

Abstract
Cytotoxicity-guided fractionation of a methanol extract of the leaves and twigs of Rolandra fruticosa using the HT-29 human colon cancer cell line led to the isolation of seven sesquiterpene lactones, including a new isorolandrolide, 13-methoxyisorolandrolide (1), a new bourbonenolide, 2α,13-diacetoxy-4α-hydroxy-8α-isobutyroyloxybourbonen-12,6α-olide (2), as well as five known compounds, 13-acetoxyrolandrolide (3), 8-desacyl-13-acetoxyrolandrolide-8-O-tiglate (4), 2-epi-glaucolide E (5), 2α,13-diacetoxy-4α-hydroxy-8α-methacryloyloxybourbonen-12,6α-olide (6), and 2α,13-diacetoxy-4α-hydroxy-8α-tigloyloxybourbonen-12,6α-olide (7). The structures of the two new sesquiterpenes were elucidated on the basis of spectroscopic methods. All isolates were evaluated for their cytotoxicity using the HT-29 cell line, and only 13-acetoxyrolandrolide (3) was found to possess a potent inhibitory effect against this cell line. Compounds 3, 5 and 6 were also tested in a NF-κB (p65) inhibition assay, and 3 was assessed in an in vivo hollow fiber assay.
Keywords: Rolandra fruticosa (Asteraceae), germacrane-type sesquiterpene lactones, isorolandrolide, rolandrolide, glaucolide, bourbonenolide, cytotoxicity, HT-29 human colon cancer cells, NF-κB (p65) inhibition assay, in vivo hollow fiber assay
1. Introduction
Rolandra fruticosa (L.) Kuntze (Asteraceae), the only species of the monotypic genus Rolandra Rottb., has been separated from the large subtribe Vernonieae and placed in the subtribe Rolandrinae (Robinson et al., 1980). This species, with the common name “yerba de plata”, is an evergreen shrub widespread throughout tropical areas of South America, and it has been introduced to Indonesia and Japan in Asia (Woodson et al., 1975; Smithsonian Tropical Research Institute Herbarium, 2009). This plant has been used in certain religious rituals by members of some indigenous populations living in Colombia and Panama (Duke, 1975; Woodson et al., 1975; Van Andel et al., 2007). According to the literature, species of the subtribe Vernonieae that are taxonomically closely related to the subtribe Rolandrinae have use as folk medicines for the treatment of various diseases such as arthritis (Latha et al., 1998), diabetes (Uhegbu et al., 2004; Okolie et al., 2008), edema (Awaad et al., 2000; Mazumder et al., 2003), HIV-related symptoms (Bessong et al., 2006; Njoroge and Bussmann, 2007), and malaria (Abosi and Raseroka, 2003; Walewa et al., 2003; Njan et al., 2008). Previous phytochemical studies on R. fruticosa have revealed the occurrence of germacrane-type sesquiterpene lactones including the bourbonenolides, glaucolides, isorolandrolides, and rolandrolides (Herz et al., 1981; Jakupovic et al., 1989). These sesquiterpenes are structurally close to germacrane-type sesquiterpenes that occur extensively in plants belonging to the tribe Vernonieae (Bohlmann and Jakupovic, 1990; Da et al., 2005). Rolandrolides and isorolandrolides have been identified only from R. fruticosa thus far. Information on the bioactivitiy of these principles is very limited, with the weak in vivo antitumor activity of 13-acetoxyrolandrolide (3) against P-388 lymphocytic murine leukemia being the only report of this type (Herz et al., 1981).
As part of an effort to discover naturally occurring anticancer agents from diverse organisms (Kinghorn et al., 2009), the chloroform-soluble partition of a methanol extract from the combined leaves and twigs of R. fruticosa collected in Indonesia was found to exhibit an inhibitory effect against the HT-29 human colon cancer cell line. In the present work, activity-guided fractionation of the chloroform-soluble extract using HT-29 cells led to the isolation of seven sesquiterpene lactones (1-7). Among these compounds, 1 and 2 were identified as two new sesquiterpenes, while compounds 3-7 were identified as the previously known 13-acetoxyrolandrolide (3) (Herz et al., 1981; Jakupovic et al., 1989), 8-desacyl-13-acetoxyrolandrolide-8-O-tiglate (4) (Jakupovic et al., 1989), 2-epi-glaucolide E (5) (Jakupovic et al., 1989), 2α,13-diacetoxy-4α-hydroxy-8α-methacryloyloxybourbonen-12,6α-olide (6) (Jakupovic et al., 1989), and 2α,13-diacetoxy-4α-hydroxy-8α-tigloyloxybourbonen-12,6α-olide (7) (Jakupovic et al., 1989), respectively, by comparison of their spectroscopic data with published values. Herein, we present the isolation and structure elucidation of new compounds 1 and 2, as well as the in vitro biological evaluation of all isolates obtained in two bioassays, and the evaluation of the previously known compound, 13-acetoxyrolandrolide (3) in an in vivo hollow fiber assay.
2. Results and discussion
2.1 Structure elucidation of new compounds
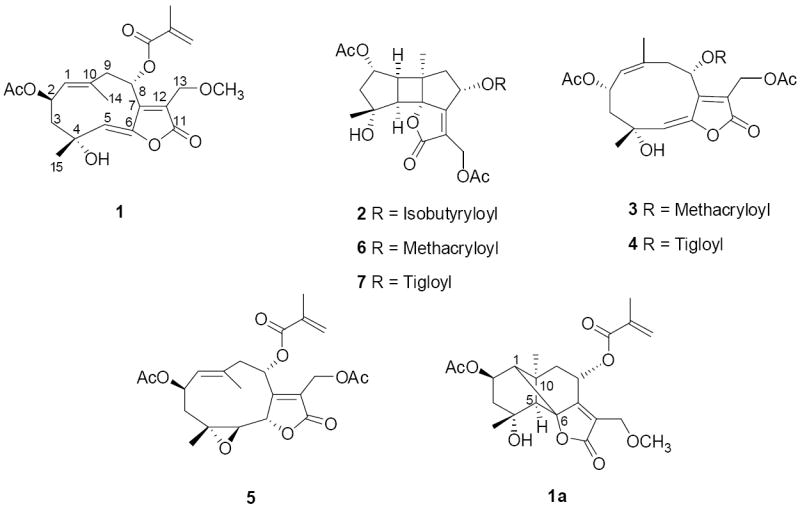
Compound 1 was obtained as a white amorphous powder. The HRESIMS analysis of compound 1 gave a sodiated molecular ion peak at m/z 443.1702 [M+Na]+ (calcd for C22H28O8Na), consistent with a molecular formula of C22H28O8. The IR absorptions indicated the occurrence of hydroxy group (3468 cm-1), double bond (3040, 954, 795 cm-1), and ester carbonyl (1750, 1731 cm-1) functionalities. The strong absorption at 211 nm (log ε 4.60) and a broad band around 313 nm (log ε 3.89) in the UV spectrum suggested the presence of an α,β-unsaturated lactone group. In the 1H NMR spectrum (Table 1), five tertiary methyl group signals (δH 1.71, 1.97, 2.00, 2.03, and 3.41, s, each 3H) were observed, and resonances for two olefinic protons at δH 6.14 (1H, br s, H-3′) and 5.66 (1H, br s, H-3′) were ascribed to an exocyclic methylene, while another two olefinic proton signals at δH 4.76 (br d, J = 10 Hz, H-1) and 5.71 (br s, H-5) were attributed to two endocyclic double bonds. Moreover, the proton signals at δH 6.37 (dd, J = 5.0, 2.0 Hz, H-8) and δH 5.82 (ddd, J = 10.8, 10.2, 3.0 Hz, H-2) were consistent with the presence of two esterified oxymethine groups, and two signals at δH 4.36 and 4.27 (each 1H, H-13) with an AB coupling pattern (JA,B = 13.6 Hz) suggested the presence of an isolated oxymethylene. Additional analysis of the 13C NMR spectrum based on the 1H NMR data mentioned above revealed the presence of an acetyl group, a methoxy group, as well as a methacrylate group (see Tables 1 and 2). After deduction of these three substituent residues, there were 15 carbon signals remaining in the 13C NMR spectrum of 1, which were classified by DEPT and HSQC into two methyls, two methylenes, four oxygenated carbons (including one primary, two secondary, and a tertiary), a carbonyl group, two trisubstituted double bonds, and a tetrasubstituted double bond. Analysis of these data suggested that compound 1 is an isorolandrolide analog. Isorolandrolide, a bicyclic germacrane sesquiterpene based on a carbon skeleton with a cyclodeca[b]furan moiety, having a 1(10)-E double bond and a 5-Z double bond in the ten-membered macrocyclic ring, was first isolated from R. fruticosa (Herz et al., 1981), and its structure was later revised (Jakupovic et al., 1989). Comparison of the 1H-NMR data of compound 1 with those of isorolandrolide revealed that the proton signal of an acetate methyl group was absent and a methoxy group singlet appeared at δH 3.41 (s, 3H), consistent with upfield shifts of H-13 and H-13’ of 0.23 ppm and 0.27 ppm, respectively. This evidence suggested that an acetyl group attached to C-13 in isorolandrolide was substituted by a methoxy group in compound 1. In the HMBC spectrum, key correlations from H-13 to the methyl signal at δC 59.2 and from the methoxy protons to the oxygenated methylene at δC 63.9 (C-13) were found. Moreover, cross peaks between H-8 with the carbonyl of the methacrylate group at δC 166.4, and H-2 with the carbonyl of the acetyl group at δC 170.5 revealed the locations of these two ester residues. The positions of two endocyclic double bonds at C-1(10) and C-5(6) were confirmed from the HMBC correlations of H-1/C-14, H-1/C-9, H-2/C-1, H-9/C-10, H-5/C-15, H-5/C-4, and H-8/C-6.
Table 1.
1H and 13C NMR chemical shifts of compounds 1 and 2a
| position | 1 |
2 |
||
|---|---|---|---|---|
| δC, mult.b | δH, (J in Hz)c | δC, mult.b | δH, (J in Hz)c | |
| 1 | 129.1 | 4.76 (br d, 10.0) | 51.0 | 2.71 (d, 10.8) |
| 2 | 68.7 | 5.82 (ddd, 10.8, 10.2, 3.0) | 77.2 | 5.38 (d, 7.0) |
| 3α | 51.3 | 2.06 (dd, 13.0, 3.0) | 49.2 | 2.22 (d, 16.7) |
| 3β | 2.23 (dd, 13.0, 11.0) | 2.53 (dd, 16.7, 7.0) | ||
| 4 | 73.0 | 80.6 | ||
| 5 | 133.2 | 5.71 (br s) | 59.5 | 3.43 (dd, 10.2, 1.8) |
| 6 | 144.1 | 92.0 | ||
| 7 | 152.4 | 169.0 | ||
| 8 | 70.2 | 6.37 (dd, 5.0, 2.0) | 70.7 | 6.23 (dd, 9.2, 8.2) |
| 9α | 44.5 | 2.81 (dd, 14.0, 5.4) | 45.5 | 1.74 (dd, 14.0, 9.2) |
| 9β | 2.31 (br d, 14.0) | 2.89 (dd, 14.0, 8.2) | ||
| 10 | 135.6 | 47.8 | ||
| 11 | 123.6 | 123.6 | ||
| 12 | 169.0 | 170.1 | ||
| 13a | 63.9 | 4.36 (d, 13.6) | 55.2 | 4.93 (d,12.6) |
| 13b | 4.27 (d, 13.6) | 4.77 (d, 12.6) | ||
| 14 | 21.5 | 2.03 (s) | 22.6 | 1.31 (s) |
| 15 | 29.1 | 1.71 (s) | 23.2 | 1.24 (s) |
| 1′ | 166.4 | 176.0 | ||
| 2′ | 135.6 | 33.8 | 2.61 (hep, 6.9) | |
| 3′ | 127.4 | 6.14 (br s) | 19.0 | 1.20 (d, 6.9) |
| 5.66 (br s) | ||||
| 4′ | 18.7 | 1.97 (br s) | 18.5 | 1.21 (d, 6.9) |
| OAc-2 | 170.5 | 170.1 | ||
| 20.4 | 2.00 (s) | 21.3 | 2.07 (s) | |
| OAc-13 | 170.4 | |||
| 20.6 | 2.04 (s) | |||
| OMe-13 | 59.2 | 3.41 (s) | ||
1H NMR measured at 600 MHz, 13C NMR measured at 150 MHz; obtained in CDCl3 with TMS as internal standard. Assignments are based on HSQC and HMBC NMR spectra.
Multiplicity obtained from the DEPT spectra.
J values (Hz) are given in parentheses.
Table 2.
In vitro cytotoxicity and NF-κB inhibitory activity of compounds isolated from R. fruticosa.a
| Compound | Cytotoxicityb | NF-κB Inhibitionc |
|---|---|---|
| 3 | 0.16 | 7.1 |
| 4 | 1.5 | - |
| 5 | 9.4 | > 10 |
| 6 | >10 | 2.8 |
| Paclitaxeld | 0.0006 | - |
| Camptothecind | 0.06 | - |
| Rocaglamided | - | 0.08 |
Results are expressed as ED50 values (μM).
Compounds 1, 2, 6, and 7 were considered to be inactive against HT-29 cells (ED50 >10 μM).
Compounds 1, 2, 4, and 7 were not tested due to the limited amount isolated.
Used as positive control substances.
The relative configuration of compound 1 was established by NOESY analysis as well as by chemical transformation. The configuration of the 1(10)-double bond was deduced as E, based on the observed NOE effects from H-14 to H-2, H-9α, as well as from H-1 to H-3β and H-9β, and the Z configuration of 5(6)-double bond was suggested by NOE correlations between H-5 with H-14 and H-3α. In order to confirm the configuration of these two double bonds, an intramolecular [2+2] addition reaction was induced by dissolving compound 1 in deuteriobenzene and keeping the solvent exposed to daylight for three days, to yield a known bourboneolide isomer (1a) as the major product. This chemical transformation also implied that the five-membered lactone ring and H-1 of compound 1 are β-oriented, while the methyl (C-14) on C-10 and H-5 are α-oriented relative to the main plane (see Fig. 1), which were consistent with deductions made by analysis of the NOESY spectrum. Furthermore, the β orientation of the acetyl group on C-2, as well as the α orientation of the methacrylate group on C-8, were deduced from NOE effects between H-2 with H-14 and H-3α, and between H-8 with H-13, respectively. All of this information showed the relative configuration of compound 1 to be identical with that of known isorolandrolides. Thus, the structure of compound 1 was determined as 13-methoxyisorolandrolide.
Fig. 1.
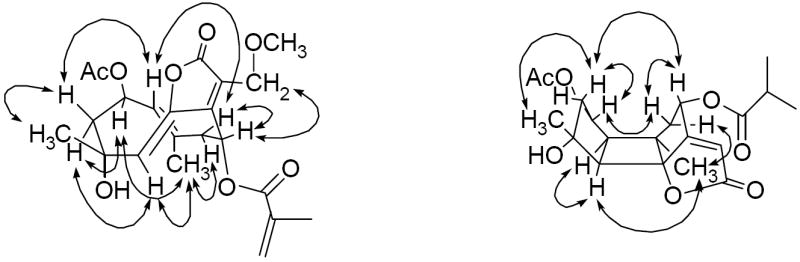
Key NOESY correlations of 1 and 2
In order to prove that compound 1 was not an artifact produced during extraction with methanol, a small amount of the plant material (10 g) was extracted with ethanol. Then, 10% water was added to the ethanol extract, and the latter was partitioned with hexane. The hexane solution was evaporated under reduced pressure to yield 200 mg of a hexane-soluble extract. A highly concentrated solution (100 mg/mL) of this hexane soluble extract was prepared and analyzed by HPLC using MeOH-H2O (50:50; 1.5 ml/min). Compound 1 (tR 24.7 min) was detected in this hexane-soluble extract. Thus, compound 1 was determined to be a naturally occurring metabolite rather than a methylated artifact.
Compound 2 was obtained as a colorless resin. The molecular formula was determined as C23H30O9, based on the sodiated molecular ion peak at m/z 473.1772 (calcd for C23H30O9Na) in the HRESIMS. The IR spectrum showed absorptions for hydroxy (3479 cm-1), carbonyl (1768, 1740 cm-1), and methyl (1367 cm-1) groups. In the 1H NMR spectrum (Table 1), two tertiary methyls (δH 1.24 and 1.31, s, each 3H), two acetyl groups (δH 2.04 and 2.07, s, each 3H), as well as an isobutyryl group (δH 1.20 and 1.21, each 3H, d, J = 6.9 Hz; δH 2.61, 1H, hep, J = 6.9 Hz) were recognized in the high-field region. Two proton signals at δH 5.38 (1H, d, J = 7.0 Hz, H-2) and δH 6.23 (1H, dd, J = 9.2, 8.2 Hz, H-8) suggested the presence of two esterified oxymethines, and resonances at δH 4.77 and 4.93 (each 1H, J = 12.6 Hz, H-13) were ascribed to an oxymethylene group. In the 13C NMR spectrum, corresponding carbonyl groups of two acetyl groups and an isobutyrate group mentioned above were observed at δC 170.1, 170.4, and 176.0, respectively. Except for these three substituents, the 15 carbon signals that remained in the 13C NMR spectrum were assigned as skeletal carbons with the aid of the DEPT and HSQC NMR data. On comparing the skeletal 13C NMR signals of compound 2 with those of compound 1, a similar α-oxymethylene-α,β-unsaturated-γ-lactone moiety was evident based on the chemical shifts of a carbonyl group at δC 170.1, a tetrasubstituted double bond at δC 169.0 and 123.6, and an oxymethylene at δC 55.2. Instead of 13C NMR resonances belonging to two trisubstituted endocyclic double bonds in compound 1, signals for two methines (δC 51.0, C-1; δC 59.5, C-5), an oxygenated carbon (δC 92.0, C-6), and a tertiary alkyl carbon (δC 47.8, C-10) were assigned for compound 2. Furthermore, considering the unsaturation requirement from the molecular formula, a three-ring carbon skeleton was necessary to complete the molecular construction. Analysis of the above data indicated that compound 2 is a bourbonenoid sesquiterpene ester (Jakupovic et al., 1989). The position of the isobutyrate group was designated at C-8 based on a key HMBC correlation between H-8 with the carbonyl carbon that resonated at δC 176.0. The observed HMBC correlations between H-2 and the protons signal of the acetyl group at δH 2.07 with the carbonyl group at δC 170.1, and H-13 and the protons signal of the acetyl group at δH 2.04 with the carbonyl group at δC 170.4, led to the location of two acetyl groups on C-2 and C-13, respectively. The relative configuration of compound 2 was the same as that of known bourbonenoid analogs, as a result of the NOE effects between H-1 with H-14 and H-5, H-2 and H-8 with H-3β and H-9β, respectively, and H-15 with H-3β (see Fig. 1). Therefore, the structure of compound 2 was determined as 2α,13-diacetoxy-4α-hydroxy-8α-isobutyroyloxybourbonen-12,6α-olide.
2.2 Biological activity evaluation
All pure isolates obtained in the present investigation were evaluated for their cytotoxic activity against the HT-29 human colon cancer cell line (Table 2). Among these compounds, 13-acetoxyrolandrolide (3), was found to be a highly active principle with an ED50 = 0.16 μM, with compound 4 (8-desacyl-13-acetoxyrolandrolide-8-O-tiglate) being considerably less cytotoxic. The glaucolide analog 5 showed only marginal cytotoxicity, while the three bourbonenolides (2, 6 and 7) were considered to be inactive (ED50 >10 μM) (Table 2). An enzyme-based ELISA NF-κB assay was employed to test the p65 (RelA) inhibitory activity of three major principles, 3, 5 and 6, and compounds 3 and 6 demonstrated moderate activities (Table 2). The major active compound, 13-acetylrolandralide (3) was evaluated in the hollow fiber assay, which is used as a secondary discriminator for substances sharing promising in vitro activity, in order to prioritize such substances for possible further testing in a relevant in vivo xenograft system (Mi et al., 2009). The human cancer cell lines evaluated using ip administration included LNCaP (hormone-dependent human prostate cancer), MCF-7 (human breast cancer) and HT29 for the in vivo hollow fiber assay. Administration of the highest two doses (10 and 5 mg/kg/day) of 3 resulted in significant body weight loss (up to 20%) in the host mice over the course of the study, and no inhibition of proliferation was observed in any of the cancer cell types. Therefore, 13-acetoxyrolandrolide (3) was not found to represent a promising oncology lead for further development on the basis of the present study.
2.3 Conclusion
The cytotoxicity screening data obtained in this investigation implied that a 1(10)-Z double bond and a 5(6)-E double bond in the cyclodecane ring, as present in the structures of compounds 3 and 4, is important for the cytotoxicity of the rolandrolide derivatives. The conversion of the configuration or the addition reaction of these two endocyclic double bonds will lead to the loss of cytotoxicity, as found for structures of the isorolandrolide analog 1, the glaucolide analog 5, and the bourbonenoid derivatives 2, 6 and 7. Moreover, the ester substituent on the α,β-unsaturated-γ-lactone moiety may have a significant impact on the cytotoxicity of rolandrolides. Thus, when the methacrylate group on C-8 in compound 3 was substituted by a tiglate group in compound 4, the potency of the inhibitory effect on HT-29 cells decreased around ten times. It may be pointed out that the ten-membered macrocyclic ring with a 1(10) double bond and a 5(6) double bond occurred in the structures of rolandrolides was found to be unstable. As a major constituent of R. fruticosa, 13-acetoxyrolandrolide (3) was partially transformed to compound 6 by a [2+2] addition, if kept in solvents such as CDCl3 and deuteriobenzene at room temperature. This might be a possible reason for the lack of activity in the in vivo hollow fiber assay observed for this compound.
3. Experimental
3.1 General experimental procedures
Melting points were measured on a Fisher Scientific melting point apparatus and are uncorrected. Optical rotations were recorded on a Perkin-Elmer 343 automatic polarimeter. UV spectra were run on a Shimadzu 160U UV-vis recording spectrophotometer. IR spectra were obtained on a ATI Mattson Genesis Series FT-IR spectrometer. CD spectra were conducted on a JASCO J-810 spectropolarimeter. HRESIMS were acquired on a 3-Tesla Finnigan FTMS-2000 Fourier transform mass spectrometer. NMR spectroscopic data were obtained on a Bruker Avance DRX-600 MHz spectrometer (with TMS as an internal standard). Column chromatography was performed with 65-250 or 230-400 mesh silica gel (Sorbent Technologies, Atlanta, GA). Analytical thin-layer chromatography was conducted on precoated 250 μm thickness silica gel plates (UV254, glass backed, Sorbent Technologies, Atlanta, GA), and preparative thin-layer chromatography was performed on precoated 20 cm × 20 cm, 500 μm thickness silica gel plates (UV254, glass backed, Sorbent Technologies, Atlanta, GA). Analytical HPLC was conducted on a 150 mm × 4.6 mm i.d. Sunfire PrepC18 column (Waters, Milford, MA), and semi-preparative HPLC was conducted on 150 mm × 10 mm i.d. or 150 mm × 19 mm i.d., 5 μm Sunfire PrepC18 columns (Waters, Milford, MA), along with a Waters system equipped with a 600 controller, a 717 Plus autosampler, and a 2487 dual wavelength absorbance detector.
3.2. Plant material
The combined leaves and twigs of Rolandra fruticosa (500 g) were collected at Pangradin village, Jasinga, West Java by S. Riswan and the late Agus Ruskandi in August, 2005. A voucher specimen (acquisition no. 2285414) has been deposited in the John G. Searle Herbarium of the Field Museum of Natural History, Chicago, Illinois, where the identification of the species was confirmed.
3.3. Extraction and isolation
The dried and finely ground plant material (500 g) of R. fruticosa was extracted with methanol at room temperature three times (3 × 24 h). The solvent was evaporated under reduced pressure to yield 34 g of a crude extract, which was suspended in a methanol-water (9:1) mixture and then extracted with hexane (3 × 1 L) and chloroform (3 × 1 L), sequentially. The hexane-soluble extract (13 g) was obtained after evaporation of the hexane solution under reduced pressure. The chloroform partition was washed with 1% aqueous NaCl to yield 4.5 g of a partially detannified chloroform-soluble extract. Both the hexane-soluble and chloroform-soluble extracts were found to be active against the HT-29 cell line, with ED50 values of 4.5 μg/ml and 1.6 μg/ml, respectively. Part of the CHCl3 extract (4.0 g) was subjected to separation over a Si gel column and eluted with a CH2Cl2-acetone gradient solvent system (20:1 to pure acetone). Of the sub-fractions obtained, F01 and F02 were found to be the most active (both with ED50 values of 0.4 μg/mL). These two fractions were combined (600 mg) and chromatographed on a Sephadex LH-20 column to remove chlorophylls, using MeOH-H2O (90:10 to pure MeOH) for elution, then the residue (300 mg) was further purified repeatedly on a semi-preparative RP18 column (150 mm × 10 mm i.d.) by HPLC with MeOH-H2O (50:50; 6 mL/min) as solvent system, to give compounds 3 (tR 18.5 min; 50 mg), 5 (tR 11.7 min; 25 mg), 6 (tR 7.8 min; 10 mg), and a subfraction (F0104, tR 13.2 min; 4.0 mg), which was further purified by HPLC using CH3CN-H2O (45:55; 6 mL/min) as mobile phase, to give compound 2 (tR 11.4 min; 1.2 mg). The hexane-soluble extract was purified using a similar isolation procedure to the CHCl3-soluble extract. A portion of the hexane extract (12 g) was separated over a Si gel column with a gradient system of hexane–acetone for elution. Active subfractions F02 (ED50 1.9 μg/ml) and F03 (ED50 1.5 μg/ml) were combined (500 mg) and subjected to passage over a Sephadex LH-20 column, using MeOH-H2O (90:10 to pure MeOH) for elution. The chlorophyll-free fraction (210 mg) was then purified on a semi-preparative RP18 column (150 mm × 19 mm i.d.) by HPLC using MeOH-H2O (50:50; 10 mL/min) as solvent system. Compounds 3 (tR 46.5 min; 10 mg), 5 (tR 41.5 min; 12 mg), and 6 (tR 30.2 min; 5 mg) were also found to be major constituents of the active fractions of the hexane-soluble extract. In addition to these three compounds, compounds 1 (tR 60.7 min; 2.0 mg), 4 (tR 67.2 min; 2.5 mg) and 7 (tR 36.0 min; 1.5 mg) were obtained as three minor principles.
3.4. 13-Methoxyisorolandrolide (1)
White amorphous resin; [α]23 D −7.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 211 (4.60) nm, 313 (br, 3.89) nm; IR νmax (film) 3468, 3040, 2934, 1750, 1731, 1671, 1452, 1371, 1244, 1157, 1020, 954, 795 cm-1; CD (c 1.2×10-4 M, MeOH) λmax (Δε) 214 (+16.16), 240 (+14.14), 320 (−6.01) nm; HRESIMS m/z 443.1702 [M+Na]+, calcd for C22H28O8Na, 443.1682; 1H and 13C NMR data, see Table 1.
3.5. 2α,13-Diacetoxy-4α-hydroxy-8α-isobutyroyloxybourbonen-12,6α-olide (2)
Corlorless resin; [α]23 D −83 (c 0.04, MeOH); UV (MeOH) λmax (log ε) 218 (br, 4.53) nm; IR νmax (film) 3479, 2966, 2923, 1768, 1740, 1455, 1367, 1246, 1148, 1025, 923 cm-1; CD (c 1.1×10-4 M, MeOH) λmax (Δε) 205 (+11.2), 240 (−36.98), 320 (+1.86) nm; HRESIMS obsd m/z 473.1772 [M+Na]+, calcd for C23H30O9Na, 473.1788; 1H and 13C NMR data, see Table 1.
3.6. 13-Acetoxyrolandrolide (3)
Colorless crystals; mp 169-170 °C (lit. 174-175 °C); [α]23 D +63.0 (c = 0.1, MeOH) (lit. +60.6); HRESIMS obsd m/z 471.1643 [M+Na]+, calcd for C23H28O9Na 471.1631; the spectroscopic data (1H and 13C NMR) were comparable with published values (Herz et al., 1981; Jakupovic et al., 1989).
3.7. 8-Desacyl-13-acetoxyrolandrolide-8-O-tiglate (4)
Colorless gum; [α]23 D +52.0 (c = 0.04, MeOH); HRESIMS obsd m/z 485.1773 [M+Na]+, calcd for C24H30O9Na 485.1788; the spectroscopic data (1H and 13C NMR) were comparable with published values (Jakupovic et al., 1989).
3.8. 2-epi-Glaucolide E (5)
Colorless crystals; mp 156-157 °C (lit. 159 °C); [α]23 D −19.0 (c = 0.1, CHCl3) (lit. −25.0); HRESIMS obsd m/z 471.1646 [M+Na]+, calcd for C23H28O9Na 471.1631; the spectroscopic data (1H and 13C NMR) were comparable with published values (Jakupovic et al., 1989).
3.9. 2α,13-Diacetoxy-4α-hydroxy-8α-methacryloyloxybourbonen-12,6α-olide (6)
White amorphous powder; [α]23 D −60.0 (c = 0.1, MeOH) (lit. –70.0); HRESIMS obsd m/z 471.1637 [M+Na]+, calcd for C23H28O9Na 471.1631; the spectroscopic data (1H and 13C NMR) were comparable with published values (Jakupovic et al., 1989).
3.10. 2α,13-Diacetoxy-4α-hydroxy-8α-tigloyloxybourbonen-12,6α-olide (7)
White amorphous powder; [α]23 D −12.0 (c = 0.05, MeOH); HRESIMS obsd m/z 485.1789 [M+Na]+, calcd for C24H30O9Na 485.1788; the spectroscopic data (1H and 13C NMR) were comparable with those published values (Jakupovic et al., 1989).
3.11. 2β-Acetoxy-4α-hydroxy-13-methoxy-8α-methacryloyloxycopa-7(11)-en-12,6α-olide (1a)
Compound 1 (0.8 mg) was dissolved in 2 mL of deuteriobenzene, and the solution was kept in a small glass vial and exposed to daylight for 72 hours at room temperature. Deuteriobenzene was evaporated to give 0.8 mg of residue, with bourboneolide isomer 1a obtained as a major compound (yield >90%). Pale yellow oil; [α]23 D −40.0 (c = 0.03, MeOH); HRESIMS m/z 443.1698 [M+Na]+, calcd for C22H28O8Na, 443.1682; the spectroscopic data (1H and 13C NMR) were comparable with those published values (Jakupovic et al., 1989).
3.12. Evaluation of cytotoxic activity
The cytotoxic activity of extracts, chromatographic fractions of extracts, and all pure compounds were evaluated against the HT-29 (human colon cancer) cell line, according to an established protocol (Seo et al., 2001).
3.13. Evaluation of NF-κB inhibitory activity
The NF-κB p65 subunit inhibitory activity of three major compounds (3, 5, and 6) was tested in an enzyme-based ELISA NF-κB assay, which was carried out according to an established protocol (Renard et al., 2001; Deng et al., 2009). Rocaglamide was used as a positive control.
3.14 Evaluation in the in vivo hollow fiber assay
The potential in vivo anticancer activity of 13-acetoxyrolandrolide (3) against HT-29, LNCaP, and MCF-7 human cancer cells was evaluated in the murine hollow fiber model, according to a procedure developed at the U.S. National Cancer Institute (NCI) (Hollingshead et al., 1995; Mi et al., 2009). Based on our previous MTT assay results, a dose range from 0.5 to 10 mg 13-acetylrolandrolide (3) per kg per day for four days was used. The vehicle was 5% EtOH and 5% Tween 80 in physiologic saline, and paclitaxel was used as the positive control (2.0 mg/kg per day for four days).
Acknowledgments
This study was supported by grants U19-CA52956 and P01-CA125066, funded by the National Cancer Institute, NIH, Bethesda, Maryland. We are grateful to the late Agus Ruskandi, Herbarium Bogoriense, Research Center for Biology, Indonesian Institute of Science, Bogor 16122, Indonesia, for assistance with the plant collection. We acknowledge the late Dr. Charles Cottrell, The Ohio State University (OSU) Campus Chemical Instrument Center, and John W. Fowble, College of Pharmacy, OSU, for the provision of NMR spectroscopic equipment used in this investigation. We are also appreciative to Mr. Justin S. Williams of the OSU Campus Chemical Instrument Center, for the mass spectrometric data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abosi AO, Raseroka BH. In vivo antimalarial activity of Vernonia amygdalina. Br J Biomed Sci. 2003;60:89–91. doi: 10.1080/09674845.2003.11783680. [DOI] [PubMed] [Google Scholar]
- Awaad AS, Sokkar NM, Khafaga HS. Pharmacognostical studies and biological activity for Vernonia galamensis sub-species galamensis var. petitiana (A. Rich.) M. Gilbert. Bull Fac Pharm. 2000;8:107–113. [Google Scholar]
- Bessong PO, Rojas LB, Obi LC, Tshisikawe PM, Igunbor EO. Further screening of Venda medicinal plants for activity against HIV type 1 reverse transcriptase and integrase. Afr J Biotechnol. 2006;5:526–528. [Google Scholar]
- Bohlmann F, Jakupovic J. Progress in the chemistry of the Vernonieae (Compositae) Plant System Evol Suppl. 1990;4:3–43. [Google Scholar]
- Da C, Fernando B, Terfloth L, Gasteiger J. Sesquiterpene lactone-based classification of three Asteraceae tribes: a study based on self-organizing neural networks applied to chemosystematics. Phytochemistry. 2005;66:345–353. doi: 10.1016/j.phytochem.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Deng Y, Balunas MJ, Kim J-A, Lantvit DD, Chin Y-W, Chai H-B, Sugiarso S, Kardono LBS, Fong HHS, Pezzuto JM, Swanson SM, Carcache-Blanco EJ, Kinghorn AD. Bioactive 5,6-dihydro-α-pyrone derivatives from Hyptis brevipes. J Nat Prod. 2009;72:1165–1169. doi: 10.1021/np9001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke JA. Ethnobotanieal observations on the Cuna Indians. Econ Bot. 1975;29:278–293. [Google Scholar]
- Herz W, Govindan SV, Blount JF. Structures of the rolandrolides and isorolandrolides, unusual germacradienolides from Rolandra fruticosa. J Org Chem. 1981;46:761–765. [Google Scholar]
- Jakupovic J, Grenz M, Bohlmann F, Wasshausen DC, King RM. Sesquiterpene lactones from Rolandra fruticosa. Phytochemistry. 1989;28:1937–1941. [Google Scholar]
- Hollingshead MG, Alley MC, Camalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. In vivo cultivation of tumor cells in hollow fibers. Life Sci. 1995;57:131–141. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- Kinghorn AD, Carcache-Blanco EJ, Chai H-B, Orjala J, Farnsworth NR, Soejarto DD, Oberlies NH, Wani MC, Kroll DJ, Pearce CJ, Swanson SM, Kramer RA, Rose WC, Emanuel S, Vite GD, Jarjoura J, Cope FO. Discovery of anticancer agents of diverse natural origin. Pure Appl Chem. 2009;81:1051–1063. doi: 10.1351/PAC-CON-08-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latha RM, Geetha T, Varalakshmi P. Effect of Vernonia cinerea Less. flower extract in adjuvant-induced arthritis. Gen Pharmacol. 1998;31:601–606. doi: 10.1016/s0306-3623(98)00049-4. [DOI] [PubMed] [Google Scholar]
- Mazumder UK, Gupta M, Manikandan L, Bhattacharya S, Haldar PK, Roy S. Evaluation of anti-inflammatory activity of Vernonia cinerea Less. extract in rats. Phytomedicine. 2003;10:185–188. doi: 10.1078/094471103321659915. [DOI] [PubMed] [Google Scholar]
- Mi Q, Pezzuto JM, Farnsworth NR, Wani MC, Kinghorn AD, Swanson SM. Use of the in vivo hollow fiber assay in natural products anticancer drug discovery. J Nat Prod. 2009;72:573–580. doi: 10.1021/np800767a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njan AA, Adzu B, Agaba AG, Byarugaba D, Diaz-Llera S, Bangsberg DR. The analgesic and antiplasmodial activities and toxicology of Vernonia amygdalina. J Med Food. 2008;11:574–581. doi: 10.1089/jmf.2007.0511. [DOI] [PubMed] [Google Scholar]
- Njoroge GN, Bussmann RW. Ethnotherapeutic management of skin diseases among the Kikuyus of Central Kenya. J Ethnopharmacol. 2007;111:303–307. doi: 10.1016/j.jep.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Okolie UV, Okeke CE, Oli JM, Ehiemere IO. Hypoglycemic indices of Vernonia amygdalina on postprandial blood glucose concentration of healthy humans. Afr J Biotechnol. 2008;7:4581–4585. [Google Scholar]
- Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29:e21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson H, Bohlmann F, King RM. Chemosystematic notes on the Asteraceae III. Natural subdivisions of the Vernonieae. Phytologia. 1980;46:421–436. [Google Scholar]
- Seo E-K, Kim N-C, Mi Q, Chai H, Wall ME, Wani MC, Navarro HA, Burgess JP, Graham JG, Cabieses F, Tan GT, Farnsworth NR, Pezzuto JM, Kinghorn AD. Macharistol, a new cytotoxic cinnamylphenol from the stems of Machaerium aristulatum. J Nat Prod. 2001;64:1483–1485. doi: 10.1021/np0103158. [DOI] [PubMed] [Google Scholar]
- Smithsonian Tropical Research Institute’s Herbarium (SCZ) [January 2009]; Further information available at http://biogeodb.stri.si.edu/herbarium/species/1748/?fam=Asteraceae&page=12.
- Uhegbu FO, Ogbuehi KJ. Effect of aqueous extract (crude) of leaves of Vernonia amygdalina (Del.) on blood glucose, serum albumin and cholesterol levels in diabetic albino rats. Global J Pure Appl Sci. 2004;10:189–194. [Google Scholar]
- van Andel T, Behari-Ramdas J, Havinga R, Groenendijk S. The medicinal plant trade in Suriname. Ethnobot Res Appl. 2007;5:351–372. [Google Scholar]
- Walewa EO, Iwalewa OJ, Adeboye JO. Analgesic, antipyretic, anti-inflammatory effects of methanol, chloroform and ether extracts of Vernonia cinerea Less. leaf J Ethnopharmacol. 2003;86:229–234. doi: 10.1016/s0378-8741(03)00081-3. [DOI] [PubMed] [Google Scholar]
- Woodson RE, Schery RW, Jr, D’Arcy WG, Elias TS, Busey P, King RM, Robinson H, Stuessy TF, Canne JM, Keil DJ, Barkley TM, Gardner RC, Simpson BB, Tomb AS. Flora of Panama. Part IX. Family 184. Compositae. Ann Missouri Bot Gard. 1975;62:835–1321. [Google Scholar]