 | B | Et | H | H | 3.3 | 1.0 | 0.3 | >10 |
| 81 | B | Et | Me | Me | 2.5 | 1.2 | 1.3 | 2.3 |
| 82 | B | OMe | H | H | >20 | 3.5 | 0.6 | >10 |
Phenanthrene-Based Tylophorines: PBT-1 and Its Analogs
Tylophorine (83), antofine (84) and other phenanthroindolizidine alkaloids found in the genus Tylophora are collectively known as Tylophora alkaloids (Figure 18). Several such compounds have shown activity against various cancer cell lines, including refractory cancers. One of these compounds, tylocrebine (85), reached anticancer clinical trials, but failed due to central nervous system (CNS) toxicity.50 Structurally simplified phenanthrene-based tylophorine (PBT) analogs (86–89, Figure 18), which lack the indolizidine ring system found in the natural product leads, were synthesized through an efficient five-step route.51 The new compounds have a core phenanthrene substituted with an aminosubstituted methylene at the 9-position. The amine group could be an alkylamino, pyrrole, piperidine, or piperazine with a terminal carboxy or preferably a hydroxy group.52,53 N-(2,3-Methylenedioxy-6-methoxy-phenanthr-9-yl-methyl)-l-piperidinemethanol (86, PBT-1) emerged as the lead compound from SAR studies. It showed good cytotoxic activity against various cancer cell types with IC50 ranging from 0.04–0.07 μM. Its hydrochloride salt (87) also showed moderate in vivo activity against A549 lung cancer xenografts in mice, without CNS toxicity.52 Interestingly, mechanistic studies showed that natural Tylophora alkaloids and the structurally related synthetic PBT analogs do not share the same mechanism of action (Figure 19).53 Additional studies showed that 86 induced cell cycle arrest at the G2/M phase, accompanied by accumulation of cyclin B1 and activation of the MAPK signaling pathway, similar to paclitaxel.54 In addition, 86 induced apoptosis by inactivating Akt and inhibiting the NF-κB pathway.54 Lead PBT compound 86 or a new analog will likely be a good candidate for anticancer clinical trials.
Figure 18.

Structures of cytotoxic natural Tylophora alkaloids and synthetic PBT analogs
Figure 19.

Mechanistic comparison of Tylophora alkaloids
Conjugated Paclitaxel Analogs
Figure 20 shows examples of various conjugated paclitaxel analogs. First, a taxoid (either paclitaxel or cephalomannine) was conjugated at its 2'- and/or 7-position through an imine linkage to a 4β-amino-4'-O-demethylepipodophyllotoxin derivative.55 Several compounds (e.g., 90) showed comparable or better cytotoxic activity than the unconjugated epipodophyllotoxin, and showed enhanced activity against paclitaxel-resistant cancer cell lines.55
Figure 20.

Structures of conjugated paclitaxel analogs
Similarly, paclitaxel was also conjugated with a camptothecin derivative.56 Compared with camptothecin, all of the new conjugates (e.g., 91–93) showed higher cytotoxicity, but lower inhibition of topo I. Against HCT-8 cancer cells, the conjugates were also more active than paclitaxel, suggesting a different spectrum of activity, and thus, possibly a novel mechanism of action.56
Several different dietary antioxidants (including vitamin E, curcumin, dehydrozingerone, 4-methylumbelliferone, and others) were conjugated to the 2'-hydroxy group in the paclitaxel side chain through an ester linkage.57 Many of the compounds showed selective cytotoxicity against certain cancer cell lines, particularly the 1A9 and KB cell lines. The paclitaxel-vitamin E conjugate (94) with a glycine ester salt at the C-7 OH of paclitaxel exhibited notable inhibition against pancreatic cancer (Panc-1) cells with a lesser effect on the normal (E6E7) cell line, and is a good candidate for further studies.57
Chromenones: Desmosdumotin and Protoapigenone Analogs
Desmosdumotins B (95) and C (96) are bioactive flavanoids from in Desmos dumosus. A short efficient route to both compound types was established (Scheme 1). Among 96-type chalcones, a 4-bromo-3',3',5'-tripropyl analog (97) was the most potent with EC50 values of 0.9–2.3 μg/mL against seven different cancer cell lines, compared with 3.0–11.1 μg/mL for desmosdumotin C.58 Among 95-type flavones, the naphthyl substituted 98 showed potent cytotoxicity against all six cancer cell lines tested, with EC50 values of 0.2–0.6 μg/mL. 6,8,8-Triethyl substituted compounds (e.g., 99), particularly when coupled with 4'-methyl or -ethyl substituents on the pendant phenyl ring, showed notable cytotoxicity (EC50 0.03 and 0.025 μg/mL, respectively) and selectivity against vincristine-resistant KB cell lines (KB/KB-vin >460 and 320, respectively).59 Protoapigenone (100) is structurally related to 95, but has an unusual non-aromatic B-ring with a hydroxy group on the C-1' position. The first total synthesis of this compound also allowed for preparation of modified analogs (Scheme 2). Initial SAR study showed that changing the C-5 and C-7 A-ring hydroxy groups to methoxy groups (e.g., 101) or changing the phenyl A-ring to a naphthyl ring system (102) increased the cytotoxicity when compared to 100. Analogs 101 and 102 had comparable potency to that of doxorubicin against liver and breast cancer cell lines, respectively.60
Scheme 1.

Synthesis of desmosdumotins B and C and analogs Reagents: a) NaOMe, RI; b) TMSCHHN2; c) ArCHO, KOH; d) I2, DMSO then BBr3
Scheme 2.

Synthesis of protoapigenone and analogs Reagents: a) MOMCl, K2CO3; b) KOH, 4-OBn-PhCHO; c) I2, DMSO or Py; d) 10% Pd/CC, H2; e) TAIB, CH3CN/H2O for R3=OH or MeOH for R3=OMe; f) 15% HCl/i-PrOH
Kalanchosides
Three new bufadienolides (kalanchosides A–C, 103–105) were isolated from the medicinal herb Kalachoe gracilis. These compounds showed remarkably potent cytotoxicity against a HTCL panel, particularly against the A549 lung cancer cell line, where the EC50 value of 103 was 0.5 ng/mL (Table 5).61
Table 5.
Cytotoxicity of Kalanchosides
 | |||||||
|---|---|---|---|---|---|---|---|
| cmpd | IC50 (μg/mL) for 3 days continuous exposure | ||||||
| KB | KB-VIN | A549 | 1A9 | PC-3 | HCT-8 | A431 | |
| 103 | 0.003 | 0.003 | 0.0005 | 0.0008 | 0.002 | 0.006 | 0.007 |
| 104 | 0.005 | 0.013 | 0.001 | 0.007 | 0.010 | 0.015 | 0.022 |
| 105 | 0.016 | 0.026 | 0.006 | 0.012 | 0.025 | 0.045 | 0.055 |
Summary of the Highlights of Antitumor Agents Discovered by NPRL in Clinical Trials and in Preclinical Studies
Numerous compounds with potent cytotoxicity have been discovered as new drug candidates through studies in the author's NPRL, and have been reviewed before in this journal.62 A summary and highlights in regard to the above compounds are given in Figure 21.
Figure 21.

Summary of the highlights of antitumor agents discovered by NPRL in clinical trials and preclinical studies
Antimalarial Agents
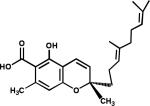
Artemisia annua was long used as a medicinal plant for malaria fever, and artemisinin (106) (“Qing Hao Su”), isolated from this plant, was used as a safe and effective cure for malaria in mainland China.63 Artemether (107) and arteether (108) are semi-synthetic derivatives that are used clinically to treat drug-resistant malaria and as a second-line therapy for severe malaria cases, respectively.64 A simpler endoperoxide (C-O-O-C-C=C) analog (109) synthesized from α-santonin was not active, which showed that the unique 1',2',4'-trioxane ring (C-O-O-C-O-C) could be quite specific for activity, because a simpler cyclic epoxide ring was not adequate.65 Subequent synthetic studies to explore this issue resulted in several desethanoqinghaosu analogs, both cyclic peroxide lactone (110) (C-O-O-C-O-C-O-C=O),66 as well as non-lactone (111, 112) (C-O-O-C-O-C-O) compounds,66 and 12-deoxo (113, 114) (C-O-O-C-O-C-O-C)67 analogs. All of these compounds were less potent than 106. However, tricyclic 1',2',4'-trioxane acid hydrolysis products (115, 116) of 106, in which the lactone ring was opened, were equipotent to the tricyclic parent compound. Thus, the ethane bridge forming the fourth ring is essential for potent activity, and likely imposes a strict steric stricture on the 1',2',4'-trioxane ring.68 More recent studies by Posner et al. with a structurally related compound (117) have supported these earlier findings.69 Compounds of interest are shown in Figure 22.
Figure 22.

Structures of artemisinin and inactive, less active, and equipotent analogs
Antifungal Agents
Anthracenediones substituted with 2',3'-epoxypropylamino groups were found to be potent antibacterial and antifungal agents. The compounds combined structural features of mitoxantrone (118), an anthracenedione antineoplastic agent, and teroxirone (119), an experimental triepoxide antitumor agent (Figure 23). 1,4-Di-(2,3-epoxy-propylamino)anthracenedione (120) had a minimum inhibitory concentration (MIC) of less than 0.13 ppm against Pseudomonas fluorescens, Staphylococcus aureus, Aspergillus niger, and Aureobasidum pullulans, but was less active against Ps. aerugenosa (MIC = 63 ppm) and Escherichia coli (MIC = 250 ppm).70 Compound 120 was licensed by Rohm and Haas Company (Philadephia, PA) for use as an antifungal agent.
Figure 23.

Structure of antibacterial/antifungal agent licensed by Rohm & Haas
Anti-AIDS Agents
The life cycle of human immunodeficiency virus (HIV), the causative agents of AIDS, offers various points for chemotherapeutic attack. Many anti-HIV agents inhibit the actions of the viral enzymes, including reverse transcriptase (nucleoside and non-nucleoside reverse transcriptase inhibitors, NRTIs and NNRTIs), protease (protease inhibitors, PIs), and integrase (integrase inhibitors, IN). Drug/drug candidates that inhibit other viral processes include maturation inhibitors (MIs), which inhibit virus budding/maturation, and entry inhibitors, which inhibit viral adsorption, chemokine co-receptor binding, or virus-cell fusion. The NPRL's anti-AIDS research program focuses on discovering lead natural products with promising anti-HIV activity, which can offer new structural and mechanistic classes of drug/drug candidates, particularly as viral resistance to currently used agents is a growing problem and limits therapeutic options. The earliest work led to the discovery of four new tetragalloylquinic acids, which at least in part inhibited HIV RT transcriptase activity.71 Continued studies have led to numerous natural products from various chemical classes with promising anti-HIV activity, based primarily on initial results from a screening to determine the level of HIV infection in treated cells. Examples (121–131) are given in Table 6.72-92 More detailed discussion will be given on two compound classes: betulinic acid derivatives and suksdorfin derivatives.
Table 6.
| cmpd | name | structure | plant source | chemical class | anti-HIV EC50 (μM) | TI (IC50 ÷ EC50) |
|---|---|---|---|---|---|---|
| 121 | (+)-1R-cocluaurine72 |

|
Nelumbo nucifera | alkaloid | 2.8 | 125 |
| 122 | triptonine B73 |
|
Tripterygium hypoglaucum | sesquiterpene alkaloid | <0.1 (μg/mL) | >1,000 |
| 123 | daurichromenic acid78 |
|
Rhododendron dauricum | chromane | 0.015 | 3,710 |
| 124 | 2-methoxy-3-methyl-4,6-dihydroxy-5-(3′-hydroxy)cinnamoyl-benzaldehyde79 |

|
Desmos spp | flavonoid | 0.067 | 489 |
| 125 | gomisin G83 |

|
Kadsura interior | lignan | 0.011 | 600 |
| 126 | sodium and potassium salts of caffeic acid tetramers85 |

|
Arnebia euchroma | caffeic acid tetramer | 1.9 | 33.3 |
| 127 | 8-C-ascorbyl-(-)-epigallocatechin86 |

|
tea polyphenol | 4 | 9.5 | |
| 128 | neotripterifordin87 |

|
Tripterygium wilfordii | diterpene | 0.025 | 125 |
| 129 | moronic acid88 |

|
Brazilian Propolis | triterpene | <0.22 | >186 |
| 130 | betulinic acid91 |

|
Syzygium claviflorum | triterpene | 1.4 | 9.3 |
| 131 | heraclenol75 |

|
Ferula sumbul | coumarin | 0.38 | 870 |
| 132 | suksdorfin77 |

|
Lomatium suksdorfii | coumarin | 1.3 | 40 |
Betulinic Acid Derivatives
3-O-(3',3'-Dimethylsuccinyl)betulinic Acid (DSB, Bevirimat): Maturation Inhibitors
A prior review93 described the identification of the triterpene betulinic acid (130) as an anti-HIV lead compound, followed by the identification of its ester analog, 3-O-(3',3'-dimethylsuccinyl)-betulinic acid (DSB, 133) (Figure 24), as the first HIV maturation inhibitor, through SAR modification, mechanism of action, and preclinical studies. A brief description of these studies will be given here.
Figure 24.

Structures of betulinic acid, its natural precursor betulin, and its synthetic analog DSB
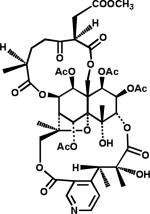
In 1994, 130 was isolated as anti-HIV principle from leaves of Syzigium claviflorum (“Pang Hua Chih Nan”).91 It is also found in the bark of the London plane tree (Platanus acerifolia), and its precursor betulin (134) in the bark of white birch (Betula alba) (Figure 24). Among a series of 3-acyl derivatives of 130, 133 was found to be the most potent compound (Table 7, 135–140).94 It was licensed to Panacos Inc. (Watertown, MA), renamed PA-457 and then Bevirimat, and subjected to intensive preclinical studies. It was a potent inhibitor of primary HIV-1 isolates in vitro, retained activity against virus isolates resistant to NRTIs, NNRTIs, and PIs, and also showed synergistic effects with other AIDS drugs. In mechanistic studies, 133-treated HIV virions showed immature, spherical cores.95 Studies suggested that 133 interferes with normal gag processing, which is necessary to form mature infectious virus particles. Indeed, further research proved that 132 disrupts cleavage at the CA-SP1 junction of the gag precursor protein, and disrupts normal capsid condensation. The resulting virus particles are defective and noninfectious. In summary, 132 was found to be the first-in-class HIV maturation inhibitor, targeting the CA-SP1 region of gag.88,95 Compound 133 was easily formulated as in a salt form with good oral availability, was eliminated via glucuronidation, and was active in a SCID mouse model.96 In Phase I clinical trials, it was safe and well tolerated with a good half-life and plasma levels.97 In Phase II clinical trials, 133 reduced viral load significantly (mean of -1.18 log10 copies/mL after 14 days of treatment) with good plasma levels found with a tablet formulation.97 It has now been licensed to Myriad Pharmaceuticals (Salt Lake City, UT), which has renamed the compound “MPC-4326” and is planning Phase III clinical trials for 2009/2010. The US FDA granted “Fast-Track” new drug development status for 133, for treatment of HIV in combination with already approved drugs or possibly as a first-line therapy. Figure 25 summarizes the status of the clinical development of 133.
Table 7.
Anti-HIV Activity of 130-analogs in H9 Cell
 | ||||
|---|---|---|---|---|
| cmpd | R | IC50 (μM) | EC50 (μM) | TI |
| 130 | H | 13.0 | 1,4 | 9.3 |
| 133 (DSB) |

|
7 | 0.00035 | 20,000 |
| 135 |

|
16 | 4.0 | 4 |
| 136 |

|
15.9 | 2.7 | 6.7 |
| 137 |
|
12.8 | 0.044 | 292 |
| 138 |
|
11.7 | 0.01 | 1,170 |
| 139 |
|
4.5 | 0.003 | 2,000 |
| 140 |

|
48 | 19 | 2.5 |
Figure 25.

Summary of clinical development of 133, an anti-AIDS compound discovered by NPRL
(3'-Monomethylsuccinyl)betulinic Acid (MSB) Analogs
More recently, the 3' S-methyl group was found to be the main contributor to the extremely high anti-HIV potency, because 3'S-MSB (S-141) was much more active than 3'R-MSB (R-141) (Table 8).98
Table 8.
Anti-HIV Activity of Monomethylsuccinyl Analogs of 130
 | |||
|---|---|---|---|
| cmpd | IC50 (μM) | EC50 (μM) | TI |
| S-141 | 33 | 0.0087 | 6,274 |
| R-141 | >40 | 0.12 | >961 |
| S-141+R-141 | >40 | 0.016 | >5,323 |
| 133 | >40 | 0.0013 | >30,555 |
| AZT | 1,870 | 0.034 | 55,330 |
C-28 Amide Substituted Analogs: Entry Inhibitors
For anti-HIV maturation activity, the C-3 position of betulinic acid analogs was found to be the pharmacophore for anti-HIV maturation activity (Figure 26). For optimal potency, the analog should have a C-3 acyl side with the proper length, terminal carboxylic acid moiety, and dimethyl substitution at the C-3' position for optimal potency.93,97 However, prior studies by Soler et al. showed that betulinic acid derivatives substituted with ω-aminoalkanoic acid at the C-28 position, e.g., RPR103611 (142, Figure 27), showed anti-HIV activity in the nanomolar range, by interfering with the virus-cell fusion process.99 Thus, the C-28 position of betulinic acid is the pharmacophore for anti-HIV entry activity (Figure 26).99,100 Addition of two ω-aminoalkanoic acids (m=7-10, n=3 or 4) resulted in optimal activity. Both statine and l-leucine, as the terminal amino acid, gave potent analogs, as evaluated in the MAGI assay for HIV-1 entry inhibitors.101 The exact target of these triterpene entry inhibitors has not been determined, although viral mutations that confer resistance were found in gp41 for RPR103611 and gp120 for its stereoisomer IC9564 (143, Figure 27).100
Figure 26.

Pharmacophores for anti-HIV maturation versus entry inhibitory 130-analogs
Figure 27.

130-Derived HIV-1 entry inhibitors
C-3, C-28 Disubstituted Inhibitors: Bifunctional Inhibitors
Addition of an appropriate acyl side chain at C-3 and amide side chain at C-28 to betulinic acid results in derivatives that inhibit both HIV entry and maturation.100 In SAR studies, the lead compound A12-2 (144; dimethylsuccinyl at C-3, 7-aminoheptanoic acid at C-28) was at least 20 times more potent at inhibiting HIV replication than either 130 (C-3, but not C-28 substituted; maturation inhibitor only) or 143 (C-28, but not C-3 substituted; entry inhibitor only), as well as demonstrating both anti-maturation and anti-entry activities (Figure 28).102 In SAR studies, smaller and bulkier C-3 acyl side chains were detrimental to activity.103 At the C-28 position, forming the amide bond by using a cyclic secondary amine, such as piperidine, increased metabolic stability.104 Regarding the triterpene molecular scaffold, moronic, ursolic, and oleanolic acid analogs were active, while glycyrrhetinic and lithocholic acid analogs were not.103,105 In fact, moronic analog 145, which is substituted with a 3',3'-dimethylsuccinyl ester at C-3 and l-leucine amide at C-28, showed better potency than bevirimat against several HIV strains (EC50 values against NL4-3 were 0.0085 μM/0.096 μM and against PI-R were 0.021 μM/0.43 μM). Thus, 145 could be another promising clinical trial candidate in the triterpene class.
Figure 28.

Comparison of C-3 mono-, C-28 mono-, and C-3,28 di-substituted triterpene HIV-1 inhibitors
Suksdorfin Derivatives
Dicamphanoylkhellactone (DCK) Analogs
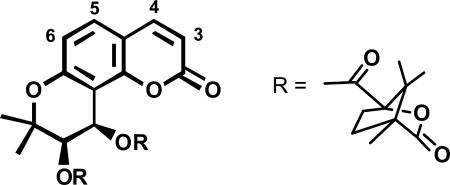
Bioactivity-directed fractionation using a p24 antigen ELISA assay for HIVIIIB replication in H9 lymphocytes led to the isolation of the natural coumarin suksdorfin (132) as an anti-HIV principle (EC50 = 1.3 μM, TI >40) from Lomatium suksdorfii.106 Replacing the two natural acyl groups with various esters led to the synthesis of the new anti-HIV lead 3',4'-di-O-(-)-camphanoyl-(+)-cis-khellactone (146, DCK) (Figure 29), which had greatly increased potency (EC50 = 0.049 μM, TI >328).77,106 Compound 146 and its derivatives could be prepared efficiently through a synthetic route developed using a Sharpless asymmetric dihydroxylation (Scheme 3).107,108 In initial SAR studies, methylation at the 4- or 5-position of the coumarin ring greatly increased potency, resulting in EC50 and TI values of 0.006 μM/6,600 (4-MeDCK, 148) and 0.0086 μM/>2,000 (5-MeDCK, 149) (Table 9).109 6-MeDCK (150) was much less active, and 3-MeDCK (147) was inactive. Adding a methoxy group at position-3, -4, or -5 (151–153, respectively) resulted in retained activity relative to 146, while 6-OMeDCK (154) was inactive (Table 9). Di-substitution or mono-substitution with larger alkyl or phenyl groups was less favorable or unfavorable.109 In an effort to enhance water solubility and oral bioavailability of DCK analogs, methyl groups substituted with various polar groups (-CH2X) were added.110 While addition of aminomethyl or diethylaminomethyl groups at the C-3 position of 4-methylDCK decreased potency, both bromomethyl and hydroxymethyl groups were favorable. Indeed, 3-bromomethyl-4-methylDCK (155) showed impressive EC50 and TI values of 0.00011 μM / 186,000 (Figure 30).110 However, 3-hydroxymethyl-4-methylDCK (156, HMDCK) (EC50 0.0042 μM, TI 6,000) was selected as a clinical trial candidate and licensed by Panacos Pharmaceuticals, based on better oral bioavailability in rats. In mechanistic studies, 146 and 156 were found to inhibit DNA-dependent DNA polymerase activity of the viral reverse transcriptase (RT). However, unlike traditional RT inhibitors that block generation of single stranded DNA from the RNA template, 146 and its analogs inhibit the production of double-stranded vial DNA from the single-stranded DNA intermediates. The exact binding site of these compounds has not yet been determined, but is possibly in the p51 subunit of HIV RT, where 146 binding could interfere with second strand transfer.111 Regardless, the unique mechanism of action makes 146 or future analogs potentially clinically useful.
Figure 29.

Structures of anti-HIV natural coumarin suksdorfin and synthetic analog DCK
Scheme 3.

Synthesis of DCK analogs
Table 9.
Effect of Mono-methylation or –methoxylation on Activity of DCK
 | |||
|---|---|---|---|
| cmpd | IC50 (μM) | EC50 (μM)a | TI |
| DCK (146) | >16.1 | 0.05 | >328 |
| 3-Me (147) | -- | No suppression | -- |
| 4-Me (148) | >39 | 0.006 | 6,600 |
| 5-Me (149) | >16 | 0.008 | >2,000 |
| 6-Me (150) | >16 | 0.21 | >72 |
| 3-OMe (151) | >15 | 0.03 | >533 |
| 4-OMe (152) | >15 | 0.05 | >300 |
| 5-OMe (153) | >15 | 0.044 | >350 |
| 6-OMe (154) | -- | No suppression | -- |
| AZT | -- | 0.044 | -- |
HIV replication in H9 lymphocytes
Figure 30.

Potent disubstituted analogs of 146
In SAR studies, replacing an oxygen atom in the lactone ring of 146 with sulfur or nitrogen led to analogs with significant potency, with EC50 values in the lower micromolar to nanomolar range.112-114 For example, replacing the carbonyl oxygen of 4-methyl-DCK with sulfur (-OC=S) giving 158 (4-MeDCK thiolactone) led to no loss or improvement in potency when assayed in CEM-SS cells (Figure 31).112 Similarly, replacing the alcoholic oxygen of the lactone with nitrogen (-NHC=O, 159)113 or sulfur (-SC=O, 160)114 led to equipotent or slightly more potent compounds, compared with 146 (-OC=O).
Figure 31.

Biostereo-isomeric analogs of 146
Dicamphanoylpyranochromone (DCP) Analogs
Another significant structural variation was to synthesize 3’R,4’R-di-O-(-)-camphanoyl-2’,2’-dimethyldihydropyrano[2,3-f]chromone (DCP) analogs, which are positional isomers of DCK.115 Among a series of mono- and di-substituted DCP derivatives, several compounds (161–166) exhibited extremely high anti-HIV activity in the non-drug resistant strain assay, with EC50 values ranging from 0.00032 to 0.0057 μM and remarkable therapeutic indexes (TI) ranging from 5.6×103 to 1.16×105 (Table 10), which were similar to those of 148 (EC50 0.0059 μM, TI 6.6×103) and better than those of 146 (EC50 0.049 μM, TI 328). Even more promisingly, some DCP analogs also showed activity against a multi-RT inhibitor resistant strain, HIV-1 RTMDR1, whereas most DCK analogs did not. An ethyl group was the optimal C-2 substituent for activity against non-drug resistant and multi-drug resistant HIV strains. Thus, the most significant compound was 2-Et-DCP (162), which showed the best anti-HIV activity in both assays, including an EC50 value of 0.06 μM and TI of 718 against the multi-RT inhibitor resistant HIV-1 strain (resistant to AZT, ddI, nevirapine, and other NNRTIs). In addition, 2-substituted DCPs were less toxic to cells than the unsubstituted or 3-substituted compounds. Due to their activity against drug-resistant HIV strains, DCP analogs may well be more promising than DCK analogs for further development as clinical trial candidates. Figure 32 summarizes the status of the preclinical development of DCK and DCP derivatives.
Table 10.
Anti-HIV Activity of 2-Substituted DCPs
 | |||||||
|---|---|---|---|---|---|---|---|
| cmpd | R2 | HIV-1 IIIB |
HIV-1 RTMDR1 |
||||
| IC50 μM | EC50 μMa | TI | IC50 μM | EC50 μMa | TI | ||
| 161 | Me | 27.3 | 0.0031 | 8,600 | 11.8 | 0.19 | 63 |
| 162 | Et | 37.2 | 0.00032 | 116,200 | 43.1 | 0.06 | 718 |
| 163 | n-Pr | >37.7 | 0.02 | 1,860 | 37.7 | 0.14 | 272 |
| 164 | i-Pr | 33.4 | 0.07 | 483 | >15 | 0.14 | >111 |
| 165 | CH2OEt | 15.1 | 0.1 | 151 | 12.5 | 0.37 | 34 |
| 166 | C6H5 | 36 | 0.13 | 277 | 12.2 | 0.17 | 71 |
| 146 (DCK) | >16 | 0.049 | >328 | >16.1 | 12.1 | 1.3 | |
| 148 (4-MeDCK) | >39 | 0.0059 | >6600 | >16 | 9.43 | 1.7 | |
HIV replication in H9 cells
Figure 32.

Summary of preclinical development of DCK and DCP derivatives, anti-HIV compounds discovered by NPRL
Summary
Plant products still serve as an excellent source for modern drug discovery and development. Through a medicinal chemistry approach, natural products with low bioactivity or known compounds can be modified synthetically to improve their pharmacological profiles. Synthesis of new compounds must be accompanied by appropriate biological assay screening to successfully optimize a lead compound into a clinical trial candidate. As shown by the work described above, academic laboratories can indeed successfully accomplish the goals of bringing compounds into clinical trials.
Acknowledgements
Drs. Keduo Qian and Susan L. Morris-Natschke helped me to prepare my award presentation and this review paper. Many thanks are due to a large number of research collaborators and co-authors, especially for those who are listed in my published research articles. I would also like to particularly thank Drs. T. H. Yang, S.T. Lu, M. Tomita, T. Nakano, T.O. Soine, T. A. Geissman, A. Brossi, G. Cragg, and H. Floss, who have been my mentors during my research career. I would also like to recognize my wife, Lan-Huei Lee, for her great support through these many years. Research funding support was provided by NIH grants CA-17625-32, AI-33066-17, GM-076152-3, and AI-077417-2.
Footnotes
Dedicated to the late Dr. John W. Daly of NIDDK, NIH, Bethesda, Maryland and to the late Dr. Richard E. Moore of the University of Hawaii at Manoa for their pioneering work on bioactive natural products.
Antitumor Agents 275 and Anti-AIDS Agents 80. Adapted from a Norman R. Farnsworth Research Achievement Award address presented at the 50th Annual Meeting of the American Society for Pharmacognosy, Honolulu, HI, June 27–July 1, 2009.
References and Notes
- 1.Lee KH. Public Health Nutrition. 2000;3:515–522. doi: 10.1017/s1368980000000604. [DOI] [PubMed] [Google Scholar]
- 2.Lee KH, Furukawa H, Kozuka M, Huang HC, Luhan PA, McPhail AT. J. Chem. Soc., Chem. Commun. 1973:476–477. [Google Scholar]
- 3.Lee KH, Ibuka T, Huang HC, Harris DL. J. Pharm. Sci. 1976;62:1077–1078. doi: 10.1002/jps.2600640656. [DOI] [PubMed] [Google Scholar]
- 4.McPhail AT, Onan KD, Lee KH, Ibuka T, Kozuka M, Shingu T, Huang HC. Tetrahedron Lett. 1974;32:2739–2741. [Google Scholar]
- 5.Lee KH, Imakura Y, Sims D, McPhail AT, Onan KD. J. Chem. Soc., Chem. Commun. 1976:341–342. [Google Scholar]
- 6.Lee KH, Furukawa H, Huang ES. J. Med. Chem. 1972;15:609–611. doi: 10.1021/jm00276a010. [DOI] [PubMed] [Google Scholar]
- 7.Lee KH, Huang ES, Piantadosi C, Pagano JS, Geissman TA. Cancer Res. 1971;31:1649–1654. [PubMed] [Google Scholar]
- 8.Lee KH, Hall IH, Mar EC, Starnes CO, ElGebaly SA, Waddell TG, Hadgraft RI, Ruffner CG, Weidner I. Science. 1977;196:533–536. doi: 10.1126/science.191909. [DOI] [PubMed] [Google Scholar]
- 9.Lee KH, Ibuka T, Sims D, Muraoka O, Kiyokawa H, Hall IH. J. Med. Chem. 1981;24:924–927. doi: 10.1021/jm00140a003. [DOI] [PubMed] [Google Scholar]
- 10.Lee KH, Imakura Y, Sumida Y, Wu RY, Hall IH, Huang HC. J. Org. Chem. 1979;44:2180–2185. [Google Scholar]
- 11.Furamiya N, Okano M, Miyamoto M, Tagahara K, Lee KH. J. Nat. Prod. 1992;55:468–475. doi: 10.1021/np50082a011. [DOI] [PubMed] [Google Scholar]
- 12.Ohnishi S, Fukamiya N, Okano M, Tagahara K, Lee KH. J. Nat. Prod. 1993;58:1032–1038. doi: 10.1021/np50121a007. [DOI] [PubMed] [Google Scholar]
- 13.Kupchan SM, Britton RW, Lacadie JA, Ziegler MF, Sigel CW. J. Org. Chem. 1975;40:648–654. doi: 10.1021/jo00893a023. [DOI] [PubMed] [Google Scholar]
- 14.Okano M, Lee KH. J. Org. Chem. 1981;46:1138–1141. [Google Scholar]
- 15.Lee KH, Okano M, Hall IH, Brent DA, Soltmann B. J. Pharm. Sci. 1982;71:338–345. doi: 10.1002/jps.2600710320. [DOI] [PubMed] [Google Scholar]
- 16.Lee KH, Yamagushi T. Abs. Chin. Med. 1987;1:606–625. [Google Scholar]
- 17.Okano M, Fukamiya N, Lee KH. In: Studies in Natural Product Chemistry. Atta-ur-Rahman, editor. Vol. 7. Elsevier Science; Amsterdam: 1990. pp. 369–404. [Google Scholar]
- 18.Ohno N, Fukamiya N, Okano M, Tagahara K, Lee KH. Bioorg. Med. Chem. 1997;5:1489–1495. doi: 10.1016/s0968-0896(97)00095-3. [DOI] [PubMed] [Google Scholar]
- 19.Lee KH, Tagahara K, Suzuki H, Wu RY, Haruna M, Hall IH, Huang HC, Ito K, Iida T, Lai JS. J. Nat. Prod. 1981;44:530–535. doi: 10.1021/np50017a003. [DOI] [PubMed] [Google Scholar]
- 20.Xia Y, Yang ZY, Xia P, Hackl T, Hamel E, Mauger A, Wu JH, Lee KH. J. Med. Chem. 2001;44:3932–3936. doi: 10.1021/jm0101085. [DOI] [PubMed] [Google Scholar]
- 21.Wang SW, Pan SL, Peng CY, Huang DY, Tsai AC, Chang YL, Guh JH, Kuo SC, Lee KH, Teng CM. Cancer Lett. 2007;257:87–96. doi: 10.1016/j.canlet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Chen K, Kuo SC, Hsieh MC, Mauger A, Lin CM, Hamel E, Lee KH. J. Med. Chem. 1997;40:3049–3056. doi: 10.1021/jm970146h. [DOI] [PubMed] [Google Scholar]
- 23.Lee KH. Med. Res. Rev. 1999;19:569–596. doi: 10.1002/(sici)1098-1128(199911)19:6<569::aid-med7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Chao YH, Kuo SC, Ku K, Chiu IP, Wu CH, Mauger A, Wang HK, Lee KH. Bioorg. Med. Chem. 1999;7:1025–1031. doi: 10.1016/s0968-0896(98)00241-7. [DOI] [PubMed] [Google Scholar]
- 25.Wen YF, Lee KH, Huang PT, Chen MH, Shin WC, Huang LJ, Hsu MH, Chen CJ, Kuo SC. Bioorg. Med. Chem. Lett. 2007;17:2908–2912. doi: 10.1016/j.bmcl.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 26.Capraro HG, Brossi A. In: The Alkaloids. Brossi A, editor. Vol. 23. Academic Press; New York: 1984. pp. 1–70. [Google Scholar]
- 27.Shi Q, Verdier-Pinard P, Brossi A, Hamel E, McPhail AT, Lee KH. J. Med. Chem. 1997;40:961–966. doi: 10.1021/jm960663k. [DOI] [PubMed] [Google Scholar]
- 28.Guan J, Zhu XK, Brossi A, Tachibana Y, Bastow KF, Verdier-Pinard P, Hamel E, McPhail AT, Lee KH. Collect. Czech. Chem. Commun. 1999;64:217–228. [Google Scholar]
- 29.Keller-Juslén C, Kuhn M, von Wartburg A, Stähelin H. J. Med. Chem. 1971;14:936–940. doi: 10.1021/jm00292a012. [DOI] [PubMed] [Google Scholar]
- 30.van Maanen JMS, Retèl J, de Vries J, Pinedo HM. J. Natl. Cancer Inst. 1988;80:1526–1533. doi: 10.1093/jnci/80.19.1526. [DOI] [PubMed] [Google Scholar]
- 31.Lee KH, Imakura Y, Haruna M, Beers SA, Thurston LS, Dai HJ, Chen CH, Liu SY, Cheng YC. J. Nat. Prod. 1989;52:606–613. doi: 10.1021/np50063a021. [DOI] [PubMed] [Google Scholar]
- 32.Zhou XM, Wang ZQ, Chang JY, Chen HX, Cheng YC, Lee KH. J. Med. Chem. 1991;34:3346–3350. doi: 10.1021/jm00116a001. [DOI] [PubMed] [Google Scholar]
- 33.Chang JY, Han FS, Liu SY, Wang ZQ, Lee KH, Cheng YC. Cancer Res. 1991;51:1755–1759. [PubMed] [Google Scholar]
- 34.Wang ZQ, Kuo YH, Schnur D, Bowen JB, Liu SY, Han FS, Chang JY, Cheng YC, Lee KH. J. Med. Chem. 1990;33:2860–2666. doi: 10.1021/jm00171a050. [DOI] [PubMed] [Google Scholar]
- 35.Cho SJ, Tropsha A, Suffness M, Cheng YC, Lee KH. J. Med. Chem. 1996;39:1383–1395. doi: 10.1021/jm9503052. [DOI] [PubMed] [Google Scholar]
- 36.Ji Z, Wang HK, Bastow KF, Zhu XK, Cho SJ, Cheng YC, Lee KH. Bioorg. Med. Chem. Lett. 1997;7:607–612. [Google Scholar]
- 37.Xiao Z, Bastow KF, Vance JR, Sidwell RS, Wang HK, Chen MS, Shi Q, Lee KH. J. Med. Chem. 2004;47:5140–5148. doi: 10.1021/jm030609l. [DOI] [PubMed] [Google Scholar]
- 38.Xiao Z, Vance JR, Bastow KF, Brossi A, Wang HK, Lee KH. Bioorg. Med. Chem. 2004;12:3363–3369. doi: 10.1016/j.bmc.2004.03.056. [DOI] [PubMed] [Google Scholar]
- 39.Lin L, Shi Q, Nyarko AK, Bastow KF, Wu CC, Su CY, Shih CCY, Lee KH. J. Med. Chem. 2006;49:3963–3972. doi: 10.1021/jm051043z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin L, Shi Q, Su CY, Shih CCY, Lee KH. Bioorg. Med. Chem. 2006;14:2527–2534. doi: 10.1016/j.bmc.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 41.Itokawa H, Shi Q, Akiyama T, Morris-Natschke SL, Lee KH. Chin. Med. 2008;3:11–23. doi: 10.1186/1749-8546-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee KH, Lin L, Shih CCY, Su CY, Ishida J, Ohtsu H, Wang HK, Itokawa H, Chang CS. 2008 U.S. Patent 7,355,031B2.
- 43.Shi Q, Shih CC, Lee KH. Anticancer Agents Med. Chem. 2009;9:904–912. doi: 10.2174/187152009789124655. [DOI] [PubMed] [Google Scholar]
- 44.Wang X, Morris-Natschke SL, Lee KH. Med. Res. Rev. 2007;27:133–148. doi: 10.1002/med.20077. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Bastow KF, Sun CM, Lin YL, Yu HJ, Don MJ, Wu TS, Nakamura S, Lee KH. J. Med. Chem. 2004;47:5816–5819. doi: 10.1021/jm040112r. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Nakagawa-Goto K, Bastow KF, Don MJ, Lin YL, Wu TS, Lee KH. J. Med. Chem. 2006;49:5631–5634. doi: 10.1021/jm060184d. [DOI] [PubMed] [Google Scholar]
- 47.Dong Y, Shi Q, Liu YN, Wang X, Bastow KF, Lee KH. J. Med. Chem. 2009;52:3586–3590. doi: 10.1021/jm9001567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee KH, Dong Y, Shi Q, Bastow KF. US Provisional Patent Application No. 61/139,208.
- 49.Dong Y, Shi Q, Nakagawa-Goto K, Yu D, Liu YN, Wu PC, Bastow KF, Morris-Natschke SL, Brossi A, Pai HC, Peng CY, Pan SL, Teng CM, Hung MC, Lee EYHP, Lee KH. J. Med. Chem. doi: 10.1021/jm1000858. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei L, Brossi A, Morris-Natschke SL, Bastow KF, Lee KH. In: Studies in Natural Products Chemistry. Atta-ur-Rahman, editor. Vol. 34. Elsevier; New York: 2008. pp. 3–34. [Google Scholar]
- 51.Wei L, Brossi A, Kendall R, Bastow KF, Morris-Natschke SL, Shi Q, Lee KH. Bioorg. Med. Chem. 2006;14:6560–6509. doi: 10.1016/j.bmc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Wei L, Shi Q, Bastow KF, Brossi A, Morris-Natschke SL, Nakagawa-Goto K, Wu TS, Pan SL, Teng CH, Lee KH. J. Med. Chem. 2007;50:3674–3680. doi: 10.1021/jm061366a. [DOI] [PubMed] [Google Scholar]
- 53.Gao W, Chen APC, Leung CH, Gullen EA, Fürstner A, Shi Q, Wei L, Lee KH, Cheng YC. Bioorg. Med. Chem. Lett. 2008;18:704–709. doi: 10.1016/j.bmcl.2007.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin JC, Yang SC, Hong TM, Yu SL, Shi Q, Wei L, Chen HY, Yang PC, Lee KH. J. Med. Chem. 2009;52:1903–1911. doi: 10.1021/jm801344j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi Q, Wang HK, Bastow KF, Tachibana Y, Chen K, Lee FY, Lee KH. Bioorg. Med. Chem. 2001;9:2999–3004. doi: 10.1016/s0968-0896(01)00206-1. [DOI] [PubMed] [Google Scholar]
- 56.Ohtsu H, Nakanishi Y, Bastow KF, Lee FY, Lee KH. Bioorg. Med. Chem. 2003;11:1851–1857. doi: 10.1016/s0968-0896(03)00040-3. [DOI] [PubMed] [Google Scholar]
- 57.Nakagawa-Goto K, Yamada K, Nakamura S, Chen TH, Chiang PC, Bastow KF, Wang SC, Spohn B, Hung MC, Lee FY, Lee FC, Lee KH. Bioorg. Med. Chem. Lett. 2007;17:5204–5209. doi: 10.1016/j.bmcl.2007.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakagawa-Goto K, Chen TH, Peng CY, Bastow KF, Wu JH, Lee KH. J. Med. Chem. 2007;50:3354–3358. doi: 10.1021/jm0702534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakagawa-Goto K, Bastow KF, Chen TH, Morris-Natschke SL, Lee KH. J. Med. Chem. 2008;51:3297–3303. doi: 10.1021/jm701208v. [DOI] [PubMed] [Google Scholar]
- 60.Lin AS, Nakagawa-Goto K, Chang FR, Yu D, Morris-Natschke SL, Wu CC, Chen SL, Wu YC, Lee KH. J. Med. Chem. 2007;50:3921–3927. doi: 10.1021/jm070363a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu PL, Hsu YL, Wu TS, Bastow KF, Lee KH. Org. Lett. 2006;8:5207–5210. doi: 10.1021/ol061873m. [DOI] [PubMed] [Google Scholar]
- 62.Lee KH. J. Nat. Prod. 2004;67:273–283. doi: 10.1021/np030373o. [DOI] [PubMed] [Google Scholar]
- 63.Qinghaosu Antimalaria Coordinating Research Group Chin. Med. J. Beijing, Engl. Ed.) 1979;92:811. [PubMed] [Google Scholar]
- 64.Meshnick SR. In: Antimalarial Chemotherapy. Rosenthal PJ, editor. Humana Press; Totowa, NJ: 2001. pp. 191–200. [Google Scholar]
- 65.Tani S, Fukamiya N, Kiyokawa H, Musallam HA, Pick RO, Lee KH. J. Med. Chem. 1985;28:1743–1744. doi: 10.1021/jm00149a034. [DOI] [PubMed] [Google Scholar]
- 66.Imakura Y, Yokoi T, Yamagishi T, Koyama J, Hu H, McPhail DR, McPhail AT, Lee KH. J. Chem. Soc., Chem. Commun. 1988:372–374. [Google Scholar]
- 67.Imakura Y, Hachiya K, Ikemoto T, Kobayashi S, Yamashita S, Sakakibara J, Smith FT, Lee KH. Heterocycles. 1990;31:2125–2129. [Google Scholar]
- 68.Imakura Y, Hachiya K, Ikemoto T, Yamashita S, Kihara M, Kobayashi S, Shingu T, Milhous WK, Lee KH. Heterocycles. 1990;31:1011–1016. [Google Scholar]
- 69.Posner GH, O'Neill PM. Acc. Chem. Res. 2003;46:987–994. [Google Scholar]
- 70.Mehta RJ, Swithenbank C, Lidert Z, Bowers-Daines MM, Young DH, Lange BC. 1990 U.S. Patent 4,975,459.
- 71.Nishizawa M, Yamagishi T, Dutschman GE, Parker WB, Bodner AJ, Kilkuskie RE, Cheng YC, Lee KH. J. Nat. Prod. 1989;52:762–768. doi: 10.1021/np50064a016. [DOI] [PubMed] [Google Scholar]
- 72.Yu D, Morris-Natschke SL, Lee KH. Med. Res. Rev. 2006;27:108–132. doi: 10.1002/med.20075. [DOI] [PubMed] [Google Scholar]
- 73.Duan H, Takaishi Y, Imakura Y, Jia Y, Li D, Cosentino LM, Lee KH. J. Nat. Prod. 2000;63:357–361. doi: 10.1021/np990281s. [DOI] [PubMed] [Google Scholar]
- 74.Wu TS, Tsang ZJ, Wu PL, Lin FW, Li CY, Teng CM, Lee KH. Bioorg. Med. Chem. 2001;9:77–83. doi: 10.1016/s0968-0896(00)00225-x. [DOI] [PubMed] [Google Scholar]
- 75.Zhou P, Takaishi Y, Duan H, Chen B, Honda G, Itoh M, Takeda Y, Kodzhimatov OK, Lee KH. Phytochemistry. 2000;53:689–697. doi: 10.1016/s0031-9422(99)00554-3. [DOI] [PubMed] [Google Scholar]
- 76.Shikishima Y, Takaishi Y, Hondo G, Ito M, Takeda Y, Kodzhimatov O, Ashurmetov O, Lee KH. Chem. Pharm. Bull. 2001;49:877–880. doi: 10.1248/cpb.49.877. [DOI] [PubMed] [Google Scholar]
- 77.Lee TTY, Kashiwada Y, Huang L, Snider J, Cosentino M, Lee KH. Bioorg. Med. Chem. 1994;2:1051–1056. doi: 10.1016/s0968-0896(00)82054-4. [DOI] [PubMed] [Google Scholar]
- 78.Kashiwada Y, Yamazaki K, Ikeshiro Y, Yamagishi T, Fujioka T, Mihashi K, Mizuki K, Cosentino LM, Fowke K, Morris-Natschke SL, Lee KH. Tetrahedron. 2001;57:1559–1563. [Google Scholar]
- 79.Wu JH, Wang XH, Yi YH, Lee KH. Bioorg. Med. Chem. Lett. 2003;11:1813–1815. doi: 10.1016/s0960-894x(03)00197-5. [DOI] [PubMed] [Google Scholar]
- 80.Tang R, Chen K, Cosentino M, Lee KH. Bioorg. Med. Chem. Lett. 1994;4:455–458. [Google Scholar]
- 81.Lee KH, Kashiwada Y, Nonaka G, Nishioka I, Nishizawa M, Yamagishi T, Bodner AJ, Kilkuskie RE, Cheng YC. In: Natural Products as Antiviral Agents. Chu CK, Cutler HG, editors. Plenum Press; New York: 1992. pp. 171–193. [Google Scholar]
- 82.Xie L, Xie JX, Kashiwada Y, Cosentino LM, Liu SH, Pai RB, Cheng YC, Lee KH. J. Med. Chem. 1995;38:3003–3008. doi: 10.1021/jm00016a002. [DOI] [PubMed] [Google Scholar]
- 83.Chen DF, Zhang SX, Xie L, Xie JX, Chen K, Kashiwada Y, Zhou BN, Wang P, Cosentino LM, Lee KH. Bioorg. Med. Chem. 1997;5:1715–1723. doi: 10.1016/s0968-0896(97)00118-1. [DOI] [PubMed] [Google Scholar]
- 84.Yang LM, Lin SJ, Yang TH, Lee KH. Bioorg. Med. Chem. Lett. 1996;6:941–944. [Google Scholar]
- 85.Kashiwada Y, Nishizawa M, Yamagishi T, Tanaka T, Nonaka GI, Cosentino LM, Snider JV, Lee KH. J. Nat. Prod. 1995;58:392–400. doi: 10.1021/np50117a007. [DOI] [PubMed] [Google Scholar]
- 86.Hashimoto F, Kashiwada Y, Nonaka GI, Nishioka I, Nohara T, Cosentino LM, Lee KH. Bioorg. Med. Chem. Lett. 1996;6:695–700. [Google Scholar]
- 87.Wu YC, Hung YC, Chang FR, Cosentino M, Wang HK, Lee KH. J. Nat. Prod. 1996;59:635–637. doi: 10.1021/np960416j. [DOI] [PubMed] [Google Scholar]
- 88.Yu D, Morris-Natschke SL, Lee KH. Med. Res. Rev. 2007;27:108–132. doi: 10.1002/med.20075. [DOI] [PubMed] [Google Scholar]
- 89.Konoshima T, Kashiwada Y, Takasaki M, Kozuka M, Yasuda I, Cosentino LM, Lee KH. Bioorg. Med. Chem. Lett. 1994;4:1323–1326. [Google Scholar]
- 90.Sakurai N, Wu JH, Sashida Y, Mimaki Y, Nikaido T, Koike K, Itokawa H, Lee KH. Bioorg. Med. Chem. Lett. 2004;14:1329–1332. doi: 10.1016/j.bmcl.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 91.Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. J. Nat. Prod. 1994;57:243–247. doi: 10.1021/np50104a008. [DOI] [PubMed] [Google Scholar]
- 92.Ito J, Chang FR, Wang HK, Park YK, Ikegaki M, Kilgore N, Lee KH. J. Nat. Prod. 2001;64:1278–1281. doi: 10.1021/np010211x. [DOI] [PubMed] [Google Scholar]
- 93.Yu D, Wild CT, Martin DE, Morris-Natschke SL, Chen CH, Allaway GP, Lee KH. Expert Opin. Investig. Drugs. 2005;14:681–693. doi: 10.1517/13543784.14.6.681. [DOI] [PubMed] [Google Scholar]
- 94.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PA, Lee KH. J. Med. Chem. 2006;39:1016–1017. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 95.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JL, Allaway GP, Freed EO, Wild CT. Proc. Natl. Acad. Sci. USA. 2003;100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stoddart CA, Joshi P, Sloan B, Bare JC, Smith PC, Allaway GP, Wild CT, Martin DE. PloS One. 2007;2:e1251. doi: 10.1371/journal.pone.0001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qian K, Nitz TJ, Yu D, Allaway GP, Morris-Natschke SL, Lee KH. In: Natural Product Chemistry for Drug Discovery. Buss AD, Butler M, editors. RSC Publishing; Cambridge, UK: 2010. pp. 374–391. Chapter 13. [Google Scholar]
- 98.Qian K, Nakagawa-Goto K, Yu D, Morris-Natschke SL, Nitz TJ, Kilgore N, Allaway GP, Lee KH. Bioorg. Med. Chem. Lett. 2007;17:6553–6557. doi: 10.1016/j.bmcl.2007.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soler F, Poujade C, Evers M, Carry JC, Hénin Y, Bousseau A, Huet T, Pauwels R, De Clercq E, Mayaux JF, Le Pecq JB, Dereu N. J. Med. Chem. 1996;39:1069–1083. doi: 10.1021/jm950669u. [DOI] [PubMed] [Google Scholar]
- 100.Qian K, Morris-Natschke SL, Lee KH. Med. Res. Rev. 2009;29:369–393. doi: 10.1002/med.20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun IC, Chen CH, Kashiwada Y, Wu JH, Wang HK, Lee KH. J. Med. Chem. 2002;45:4271–4275. doi: 10.1021/jm020069c. [DOI] [PubMed] [Google Scholar]
- 102.Huang L, Ho P, Lee KH, Chen CH. Bioorg. Med. Chem. 2006;14:2279–2289. doi: 10.1016/j.bmc.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 103.Huang L, Yu D, Ho P, Lee KH, Chen CH. Lett. Drug Discov. 2007;4:471–478. [Google Scholar]
- 104.Qian K, Yu D, Chen CH, Huang L, Morris-Natschke SL, Nitz TJ, Salzwedel K, Reddick M, Allaway GP, Lee KH. J. Med. Chem. 2009;52:3248–3258. doi: 10.1021/jm900136j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu D, Sakurai Y, Chen CH, Chang FR, Huang L, Kashiwada Y, Lee KH. J. Med. Chem. 2006;49:5462–5469. doi: 10.1021/jm0601912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang L, Kashiwada Y, Cosentino LM, Fan S, Chen CH, McPhail AT, Fujioka T, Mihashi K, Lee KH. J. Med. Chem. 1994;37:3947–3955. doi: 10.1021/jm00049a014. [DOI] [PubMed] [Google Scholar]
- 107.Xie L, Crimmins MT, Lee KH. Tetrahedron Lett. 1995;36:4529–4532. [Google Scholar]
- 108.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 109.Xie L, Takeuchi Y, Cosentino LM, Lee KH. J. Med. Chem. 1999;42:2662–2672. doi: 10.1021/jm9900624. [DOI] [PubMed] [Google Scholar]
- 110.Xie L, Yu D, Wild C, Allaway G, Turpin J, Smith P, Lee KH. J. Med. Chem. 2004;47:756–760. doi: 10.1021/jm030416y. [DOI] [PubMed] [Google Scholar]
- 111.Huang L, Yuan X, Yu D, Lee KH, Chen CH. Virology. 2005;332:623–628. doi: 10.1016/j.virol.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 112.Yang ZY, Xia Y, Xia P, Cosentino LM, Lee KH. Bioorg. Med. Chem. Lett. 1998;8:1483–1486. doi: 10.1016/s0960-894x(98)00254-6. [DOI] [PubMed] [Google Scholar]
- 113.Yang ZY, Xia Y, Brossi A, Cosentino LM, Lee KH. Bioorg. Med. Chem. Lett. 2000;10:1003–1005. doi: 10.1016/s0960-894x(00)00126-8. [DOI] [PubMed] [Google Scholar]
- 114.Xia P, Yin ZJ, Chen Y, Zhang Q, Zhang B, Xia Y, Yang ZY, Kilgore N, Wild C, Morris-Natschke SL, Lee KH. Bioorg. Med. Chem. Lett. 2004;14:3341–3343. doi: 10.1016/j.bmcl.2004.03.051. [DOI] [PubMed] [Google Scholar]
- 115.Yu D, Chen CH, Brossi A, Lee KH. J. Med. Chem. 2004;47:4072–4082. doi: 10.1021/jm0400505. [DOI] [PubMed] [Google Scholar]