

Abstract
Smooth muscle myosin light chain kinase (smMLCK) is a calcium/calmodulin dependent enzyme that activates contraction of smooth muscle. The polypeptide chain of rabbit uterine smMLCK (Swiss-Prot: P29294) contains the catalytic/regulatory domain, three immunoglobulin related motifs (Ig), one fibronectin related motif (Fn3), a repetitive, proline rich segment (PEVK) and, at the N-terminus, a unique F-actin binding domain. We have evaluated the spatial arrangement of these domains in a recombinant 125 kDa full-length smMLCK and its two catalytically active C-terminal fragments (77 kDa, residues 461-1147 and 61 kDa, residues 461-1002). Electron microscopic images of smMLCK cross-linked to F-actin show particles at variable distance (11-55 nm) from the filament, suggesting that a well-structured C-terminal segment of smMLCK is connected to the actin-binding domain by a long, flexible tether. We have used structural homology and molecular dynamics methods to construct various all-atom representation models of smMLCK and its two fragments. The theoretical sedimentation coefficients computed with the program HYDROPRO were compared with those determined by sedimentation velocity. We found agreement between the predicted and observed sedimentation coefficients for models in which the independently folded catalytic domain, Fn3 and Ig domains are aligned consecutively on the long axis of the molecule. The PEVK segment is modeled as an extensible linker that enables smMLCK to remain bound to F-actin and simultaneously activate the myosin heads of adjacent myosin filaments at a distance of 40 nm or more. The structural properties of smMLCK may contribute to the elasticity of smooth muscle cells.
Keywords: smooth muscle regulation, analytical ultracentrifugation, electron microscopy, actin binding, homology modeling, zero length cross-linking, Ig domain, Fn3 domain
The primary pathway for the regulation of contraction in smooth muscles involves the Ca2+/calmodulin dependent activation of myosin light chain kinase (MLCK). Upon stimulation of a smooth muscle cell, Ca2+ is transiently elevated in the cytoplasm by entering from the extracellular fluid or from intracellular stores via the action of inositol 1,4,5-trisphosphate. The Ca2+/CaM complex binds to and activates MLCK, which in turn phosphorylates the 20 kDa regulatory light chain of myosin (LC20) at Ser19 and Thr18, leading to the activation of myosin ATPase by actin and to muscle contraction.
The phospho-transferase catalytic function of MLCK is localized in a ∼300 residues long (∼35 kDa) structurally conserved domain (1, 2) that exhibits high sequence similarity to other protein kinases (3). The function of this domain is regulated by the intra-steric inhibition involving a pseudo-substrate segment located at the C-terminus of the domain. The inhibition is released upon Ca2+/CaM binding to the adjacent specific CaM binding site (4-7)). The polypeptide chain of rabbit skeletal MLCK (Swiss-Prot: P07313) contains 607 residues (8), which is twice as long as the minimum required for the catalytic function. Much longer are the polypeptide chains of smooth muscle MLCK, e.g. chicken gizzard (P11799-2) (9) or rabbit uterine MLCK (P29294) (10), which contain 972 and 1147 amino acids, respectively. The largest known MLCK is the 210 kDa nonmuscle isoform (e.g. Q15746-1, P11799-1), (see Gallagher et al. for a review (7)). The functional significance of such complex structures is unclear.
Analysis of the amino acid sequence of rabbit uterine MLCK indicates several distinct domains in the 1147 amino acid polypeptide chain of this protein (10). It has been established that smMLCK binds both F-actin (11) and unphosphorylated smooth muscle myosin with micromolar Kd (12). The F-actin-binding site has been localized at the N-terminus of smMLCK (13, 14) and shown to involve three DFRxxL motifs (15). The 3D reconstructions of F-actin decorated with the N-terminal 147 residues fragment of smMLCK showed MLCK density on the extreme periphery of subdomain-1 of each actin monomer forming a bridge to subdomain-4 of the azimuthally adjacent actin (16). This unique location enables MLCK to bind to actin without interfering with the binding of any other key actin-binding proteins, including myosin, tropomyosin, caldesmon, and calponin. Next to the actin binding domain there is a segment containing 16 repeats of a proline rich 12-residue motif TLKPV(G,A)N(A,I,T)KPAE (10). A similar sequence occurs in smooth muscle MLCK from bovine stomach and a shorter one in MLCK isolated from human brain. This segment is absent from chicken gizzard and from skeletal muscle MLCK. This region has been referred to as the PEVK region by analogy to the Pro, Glu, Val and Lys rich segment of the giant protein titin (17-19). Adjacent to the repetitive segment there is a 90 residue long immunoglobulin related domain (Ig1) followed by a linker that is readily cleaved by proteolytic enzymes (20, 21). Further downstream is another Ig domain (Ig2) and a fibronectin type III related motif (Fn3). The catalytic and regulatory domains that follow the Ig2-Fn3 tandem are by far the most studied parts of MLCK (for reviews see (7, 22)). Finally, at the C-terminus there is one more Ig motif (Ig3) terminated with a stretch of Glu residues (polyE). The 154-residue C-terminal segment of smMLCK containing the Ig3 domain is also expressed independently of the full-length MLCK and is referred to as telokin (23) or kinase related protein (KRP) (24). Telokin was found to promote filament formation of unphosphorylated myosin in the presence of ATP presumably through its binding to the head-tail junction of myosin (25). Telokin was found to decrease Km but not Vmax of myosin phosphorylation by MLCK (25). It appears that the C-terminal extension of smMLCK corresponding to telokin also interacts with myosin but the significance of this interaction for the catalytic activity of MLCK is not clear. Telokin is the only part of smMLCK whose high-resolution structure has been solved (26). The presence of the specific actin and myosin binding sites at the extreme termini of the polypeptide chain suggests that in smooth muscle smMLCK might interact simultaneously with both thin and thick filament. Stull et al. considered such a possibility in their 1998 review article (27). They pointed out that if the independently folded Ig, Fn3 and catalytic domains of smMLCK are arranged in a linear fashion, while the remaining segments are in an extended conformation, the molecule could span over 600Å and bridge readily the thick and thin filaments in smooth muscle cells. This hypothesis is consistent with the elongated shape in solution of skeletal and turkey gizzard MLCK as previously determined by analytical ultracentrifugation (2, 28, 29). It is also consistent with the observation that smMLCK is virtually immobilized in cultured smooth muscle cells (30). However, the postulated linear arrangement of the domains, the flexibility of the unstructured segments and their contribution to the overall shape and hydrodynamic properties of smMLCK have not been rigorously evaluated.
In the present studies we have used electron microscopy, analytical ultracentrifugation, structural homology and hydrodynamic modeling to obtain information about molecular shape and domain arrangement in recombinant rabbit smooth muscle MLCK and its two catalytically active fragments. We have found that the hydrodynamic properties of smMLCK are consistent with a modular structure in which the C-terminal part of the molecule is built of well-structured domains arranged in a linear fashion. This part of smMLCK is connected to the N-terminal actin-binding site through a flexible and extensible segment, the PEVK region. The structural properties of smMLCK appear to facilitate the preferential activation of those myosin heads that are in contact with F-actin filaments and to contribute to the elasticity of smooth muscle cells.
MATERIALS AND METHODS
Cloning and overexpression of the rabbit uterine MLCK in insect cells
We have overexpressed the full-length rabbit uterine smooth muscle MLCK in insect cells. A 3.4 kb cDNA comprising the coding region of smMLCK was constructed from two overlapping cDNA fragments (N-terminal 2.48 kb and C-terminal 1.52 kb in pGem vector) generously provided by Dr. P.J. Gallagher and Dr. J.T. Stull. After removing the overlapping sequence and ligation of the complementary fragments the full-length cDNA was subcloned into baculovirus transfer vector pBlueBac4 (Invitrogen) at BamH1 and Kpn1 sites and co-transfected to SF9 insect cells using BAC-N-Blue (linear AcMVNPV DNA) transfection kit (Invitrogen). The presence of smMLCK DNA was confirmed by PCR analysis of viral DNA and the expression of smMLCK was confirmed by western blot. After propagation of high titer viral stock 500 ml of High Five insect cells suspension culture was infected with 10 ml of the viral stock and after 48 hours of culture at 28 °C cells were harvested by centrifugation. Cell pellet was suspended in 30 ml of solution containing 50 mM NaCl, 50 mM MgCl2, 20 mM MOPS pH 7.0, 0.5 mM EGTA, 0.2 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 1 mM DTT, 10% glycerol, 0.04 mM leupeptin. MLCK was extracted by freeze and thaw of the cell suspension three times. After centrifugation of the extract at 100,000 × g for 30 minutes, clear supernatant was applied onto DEAE Sepharose CL 6B column equilibrated with a solution containing 50 mM NaCl, 30 mM MgCl2, 20 mM MOPS pH 7.0, 0.5 mM EGTA, 1 mM DTT, 0.2 mM AEBSF. Protein was eluted with a linear gradient of 0.05M - 0.45M NaCl. The peak containing MLCK was collected and further purified with the use of a CaM affinity column. The yield of MLCK was ∼10 mg. The enzymatic activity of MLCK was confirmed by myosin LC20 electrophoretic mobility shift assay.
In addition to the full-length MLCK we have generated and expressed in baculovirus its two fragments; a 77 kDa C-terminal fragment comprising residues 461-1147 (77k-MLCK) and a 61 kDa fragment (residues 461-1002) containing the catalytic /regulatory domain and the Ig2 and Fn3 motifs on the N-terminal side of the catalytic domain. This construct corresponds to the stable MLCK fragment that is generated by limited proteolysis from the full-length chicken gizzard MLCK (21). The 77k-MLCK and 61k-MLCK were expressed and purified in a similar manner as the full-length protein.
Other proteins
Actin from rabbit skeletal muscle was obtained from aceton-dried muscle powder essentially according to the method of Spudich and Watt (31) except for the 3 day dialysis step which was shortened to overnight dialysis with a single change of buffer. Recombinant human calmodulin was obtained as previously described (32).
Analytical Ultracentrifugation
Sedimentation velocity experiments were carried out on a Beckman Instruments Optima XL-I Analytical Ultracentrifuge equipped with a real-time video-based data acquisition system and Rayleigh optics. The cells were equipped with sapphire windows and 12 mm charcoal-filled epon centerpieces. Sedimentation velocity patterns were acquired every 8 seconds. Apparent sedimentation coefficient distribution patterns were computed by the time derivative method (33-35). All protein solutions were dialyzed against their respective buffers and dialysate was used for all dilutions. Molecular weights were computed from sedimentation velocity profiles using the software program SEDANAL, which employs a non-linear least squares curve fitting algorithm to fit to solutions of the differential equation (the Lamm equation) describing sedimentation. Fits were carried out on time difference data to remove the time independent systematic baseline components according to Stafford and Sherwood (36). Values of s and D obtained from the fitting procedure were substituted into the Svedberg equation to obtain the molar mass of the protein, M2:
where ρ is the density of the buffer and ν2 is the partial specific volume of the protein. Values of the partial specific volume, ν2, were computed from the amino acid sequence using the consensus partial volumes of Perkins (37). The axial ratios of the corresponding ellipsoids of revolution were calculated using Perrin’s equation for a prolate ellipsoid having semi-axes a, b, b: (cf. Eq. 19-14 in reference (38))
where f is the observed frictional coefficient and is computed from the experimentally observed values of M2 and so20,w as follows:
and f0 is the frictional coefficient of a sphere having the same mass and hydration as the protein and is given by:
where η0 is the viscosity of water at 20°C, δ1 is the hydration of the protein in units of grams of water per gram of protein, νo1 is the specific volume of pure water, and N is Avogadro’s number. Values of the hydration, δ1, were computed from the data of Kuntz and Kauzman (39). The dimensions of a protein molecule were obtained by equating its hydrated volume (V) to the volume of a corresponding prolate ellipsoid:
and using the axial ratio (b/a) obtained from Perrin’s equation above.
Rotary shadowing electron microscopy
Samples of smMLCK or its fragments were diluted to 4 nM concentration in solution containing 0.5 M ammonium acetate, 30% glycerol, 10 mM NaCl, and 3 mM MgCl2. smMLCK cross-linked to F-actin (5.7 μM with respect to the actin monomer) was diluted 5-fold in F-buffer, centrifuged at 3000 × g for 10 min and then the supernatant was further diluted in solution containing 0.5 M ammonium acetate (pH 7.0) and 30% glycerol. The purpose of the centrifugation was to remove aggregates and the high salt concentration was used to dissociate any un-cross-linked MLCK that might remain bound to F-actin during specimen preparation. The protein samples were adsorbed onto a freshly cleaved mica sheet, then stabilized by a treatment with uranyl acetate (40) and processed for rotary shadowing as previously described (41). EM specimens were observed under Philip 300A electron microscope at 60 kV
Zero-length cross-linking of smMLCK to F-actin
smMLCK was cross-linked to F-actin with the use of our EDC-NHS two-step zero length cross-linking procedure (42). In the activation step F-actin (1 mg/ml) in solution containing 0.1 M NaCl, 2 mM MgCl2, 0.2 mM ATP, 0.2 mM CaCl2, 20 mM MOPS pH 7.0 was incubated with 1 mM 1-ethyl-3-(dimethylaminopropyl)carbodiimide (EDC) and 2 mM N-hydroxysuccinimide (NHS) for 15 min followed by the addition of 5 mM β-mercaptoethanol to block the excess of EDC. Then, smMLCK was added from stock solution in 0.1 M NaCl, 2 mM MgCl2, 20 mM MOPS pH 7.0 to make the final molar ratio of smMLCK to actin equal 1:3. The solution was incubated for 1 hour at room temperature. Cross-linking was verified by SDS polyacrylamide gel electrophoresis. The cross-linked proteins were analyzed by rotary shadowing electron microscopy.
Molecular modeling
For modeling the catalytic domain, the Ig and Fn3 domains, we have used the Swiss Model server (http://swissmodel.expasy.org/SWISS-MODEL.html) in automatic and interactive modes facilitated by the Swiss PDB viewer program (43-47). Progressively more complex structures (61k- 77k- and smMLCK) were built manually from individual domains with the program O (48). The structure of each construct was energy minimized and subjected to molecular dynamics simulation in vacuo using the program CNS (49). The N-terminal 99-residue actin binding region and the short inter-domain linker segments were first modeled in the extended conformation and then subjected to molecular dynamics simulation for 100ps, which caused the extended chain to collapse into a random and relatively compact form. The 16x12 tandem repeat (residues 100-288) was modeled initially as a poly-proline type II helix (ϕ=-75° ψ=145° ω=180°) and also subjected to molecular dynamics for 50ps, 200ps and 300ps leading to different levels of structure collapse and randomization. These partially randomized structures were used to construct models of the full-length smMLCK. The theoretical sedimentation coefficients for the modeled proteins were calculated with the program HYDROPRO (50) using the full set of atomic coordinates as input for each model.
RESULTS
Recombinant smMLCK and its C-terminal catalytically active fragments
In this study we have used purified recombinant rabbit uterine MLCK overexpressed in insect cells with the use of a baculovirus expression system. In addition to the full-length smMLCK we have also constructed two fragments comprising the C-terminal part of the molecule: the 77k-MLCK (residues 461-1147) and the 61k-MLCK (residues 461-1002) (Figure 1). The 61k-MLCK corresponds approximately to the proteolysis resistant catalytically active core domain of smMLCK (1, 20, 51). It comprises the Ig2-Fn3 tandem and the kinase/regulatory domain including the autoinhibitory segment and the CaM binding site. The 77k-MLCK contains also the telokin region (Ig3) and the C-terminal poly-Glu segment. An illustration of typical purification steps and the quality of the obtained protein preparations is shown in Figure 2. All three proteins have the full Ca2+/CaM regulated kinase activity with respect to both smooth muscle myosin and the isolated regulatory light chain LC20. Turnover rates measured under the conditions of low temperature (4 °C) and low substrate concentration (0.5 μM LC20) are: 15.3 min−1, 22.0 min−1 and 10.0 min−1 for the full-length, 77k- and 61k–MLCK, respectively 2. These rates correspond to the extrapolated Vmax values in the range of 10-25 μ mol/min/mg (at 25 °C and infinite substrate concentration, assuming Kd(LC20)=10 μM), which is similar to the published values for smMLCK expressed in COS cells (10, 52)
Figure 1.

Structural domains of smMLCK. Domain definition based on ref. (9, 10). The central segment marked by the arrows corresponds to the trypsin resistant fragment of smMLCK, which is equivalent to the recombinant 61k-MLCK used in this study. This fragment comprises the Ig2-Fn3 tandem and the catalytic/regulatory domain. The 77k-MLCK has the same N-terminus as the 61k fragment and extends to the C-terminus of smMLCK to include the Ig3 (telokin) region. The atomic models of the Ig2-Fn3 tandem and the catalytic/regulatory domain obtained by homology modeling are shown in ribbon representation (see text and Table 2 for details).
Figure 2.
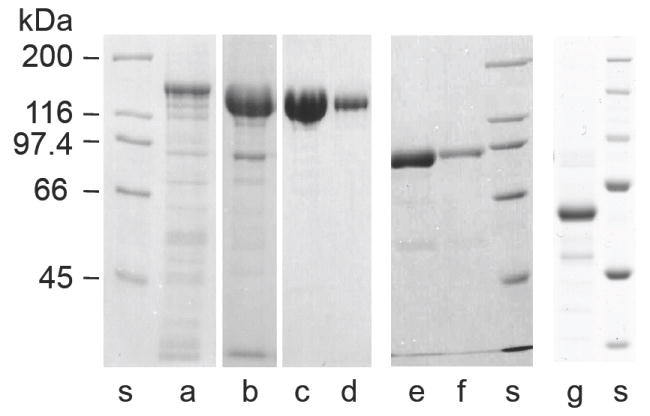
Purification of the full-length mammalian smMLCK and its C-terminal active fragments overexpressed in insect cells. Representative examples of electrophoresis on SDS-polyacrylamide gel (8%) of smMLCK samples at various steps of purification are shown: (a) - extract of soluble proteins from High Five cells expressing smMLCK, (b) - peak fraction from DEAE Sepharose CL 6B column, (c, d) - smMLCK eluted in EGTA from CaM -affinity column (two different loads are shown). The 77k-MLCK (e,f) and 61k-MLCK (g) fragments of smMLCK were obtained with similar yields. A molecular weight standard (Bio-Rad) is shown for each preparation (s).
Electron microscopic images of smMLCK bound to F-actin
We have examined the interaction of the full-length smMLCK with F-actin by rotary shadowing electron microscopy. To overcome the problem of MLCK dissociation from F-actin during specimen preparation we have cross-linked smMLCK to F-actin with the use of the two-step zero-length cross-linking procedure (42). In this method one component of the protein complex is briefly activated with a water-soluble carbodiimide (EDC) in the presence of N-hydroxysuccinimide (NHS). This converts some exposed carboxyl groups into N-succinimidyl active esters, which can cross-link to nearby Lys side chains or hydrolyze back to the original carboxyls. The second protein component of the complex is added after the activation step is terminated by the addition of β-mercaptoethanol, which blocks the remaining EDC. The cross-linking occurs during the subsequent 1-2 hour incubation. The advantage of this procedure is that only one component of the complex is exposed to the cross-linker, thus avoiding the possibility of intra-molecular cross-linking of smMLCK. We have found extensive smMLCK-actin cross-linking when F-actin was activated with EDC/NHS, whereas no cross-linking occurred when smMLCK was activated.
The F-actin cross-linked smMLCK is visible in the rotary shadowing EM images as an array of elongated and often curved or oval shaped objects positioned at variable distances from the filament (Figure 3). Apparently these objects are tethered to F-actin by a segment of the molecule that is too thin to be visualized by rotary shadowing. Clearly, the distribution of mass in smMLCK is not uniform. There is a long, thin, and flexible linker between the actin cross-linking site and the well-structured part of smMLCK. To assess the length of the linker region we have measured the distances between the edge of the F-actin filament and the most distant visible parts of the cross-linked MLCK. The measured distances fall in a broad range of 11 to 55 nm, with the average distance of 28.3±9.9 nm (n=180). The distribution appears to be bimodal with a broad peak between 16-30 nm and a second peak at ∼40 nm (Figure 4). Several reasons could account for such a broad range of the observed lengths of F-actin cross-linked smMLCK. First, the cross-linking may, in principle, involve non-specific random interaction sites distributed along the polypeptide chain of MLCK. Although we have not verified this possibility, we can exclude the C-terminal 60% of the molecule as potentially involved in the cross-linking, since the 77 kDa C-terminal fragment of MLCK does not cross-link to F-actin under the same conditions (data not shown). This observation suggests that the cross-linking requires a specific interaction site having a well-defined polarity of electrostatic contacts, i.e. F-actin contributing negatively charged groups. Thus, the cross-linking most likely involves the specific actin-binding site at the N-terminus of smMLCK identified by Smith et al. (15). The visible oval-shaped objects apparently represent the C-terminal part of smMLCK including the catalytic domain. Secondly, the thickness of F-actin and the helical distribution of the binding sites on the actin monomers along the filament may contribute to the apparent length of the bound MLCK. The measured width of the F-actin filament in Figure 3 is 15.9±1.2 nm, thus a fully extended MLCK molecule attached at the distal side of the filament may appear up to 16 nm shorter than that attached to the proximal side. Thirdly, it is impossible to assess whether a molecule extends from the filament in a perpendicular or other directions, since the linker is not visible. Thus, the measured distance from the filament may be shorter than the actual length of the molecule, if it extends at an angle with respect to the filament. Finally, the structural extensibility of the linker or other parts of smMLCK may contribute to the broad length distribution. Figure 3 also suggests that the distribution of smMLCK on actin may be nonrandom. Highly decorated filaments are visible next to segments that are free of MLCK. Also, when the filaments are highly decorated with the cross-linked MLCK the spacing between the MLCK molecules appears to be rather uniform. The reasons for such non-random distribution are not clear. In Figure 3 there are sections of F-actin, which appear to have MLCK only on one side, while the opposing site is free of MLCK. We believe such an appearance is caused by flow of solvent during specimen preparation, which orients tethered MLCK molecules on one side of the filament.
Figure 3.
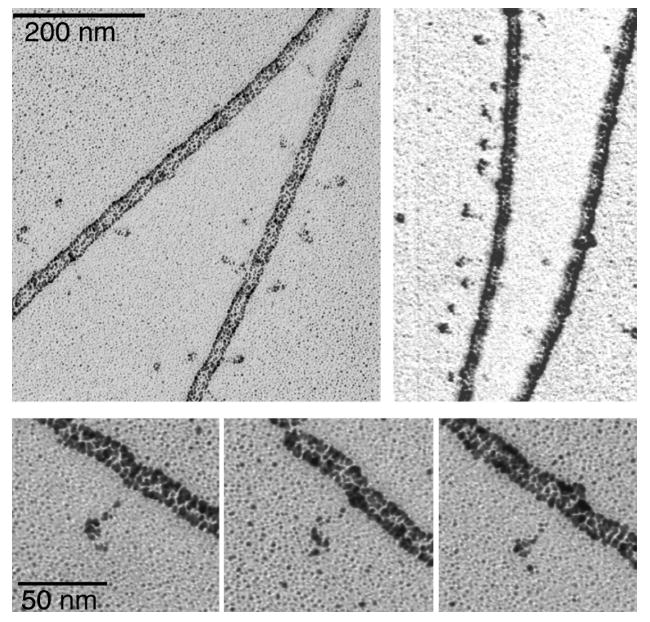
Rotary shadowing images of smMLCK zero-length cross-linked to F-actin. Note particles that appear to be tethered to the filament. The distance between these particles and the actin filament is highly variable.
Figure 4.
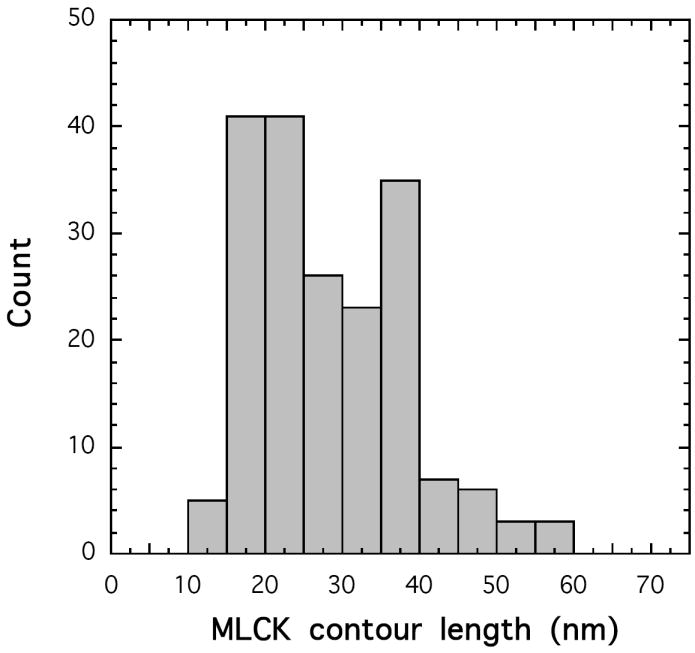
Contour length distribution of smMLCK cross-linked to F-actin. The length is measured from the edge of the actin filament to the most distant distinguishable parts of the tethered smMLCK. The distribution of distances is bimodal with a broad peak between 16-30 nm and a second peak at 40 nm. The width of the actin filament image is 15.9 ± 1.2 nm
The rotary shadowing EM technique has been used previously to visualize chicken gizzard MLCK by Numata et al (53). They reported that the molecule is flexible, since several distinct shapes can be distinguished ranging from fully extended to highly compact structures. We have also used rotary shadowing technique to visualize our recombinant rabbit smooth muscle MLCK (Figure 5) and found the results to be consistent with those of Numata et al. Images of smMLCK could be roughly subdivided into two classes: an extended conformation with the contour length of 36.4±5.0 nm (SD) and a compact conformation with the contour length of 23.9±3.2 nm. The corresponding widths are 11.9±1.5 nm and 14.8±2.4 nm for the extended and compact conformations, respectively. The rotary shadowing images of the 77k-MLCK (Figure 6) displayed significantly less variability in shape than those of smMLCK. The majority of molecules were extended with an average contour length of 25.0±2.9 nm and average width of 11.6±1.9 nm. A comparison of rotary shadowing images of smMLCK alone, whether chicken gizzard or recombinant rabbit smooth muscle, with those of smMLCK cross-linked to F-actin suggests that the N- and C-terminal parts of the molecule have very different properties and only the latter is visible in rotary shadowing images.
Figure 5.
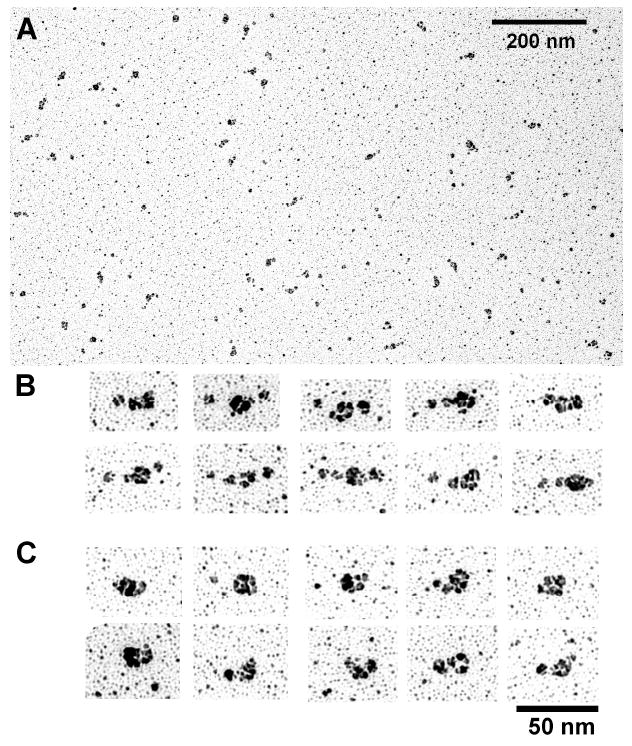
Rotary shadowing images of smMLCK. A low magnification field (A) and ensembles of high magnification images of single molecules in the extended (B) and compact (C) conformation are shown. The measured contour length for the extended conformation is Le=36.4±5.0 nm and for the compact conformation is Lc=23.9±3.2 nm. The corresponding widths are We=11.9±1.5 nm and Wc=14.8±2.4 nm.
Figure 6.
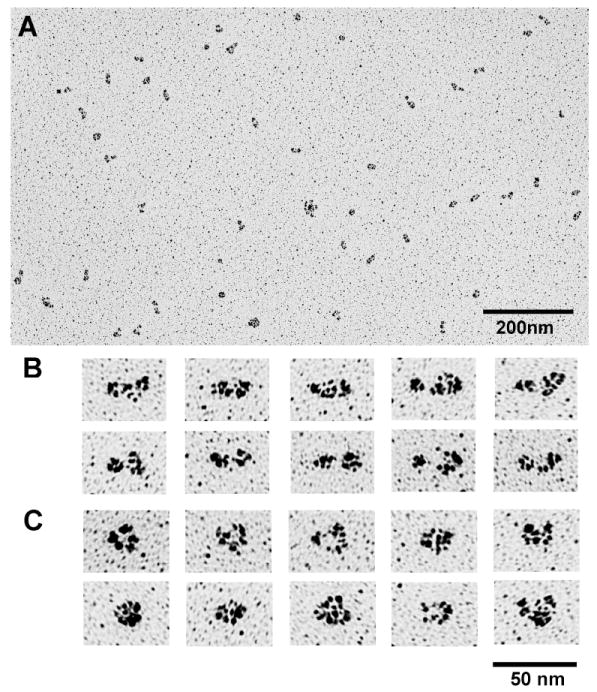
Rotary shadowing images of the 77k fragment of smMLCK. The low (A) and high (B,C) magnification images are shown. The average contour lengths are Le=25.0±2.9 nm and Lc=18.6±2.0 nm, for the extended (B) and compact (C) conformation, respectively. The corresponding widths are We=11.6±1.9 nm and Wc=15.6±1.8 nm.
It is instructive to compare the images of rabbit smMLCK cross-linked to F-actin (Figure 3) with those of gizzard MLCK cross-linked to the HMM part of myosin obtained by Numata et al. (53). In the latter case MLCK is visible as a globular particle similar in size to the myosin heads and attached to the neck region of HMM. Clearly, the site of MLCK cross-linking to actin is very different from that involved in cross-linking to HMM. The former is located next to a flexible part of the molecule while the latter is located at, or next to a well-structured part of MLCK. This conclusion is consistent with the location of the respective binding sites at the extreme N- and C- termini of smMLCK, as determined from biochemical studies, and supports the view that the cross-linking involves the respective specific interaction sites.
Molecular shape of smMLCK from sedimentation velocity
We have used sedimentation velocity to determine hydrodynamic parameters of smMLCK and its two C-terminal fragments, 77k-MLCK and 61k–MLCK. In view of the reported dimerization of chicken gizzard MLCK (54) that was modified by Ca2+/CaM (55, 56), we have tested the concentration dependence of the sedimentation velocity profile of smMLCK in the absence and presence of Ca2+/CaM. We find the sedimentation profile to be consistent with a monomeric state of the protein (Figure 7 and Table 1). Variation of concentration over a 10-fold range had no effect on the sedimentation coefficient (Figure 7a, dashed lines), indicating no self-association or non-ideality under these conditions. CaM binding causes a slight increase in the sedimentation constant consistent with the mass increase. Moreover, there is no evidence of dimer or oligomer formation. Also, no evidence of dimerization was found for the 77k-MLCK and the 61k-MLCK fragments (Figure 7).
Figure 7.
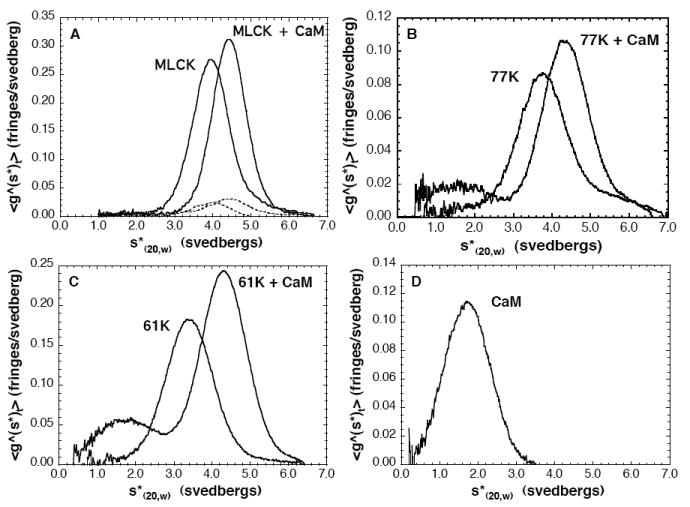
Sedimentation velocity profiles of smMLCK and its fragments in the absence and presence of CaM. A - the full-length smMLCK at 0.13 mg/ml (solid lines) and at 10× dilution (dashed lines); B - 77k-MLCK; C - 61k-MLCK; D - calmodulin. The protein samples (0.13-0.2 mg/ml) were dialyzed against a solution containing 0.1 M NaCl, 1 mM MgCl2, 20 mM MOPS pH 7.0 and 1 mM CaCl2.
Table 1.
Hydrodynamic parameters of the recombinant rabbit smooth muscle MLCK and its 61k and 77k C-terminal fragments alone and in complexes with CaM.
| MLCK | MLCK+CaM | 77k | 77k+CaM | 61k | 61k+CaM | CaM | |
|---|---|---|---|---|---|---|---|
| Number of amino acids | 1147 | 1295 | 687 | 835 | 542 | 690 | 148 |
| Hydration δ1 | 0.433 | 0.436 | 0.428 | 0.434 | 0.410 | 0.421 | 0.460 |
| Specific volume v2 | 0.737 | 0.735 | 0.731 | 0.730 | 0.735 | 0.732 | 0.723 |
| Mass calc. (kDa) | 125.7 | 142.4 | 77.3 | 94.0 | 61.2 | 77.9 | 16.7 |
| Mass obs. (kg/mole) | 126 | 142 | 77.3 | 90.8 | 63.2 | 79.7 | 17.1 |
| Sedimentation coef. s(20,w) | 3.94 | 4.42 | 3.78 | 4.32 | 3.42 | 4.30 | 1.72 |
| Frictional ratio f/fo | 1.92 | 1.86 | 1.48 | 1.44 | 1.41 | 1.32 | 1.20 |
| Axial ratio a/b | 18.0 | 16.8 | 8.8 | 8.2 | 7.7 | 6.2 | 4.2 |
| Length (Å) | 530 | 530 | 280 | 280 | 240 | 220 | 100 |
| Width (Å) | 30 | 32 | 32 | 34 | 31 | 36 | 25 |
| Stokes radius Rs (Å) | 74.3 | 75.2 | 48.5 | 50.4 | 43.4 | 44.1 | 23.8 |
The sedimentation velocity results indicate that recombinant rabbit smooth muscle MLCK is a highly asymmetric molecule with the observed mass of 126 kg/mol, sedimentation coefficient of s(20,w)=3.94 and frictional coefficient due to shape alone of f/fo=1.92 (Table 1). If modeled as a prolate ellipsoid of revolution using the Perrin equation, the molecule would have an axial ratio a/b=18.0, and the hydrodynamic length and width of 53.4 nm and 3.0 nm, respectively (Table 1). This is consistent with the results of Ausio et al. (29) who found the slightly smaller turkey gizzard MLCK (M=108 kg/mol) also to be highly asymmetric (s(20,w)=3.74, f/fo=1.95, a/b=18.9). Also asymmetric is MLCK isolated from rabbit skeletal muscle (2, 28). Much of the asymmetry of smMLCK must be attributable to the N-terminal part of the molecule since 77k-MLCK and 61k-MLCK have axial ratios of 8.8 and 7.7, respectively and the corresponding hydrodynamic lengths of 28 nm and 24 nm, respectively (Table 1). CaM binding causes a slight decrease in the axial ratios, which is attributable to an increase in the apparent widths. This is consistent with CaM binding to the catalytic domain of MLCK causing some structural rearrangement (57).
Structural homology modeling of smMLCK
Implicit in the calculation of the molecular dimensions of smMLCK from the sedimentation velocity data (Table 1) is the assumption that the shape of the molecule can be approximated by a regular geometrical shape such as a prolate ellipsoid. This approximation may yield a reasonable representation of globular proteins and those proteins that have uniform distribution of mass. Judging from the EM images in Figure 3, this is clearly not the case for smMLCK. There is a clear difference between the highly structured C-terminal and the flexible N-terminal parts of the molecule. Thus, the dimensions of smMLCK calculated from sedimentation velocity data may not represent its molecular shape in solution. To obtain a more realistic representation of the mass distribution in smMLCK, we have used the structural homology modeling approach. As described in the introduction, smMLCK has a modular structure similar in many respects to the mega-Dalton muscle protein titin. Extensive studies on titin have shown that the individual immunoglobulin related (Ig) and fibronectin related (Fn) domains are folded independently and the molecule can be represented as a linear arrangement of the individual domains (58, 59). We have modeled the domains of smMLCK and tested how their relative arrangement would affect the predicted sedimentation coefficients as calculated with the program HYDROPRO (50). As the EM images indicate, it is highly unlikely that there is a unique or even a predominant structure of smMLCK in solution. Thus, it would be futile to attempt to obtain a precise atomic structure of the entire molecule. Our goal was to combine the available biochemical, physicochemical and structural information in order to obtain a better understanding of smMLCK function. Below is a description of our approach, and the summary, including the definition of the domains and the list of structural templates used for their modeling, is given in Table 2.
Table 2.
Definition of the structural domains and segments of rabbit smooth muscle MLCK used in model building.
| Domain name | Residue range | Number of amino acids | Mass (kDa) | Template | Ref. |
|---|---|---|---|---|---|
| actin-binding domain | 1-99 | 99 | 10.668 | extended | |
| 16×12 tandem repeat (PEVK) | 100-294 | 195 | 19.881 | polyPro type II helix | |
| linker-1 | 295-327 | 33 | 3.581 | extended | |
| Ig1 | 328-419 | 92 | 10.016 | 2yr3 | (82) |
| linker-2 | 420-464 | 45 | 4.739 | extended | |
| Ig2-Fn3 tandem | 465-657 | 193 | 21.397 | 2nzi | (65) |
| linker-3 | 658-689 | 32 | 3.750 | extended/1koa | |
| kinase/regulatory domain | 690-1002 | 313 | 35.712 | 1koa, 1kob, 1tki | (83, 84) |
| linker-4 | 1003-1038 | 36 | 3.871 | 1koa | |
| Ig3 (telokin) | 1039-1134 | 96 | 10.807 | 1tlk, 1koa | (26, 83) |
| polyGlu | 1135-1147 | 13 | 1.479 | extended |
Bold type - residues modeled with high confidence - 694 amino acids total (62.0% of mass)
Italics - predicted flexible regions - 286 amino acids total (23.6% of mass). The remaining 167 residues, which include the actin binding domain, linker-3 and linker-4 are likely to have well-defined structures and to make little contribution to MLCK flexibility. Note that the 61k-MLCK, which comprises Ig2-Fn3, linker-3, and the kinase domain (residues 461-1002) is equivalent to the trypsin resistant fragment of smMLCK.
Templates used for modeling: for Ig1 - 2yr3.pdb, solution structure of the fourth Ig-like domain from myosin light chain kinase (82); for Ig2-Fn3 tandem - 2nzi.pdb, Ig(A169)-FnIII(A170) segment directly preceding the kinase domain of titin (65); for kinase domain - 1kob.pdb, the kinase domains of twitchin from Aplysia, 1koa.pdb, the kinase and telokin region of twitchin from Caenorhabditis elegans (83) and 1tki.pdb - the kinase domain of titin (84); for Ig3 - 1tlk.pdb, telokin from turkey gizzard (26)
The C-terminal region of smMLCK
The kinase/regulatory domain
The catalytic domain of smMLCK can be modeled with high confidence owing to the fact that it has a high degree of sequence similarity to other kinases and undoubtedly follows their common structural framework (3). Knighton at al. have modeled the catalytic core of smooth muscle MLCK (60) using the crystallographic coordinates of the cyclic AMP-dependent protein kinase catalytic subunit and a bound pseudo-substrate inhibitor peptide (61, 62). They have shown that despite only 30% identity of the amino acid sequence, smMLCK can be readily accommodated in that structure. Their model contains 262 amino acids corresponding to residues 689-951 of rabbit smMLCK and includes the catalytic core and the pseudosubstrate inhibitory segment. A more inclusive template for modeling smMLCK that we used here is the structure of twitchin kinase (pdb code 1KOA) (63). Twitchin is a 753 kDa protein (6,839 residues) located in the muscle A-band of the nematode Caenorhabditis elegans. The structure of twitchin kinase contains the catalytic core and a 60 residue C-terminal autoinhibitory region that extends through the active site and provides direct intra-steric inhibition of kinase activity analogous to that of smMLCK (63). The twichin kinase structure also contains at the C-terminus an Ig domain similar to the telokin region of smMLCK. Modeling of the smMLCK kinase domain required inserting two loops that were missing in twichin. Also, in the structure of twichin kinase there is an N-terminal extension, that is not present in the other kinase structures. This segment has low sequence similarity to the corresponding region of smMLCK; however, its inclusion in the model enabled us to position the N-terminally adjacent tandem Ig2-Fn3 on the opposite side of the telokin domain, thus facilitating the linear arrangement of the domains.
Immunoglobulin and fibronectin related motifs
As mentioned in the introduction, the structure of the C-terminal Ig3 region (telokin) from turkey gizzard MLCK is the only part of smMLCK whose high-resolution structure is known (26). The structure is a typical β-sandwich Ig fold containing seven β-strands (64). In view of the amino acid sequence similarity of the Ig3 domain of rabbit smMLCK and turkey gizzard MLCK, it was straightforward to model residues 1039-1136 of rabbit smMLCK using telokin as a template (pdb code 1TLK.pdb). However, there is uncertainty concerning the N-terminal part of telokin, which in the full-length MLCK makes a connection to the kinase/regulatory domain. The telokin structure begins at residue 33 (residue 1032 of smMLCK) with a stretch of six amino acids in extended conformation that does not belong to the Ig fold. These residues were not considered in our modeling and the position of the Ig3 domain with respect to the kinase domain was modeled based on the twitchin kinase structure (63) (pdb code 1KOA).
The Fn3 module comprises on average 90 residues, and has a typical double-β-sheet sandwich structure in which the two sheets have a right-handed twist and pack against each other face to face. The structure of this motif is similar to the Ig modules differing mainly in a switch of a single β-strand between the sheets (64). Structures of the remaining two Ig domains of smMLCK could be modeled with high confidence despite a relatively low amino acid sequence identity with the templates. The modeling was straightforward due to the fact the there are many high resolution Ig type structures in the PDB and the rules relating the amino acid sequence to the 3D structure of this common protein fold are well known. The relative disposition of the Ig2 and Fn3 regions of smMLCK were modeled based on the structure of one of the Ig2-Fn3 tandem repeats of titin (65) (pdb code 2NZI) (Figure 1).
Models of the 61k-MLCK and 77k-MLCK
The 61k-MLCK comprises the Ig2-Fn3 tandem, the catalytic/regulatory domain and a short linker connecting the two structures (Table 2). The linker defines the distance and the relative orientation of the Ig2-Fn3 tandem with respect to the catalytic domain. We have constructed three models of 61k-MLCK. In model 1, the long axis of the Ig2-Fn3 tandem points towards the kinase domain. The linker is structured as a helical segment projecting away from the catalytic domain, which precludes any contacts between the Ig2-Fn3 tandem and the kinase domain (Figure 8). In model 2 the orientation of the Ig2-Fn3 tandem is similar to that in model 1, but the linker region is collapsed, allowing tight contacts between the Ig2-Fn3 tandem and the kinase domain. This arrangement causes the overall length of the molecule to decrease by 14Å (9.3%). In model 3 the Ig2-Fn3 tandem is flipped back and tightly packed against the kinase domain to simulate the hypothetical compact conformation suggested by the EM. The corresponding models of 77k-MLCK were constructed by adding the Ig3 domain at the C-terminus of each of the three models of the 61k-MLCK. The orientation of the Ig3 domain with respect to the kinase domain was similar to that in the twitchin kinase. For all the models the theoretical sedimentation coefficients were calculated with the program HYDROPRO (50) (see below).
Figure 8.
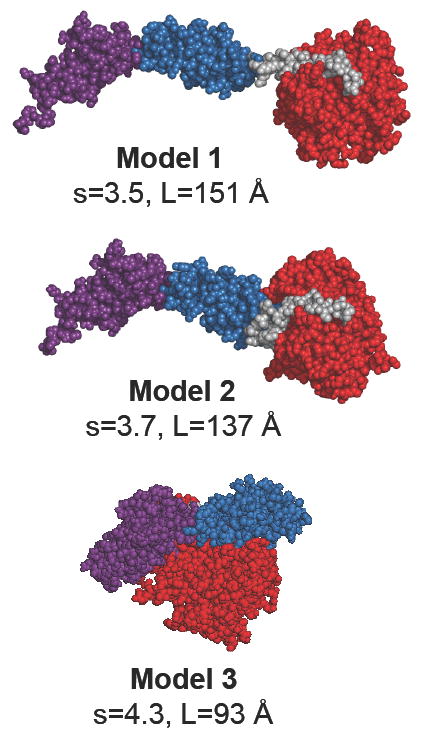
Models of the 61k fragment of smMLCK. Three different arrangements of the Ig2-Fn3 tandem with respect to the catalytic domain were tested. The domains are color coded: red - the catalytic/regulatory domain; blue - the Fn3 domain; magenta - the Ig(2) domain; gray - the linker region (linker-3) connecting the Fn3 domain with the catalytic domain. See Table 2 for the exact definition of the domains. Images of protein models were generated with the program PyMOL (85).
The N-terminal region and the linker segments of smMLCK
In contrast to the well–structured C-terminal part of smMLCK we found no templates suitable for modeling the N-terminal 327 amino acid long segment of this protein. This includes the actin-binding domain, the 16×12 tandem repeat (PEVK) and the 30-residue linker connecting the PEVK region to the first Ig domain (see Table 2). The secondary structure prediction yielded “coil” assignment to the entire region. To assess the contribution of this poorly structured segment to the hydrodynamic properties of smMLCK we used the following approach.
The actin-binding domain
Although there are many proteins that bind F-actin, the actin-binding domain of smMLCK (residues 1-99) appears to have a unique amino acid sequence and a unique binding site on F-actin. Hatch et al. (16) were able to see the density of this fragment bound to F-actin in the 3D reconstructions of EM images, suggesting that this part of smMLCK has a defined structure in the presence of the target. However, it must be an extended structure, since this segment interacts apparently with three actin monomers (15), i.e. it has to span ∼150 Å. Thus, it is likely that in solution in the absence of F-actin this region is unstructured and may be represented as random coil. Accordingly, we have initially modeled the N-terminal 99 residues of smMLCK in the extended conformation and subjected it to 100 ps of molecular dynamics in vacuo, which caused it to collapse into a relatively compact random structure. There is no significance in such a representation other than the convenience of subsequent positioning of the corresponding mass in a model of smMLCK at a defined distance from the C-terminal structured domains. Such a representation is akin to the common practice in hydrodynamic modeling of representing various parts of a protein in a form of appropriately sized beads.
The 16 × 12 tandem repeat (PEVK)
On the C-terminal side of the actin-binding region of rabbit smMLCK there is a 16-fold repeat of a 12-residue segment: TLKPV(G,A)N(A,I,T)KPAE (10). This segment has been referred to as the PEVK region, the name coined for the segments rich in Pro, Glu, Val and Lys found in the mega-Dalton muscle protein titin. An essential difference between the repetitive segment of mammalian smMLCK and the PEVK region of titin is that in the latter the sequence is less regular. An almost exact repetition of a 12-residue motif in MLCK suggests some regular structural features. Searching the Gene Bank with the BLAST program, we found sequence similarity to several prokaryotic and eukaryotic proteins including neurofilament triplet H protein, neural cell adhesion molecule, T-cell adhesion receptor CD2 domain, chicken prion protein, etc. The sequence similarity is restricted, however, to the 6-residue spacing of the Pro and/or Lys residues. It has been suggested that similar Pro rich repetitive sequences in bacterial cell wall have a poly-Pro type II helix conformation (66). There is also evidence that the poly-Pro type II helix is the predominant structure in the PEVK region of titin and that the dynamic properties of this structure make a major contribution to titin’s elasticity and extensibility (17, 67-69). The far UV CD spectrum of a synthetic peptide spanning two 12-residue repeats of smMLCK shows a strong negative peak at 198 nm (Figure 9). Similar CD spectra have been attributed to the poly-Pro type II helix (66, 67, 70, 71). Thus, we have modeled the 16 × 12 tandem repeat region of smMLCK as a poly-proline type II helix (ϕ=-75° ψ=145° ω=180°). In this conformation there are exactly three residues per turn and the carbonyl oxygen atoms of the peptide bonds point away from the helix axis allowing for stabilizing hydrogen bonding with water. In this conformation the 195 amino acids of the 16×12 tandem repeat span 590 Å (3.0 Å/residue). The initial model has been subjected to molecular dynamics simulations for various times from 50 to 300 ps, which resulted in a partial collapse of the structure. Interestingly, even though no further compaction was observed after 300 ps of MD, as judged by nearly constant count of inter-atomic contacts, the structure was not fully randomized and it retained some regular features. Most notably all i, i±3 electrostatic side chain interactions between Glu and the flanking Lys residues were retained (Figure 9C). The partially collapsed structures of the 16×12 tandem repeat region were used to construct the three models of full-length smMLCK shown in Figure 10.
Figure 9.

Structural features of the PEVK region of smMLCK. (A) - amino acid sequence showing the highly conserved repetitive nature of this segment. Note that each Glu residue is flanked on both sides with positively charged Lys side chains, which allow for the i, i±3 salt bridges in the poly-Pro helical configuration. (B) - Far UV circular dichroism spectra of a synthetic 24 residue peptide corresponding to the two 12-residue repeats marked by the rectangle in A. The negative peak at 198 nm is a characteristic of the poly-Pro helix type II. (C) - Example of the Lys-Glu-Lys salt bridges in the modeled structure of the PEVK region of smMLCK. The initial configuration (t=0) and after 50 ps molecular dynamics simulation are shown.
Figure 10.
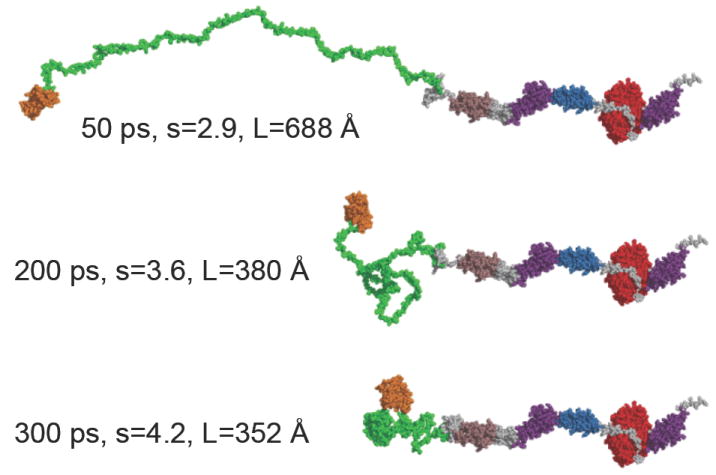
Modeling of the full-length smMLCK. The C-terminal part of the molecule starting at residue 327 modeled in the extended configuration corresponding to model 1 in Figure 8 is identical in all three models. The PEVK region (green) is subjected to MD simulation for 50ps, 200ps and 300ps, resulting in a progressive compaction of the polypeptide chain. The domains comprising the 61k-MLCK are colored in the same manner as in Figure 8. All linker regions are shown in gray, the N-terminal actin-binding domain - orange, the Ig1 domain - brown, and the C-terminal telokin region (Ig3) is shown in magenta.
Linker regions
In addition to the regions of smMLCK discussed above there are short linker segments that cumulatively account for ∼8% of the polypeptide chain. Their structure determines the relative position of the individual domains with respect to each other, but unfortunately cannot be modeled with confidence. On the other hand, variability of the shape and thus of domain arrangement in the EM images (53) strongly suggest that these linkers are flexible and may be reasonably approximated by a random coil. We have generated models of the corresponding segments in the extended conformation (3.3 Å/residue in length) and subjected them to molecular dynamics simulation. This caused the polypeptide chain to collapse into a random and relatively compact conformation. The structure was considered to be in equilibrium if no further increase in intramolecular interactions was observed during the molecular dynamics simulation. Typically 50-100 ps simulation was sufficient for the chain to reach equilibrium. These structures were joined with the other modeled segments and subjected to energy minimization in order to obtain the final models.
Validation of the models
As a method of validation of the modeled structures we have compared the theoretical sedimentation coefficients calculated from the atomic coordinates with those observed experimentally. We have used the hydrodynamic simulation procedure developed by Garcia de la Torre and colleagues (50, 72) as implemented in the program HYDROPRO. In this procedure a primary hydrodynamic model is built from atomic coordinates by replacing nonhydrogen atoms with spherical elements of some fixed radius. The resulting particle consisting of overlapping spheres is, in turn, represented by a shell model, for which a number of hydrodynamic parameters including the sedimentation coefficient are calculated. The key adjustable parameter in these calculations is the so-called atomic element radius (AER), which provides the means of volume correction for hydration by artificially inflating the surface atoms. Based on the analysis of a number of reference proteins for which the hydrodynamic parameters and the high resolution structures were known, Garcia de la Torre et al. concluded that an AER of 3.1Å yields results that are most consistent with the experiment. In view of the fact that neither the hydration nor the 3D structures of our models were certain, we tested a range of AER values for each model and used calmodulin as a control. In Figure 11 the dependence of the theoretical sedimentation coefficients on the AER for each of the structures is shown. For CaM the AER values in the range of 2.5-3.0 Å provide a good agreement with the experiment. For the 61k-MLCK and the 77k-MLCK only the most extended structures (c.f. model 1 in Fig. 8) yield the calculated sedimentation coefficients that are consistent with the experimental values. Most remarkably, the data in Figure 11 clearly show that model 2 of the 61k-MLCK, which due to the more compact conformation of linker-3 is 14 Å (9.3%) shorter than model 1, cannot be correct, since the computed sedimentation coefficients are larger than the experimental value (s(20,w)=3.4, cf. Table 1) at any reasonable level of hydration. This example illustrates the sensitivity and the discriminative power of this approach. Interestingly, model 1 of the 61k-MLCK is 151 Å long (Figure 8), which is significantly shorter (37%) than the long axis of the corresponding prolate ellipsoid calculated from the sedimentation data (240 Å, Table 1). Similarly, the length of the best model of the 77k-MLCK is only 180 Å and that of CaM is 74 Å, as opposed to 280 Å and 100 Å, respectively, for the corresponding prolate ellipsoids (Table 1). Thus, it is clear that the models of the 61k-MLCK and 77k-MLCK obtained by structural homology modeling supported by the computation of the hydrodynamic parameters with the program HYDROPRO provide a much better estimate of the molecular shape and dimensions of these molecules in solution than the commonly used prolate ellipsoid approximation.
Figure 11.

Sedimentation coefficients of CaM and MLCK models computed from their atomic coordinates. The program HYDROPRO was used for the calculations. The effect of hydration on the calculated sedimentation coefficients is tested by varying the so-called atomic element radius (AER). The dashed line in each panel shows the experimentally determined value of s(20,w) for each molecule. The abbreviations m1, m2 and m3 correspond to the three models of the 61k-MLCK shown in Fig. 8 and the respective models of the 77k-MLCK as described in the text. Note that only the most extended models of the 61k and 77k MLCK fragments are consistent with the observed sedimentation coefficients. For the full-length MLCK an extended C-terminal part combined with a moderately compacted PEVK region yields the s values consistent with the experiment.
Based on the data in Figure 11 we can conclude with reasonable confidence that the structural domains of the C-terminal part of smMLCK (residues 465-1147) are arranged in a linear fashion, i.e. the Ig2-Fn3 tandem is positioned on the opposite side of the catalytic/regulatory domain with respect to the telokin (Ig3) region. The long axis of the Ig2-Fn3 tandem is oriented in line with the long axis of the molecule, and the connecting region (linker-3, residues 658-689) has a semi-extended conformation. The linker-3 region appears to be the main, if not the only, source of flexibility in the 61k-MLCK. We used the most extended model of 77k-MLC (model 1) to construct three models of the full-length smMLCK differing in the extent of compaction of the PEVK region (Figure 10). Although for none of the models the computed sedimentation coefficient could match precisely the experimental data at any level of hydration, the model having moderately compacted PEVK region provided the best approximation. Thus, our estimate of the length of smMLCK in solution is 350-380 Å. This is significantly less than the 530Å calculated for the long axis of the corresponding prolate ellipsoid (Table 1), but both estimates are well within the broad range of the contour length distribution of smMLCK cross-linked to F-actin (Figures 3 and 4). The C-terminal 77k segment contributes approximately one half (180 Å) to the total length of smMLCK in solution. The actin binding domain, the PEVK region and linker 1 account for the remaining ∼180 Å. As pointed out earlier and experimentally documented for the PEVK region of titin, the poly-Pro type II helix is highly flexible and extensible. If stretched, the PEVK region of smMLCK could contribute up to 590 Å to the length of smMLCK (in the poly-Pro helix type II conformation) or even as much as 640 Å in the fully extended conformation. Thus, our results indicate that smMLCK molecule is not only elongated, as previously reported for turkey gizzard (29), but also exceptionally flexible and extensible.
DISCUSSION
We have applied structural homology, molecular dynamics and hydrodynamic modeling methods to obtain an approximate all-atom representation of smMLCK, a 125 kDa multi-domain protein. We have tested different spatial arrangements of the Ig and Fn3 domains with respect to the catalytic domain of smMLCK and demonstrated that in order to account for the experimentally determined sedimentation coefficients these domains must be arranged in a linear fashion with respect to the long axis of the molecule. The model of smMLCK that emerges from this work is of a highly elongated molecule with non-uniform mass distribution. It has a well-structured C-terminal part in which the catalytic domain is located, and a poorly structured, extensible segment in the N-terminal part. Such a model is consistent with our EM images of smMLCK cross-linked to F-actin and sedimentation velocity measurements on the full-length smMLCK and its two catalytically active C-terminal fragments.
The average length of smMLCK in solution obtained by the modeling approach is 350-380 Å, which is significantly less (∼30%) than the length calculated from the sedimentation velocity data using the Perrin equation for a prolate ellipsoid (530 Å, cf. Table 1). A question arises as to which approach provides a better estimate of the molecular dimensions of smMLCK in solution. Our sedimentation velocity data for the mammalian smMLCK (1147 amino acids) are consistent with those obtained by Ausio et al. (29) for the slightly smaller (972 amino acids) turkey gizzard MLCK. They have reported the sedimentation coefficient s20,w=3.74, the Stoke’s radius Rs=68.5 Å and the length L=499 Å, which compare favorably to our results for smMLCK: s20,w=3.94, Rs=74.3 Å and L=530 Å (Table 1). Thus, we are confident that our recombinant smMLCK is folded properly and our measurements are correct. Still, in our view, the HYDROPRO modeling approach provides a better estimate of the molecular dimensions of smMLCK despite the uncertainties in modeling the flexible segments and the arbitrary assumptions with respect to the relative spatial positioning of the domains. The key advantage of the modeling approach is that it takes into account the highly non-uniform mass distribution in MLCK. In contrast, the prolate ellipsoid approximation used for the calculation of molecular dimensions from the sedimentation velocity data assumes idealized uniform mass distribution, which is clearly incorrect for MLCK. Interestingly, the prolate ellipsoid approximation overestimates the length by ∼25% for our “control” protein calmodulin, which is less asymmetric than smMLCK, as revealed by the high-resolution X-ray and NMR structures. Based on these considerations we conclude that the average length of smMLCK in solution is in the range of 350-380 Å. However, it is not clear if this estimate is applicable to the conditions in smooth muscle, where the constraints of cellular environment might restrict the conformational dynamics of smMLCK causing it to assume a more compact structure. Conversely, due to the location of the actin and myosin binding sites at the extreme N- and C-termini of the molecule, respectively, smMLCK might bridge the F-actin and myosin filaments (see below) and under certain conditions undergo significant stretching. Under stretch the poorly structured repetitive segment alone could span >600 Å, which would enable the catalytic domain of smMLCK to extend up to 750 Å from F-actin filament to which it is bound by its N-terminus.
Our model of smMLCK enables us to consider from structural perspective the well known from other studies properties of this protein and to make inferences with respect to its function in the activation of smooth muscle contraction. The key properties to be considered are: 1) The elongated, flexible and extensible shape; 2) The catalytic/regulatory domain located at the opposite end of the polypeptide chain from the actin-binding site; 3) A non-catalytic myosin-binding site at the extreme C-terminus of the molecule. These properties combined with the observation that smMLCK is virtually immobilized in smooth muscle cells (30) suggest a functional model in which, in addition to its catalytic function, smMLCK provides also a structural link between the thin and thick filaments (Figure 12). In this model smMLCK is pictured as a flexible, extensible molecule bound to the actin filaments, but capable of extending to, and activating the myosin heads at a significant distance without detaching from F-actin. Such a function is clearly possible in view of the fact that the estimated distance between the thin and thick filaments in smooth muscle is only ∼150 Å (73), which is less than half of the average length of smMLCK in solution estimated in this work. Furthermore, the extensibility of the PEVK region enables the catalytic domain to extend even further from the actin filament as evident from the data in Figures 3 and 4, and from the structural arguments discussed above. In their 1998 review article Stull at al. (27) considered the possibility that the fully extended MLCK could span ∼600Å and bridge the thick and thin filaments in smooth muscle. However, no experimental evidence for the linear domain arrangement was presented, nor alternative models were considered. If indeed smMLCK bridges thin and thick filaments a question arises whether this could inhibit filament sliding during contraction. It has been shown that MLCK binding to phosphorylated myosin is much weaker than to unphosphorylated myosin heads. Thus, alternating between strong and weak binding of smMLCK to myosin might be sufficient to allow for filament sliding and muscle contraction.
Figure 12.

A hypothetical mechanism of myosin phosphorylation in mammalian smooth muscle. smMLCK is pictured as a flexible, extensible molecule bound to actin filaments and capable of bridging thin and thick filaments. Those myosin heads that are in contact with actin filaments are phosphorylated preferentially, and thus become capable of producing force. The number of activated myosin heads depends on filament overlap. Dissociation of the catalytic domain of smMLCK from phosphorylated myosin heads enables filament sliding. smMLCK might contribute to the passive tension under resting conditions by bridging the thick and thin filaments.
The binding of smMLCK to F-actin is well documented (11, 12), the interaction sites identified (13, 15, 74, 75) and even the low resolution structure determined (16). However, the question whether in smooth muscle cells smMLCK is primarily associated with F-actin or with myosin filaments is still a controversial issue. The equilibrium dissociation constant for smMLCK binding to myosin in vitro is in the micromolar range (12); however, there is also a fraction of MLCK that binds very tightly with nanomolar affinity and co-purifies with myosin (76-78) , suggesting the possibility of sub-populations of smMLCK. Recently, Cremo and her colleagues (79) explored the properties of purified smooth muscle myosin preparations that contained tightly bound MLCK. They have shown that such myosin is capable of supporting F-actin motility in vitro in a Ca2+-dependent manner without additional MLCK and calmodulin. Thus, the small amounts of MLCK associated with myosin (by their estimate as little as 1:73 MLCK/myosin ratio) was sufficient to phosphorylate myosin and to support F-actin motility. Interestingly, the maximum F-actin velocity was ∼1/2 of the velocity with pre-thiophosphorylated myosin (79). These observations suggest that each molecule of smMLCK is capable of acting on many myosin heads despite the strong binding, or perhaps owing to an unknown mechanism that enables sequential phosphorylation (e.g. that presented in Fig. 12).
An interesting feature of the hypothetical mechanism shown in Fig. 12 is that it might provide efficient means for the preferential activation of myosin heads that are in direct contact with F-actin filaments, thus upon phosphorylation can be immediately involved in force generation. This might facilitate efficient energy utilization in smooth muscle, since no ATP would be used for phosphorylation of myosin heads that are far from the actin filaments, e.g. under stretch. Our model predicts that the number of activated myosin heads, i.e. the level of phosphorylation should be proportional to the filament overlap. Such an effect was observed in permeabilized rabbit femoral artery smooth muscle strips (Toshio Kitazawa, personal communication). The fraction of phosphorylated LC20 changed with the cell length in a similar manner as the force, being the highest at Lo (43.7% LC20 phosphorylation) and significantly lower in the contracted (13.8%) and stretched (6.5%) muscles. More experiments are needed to verify this finding and demonstrate its generality.
The potential contribution of smMLCK to the structure of smooth muscle requires a comment. It has been proposed that titin, the protein structurally related to smMLCK is the major contributor to passive tension in striated muscles. Recent studies also suggest that titin plays an important role in the length-dependent activation by sensing stretch and promoting actomyosin interaction (80, 81). There is clearly a significant similarity between the properties of titin and smMLCK. In particular, due to the elasticity of the PEVK region smMLCK molecules might affect the inter-filament spacing and contribute to the passive tension in smooth muscle.
Abbreviations used
- smMLCK
smooth muscle myosin light chain kinase, 61k-MLCK and 77k-MLCK recombinant fragments of rabbit smooth muscle myosin light chain kinase containing residues 461-1002 and 461-1147, respectively
- CaM
calmodulin
- MD
molecular dynamics
Footnotes
This work was supported by the National Institutes of Health (grants AR-41637, HL-91162)
Sobieszek, A. and Grabarek, Z. unpublished data. The full account of these studies will be published elsewhere.
References
- 1.Walsh MP, Dabrowska R, Hinkins S, Hartshorne DJ. Calcium-independent myosin light chain kinase of smooth muscle. Preparation by limited chymotryptic digestion of the calcium ion dependent enzyme, purification, and characterization. Biochemistry. 1982;21:1919–1925. doi: 10.1021/bi00537a034. [DOI] [PubMed] [Google Scholar]
- 2.Mayr GW, Heilmeyer LM., Jr Skeletal muscle myosin light chain kinase. A refined structural model. FEBS Lett. 1983;157:225–231. doi: 10.1016/0014-5793(83)80552-3. [DOI] [PubMed] [Google Scholar]
- 3.Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, Sowadski JM. Structural framework for the protein kinase family. Annu Rev Cell Biol. 1992;8:429–462. doi: 10.1146/annurev.cb.08.110192.002241. [DOI] [PubMed] [Google Scholar]
- 4.Pearson RB, Wettenhall RE, Means AR, Hartshorne DJ, Kemp BE. Autoregulation of enzymes by pseudosubstrate prototopes: myosin light chain kinase. Science. 1988;241:970–973. doi: 10.1126/science.3406746. [DOI] [PubMed] [Google Scholar]
- 5.Kemp BE, Pearson RB, House C, Robinson PJ, Means AR. Regulation of protein kinases by pseudosubstrate prototopes. Cell Sign. 1989;1:303–311. doi: 10.1016/0898-6568(89)90049-1. [DOI] [PubMed] [Google Scholar]
- 6.Gallagher PJ, Herring BP, Trafny A, Sowadski J, Stull JT. A molecular mechanism for autoinhibition of myosin light chain kinases. J Biol Chem. 1993;268:26578–26582. [PMC free article] [PubMed] [Google Scholar]
- 7.Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- 8.Herring BP, Stull JT, Gallagher PJ. Domain characterization of rabbit skeletal muscle myosin light chain kinase. J Biol Chem. 1990;265:1724–1730. [PMC free article] [PubMed] [Google Scholar]
- 9.Olson NJ, Pearson RB, Needleman DS, Hurwitz MY, Kemp BE, Means AR. Regulatory and structural motifs of chicken gizzard myosin light chain kinase. Proc Natl Acad Sci USA. 1990;87:2284–2288. doi: 10.1073/pnas.87.6.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher PJ, Herring BP, Griffin SA, Stull JT. Molecular characterization of a mammalian smooth muscle myosin light chain kinase. J Biol Chem. 1991;266:23936–23944. published erratum appears in J Biol Chem 1992 May 5;267(13):9450. [PMC free article] [PubMed] [Google Scholar]
- 11.Dabrowska R, Hinkins S, Walsh MP, Hartshorne DJ. The binding of smooth muscle myosin light chain kinase to actin. Biochem Biophys Res Commun. 1982;107:1524–1531. doi: 10.1016/s0006-291x(82)80172-1. [DOI] [PubMed] [Google Scholar]
- 12.Sellers JR, Pato MD. The binding of smooth muscle myosin light chain kinase and phosphatases to actin and myosin. J Biol Chem. 1984;259:7740–7746. [PubMed] [Google Scholar]
- 13.Kanoh S, Ito M, Niwa E, Kawano Y, Hartshorne DJ. Actin-binding peptide from smooth muscle myosin light chain kinase. Biochemistry. 1993;32:8902–8907. doi: 10.1021/bi00085a023. [DOI] [PubMed] [Google Scholar]
- 14.Lin PJ, LubyPhelps K, Stull JT. Binding Of Myosin Light Chain Kinase to Cellular Actin-Myosin Filaments. J Biol Chem. 1997;272:7412–7420. doi: 10.1074/jbc.272.11.7412. [DOI] [PubMed] [Google Scholar]
- 15.Smith L, Su X, Lin P, Zhi G, Stull JT. Identification of a novel actin binding motif in smooth muscle myosin light chain kinase. J Biol Chem. 1999;274:29433–29438. doi: 10.1074/jbc.274.41.29433. [DOI] [PubMed] [Google Scholar]
- 16.Hatch V, Zhi G, Smith L, Stull JT, Craig R, Lehman W. Myosin light chain kinase binding to a unique site on F-actin revealed by three-dimensional image reconstruction. J Cell Biol. 2001;154:611–617. doi: 10.1083/jcb.200105079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tskhovrebova L, Trinick J, Sleep JA, Simmons RM. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 1997;387:308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- 18.Witt CC, Olivieri N, Centner T, Kolmerer B, Millevoi S, Morell J, Labeit D, Labeit S, Jockusch H, Pastore A. A survey of the primary structure and the interspecies conservation of I-band titin’s elastic elements in vertebrates. J Struct Biol. 1998;122:206–215. doi: 10.1006/jsbi.1998.3993. [DOI] [PubMed] [Google Scholar]
- 19.Maruyama K. Connectin/titin, giant elastic protein of muscle. Faseb J. 1997;11:341–345. doi: 10.1096/fasebj.11.5.9141500. [DOI] [PubMed] [Google Scholar]
- 20.Ikebe M, Stepinska M, Kemp BE, Means AR, Hartshorne DJ. Proteolysis of smooth muscle myosin light chain kinase. Formation of inactive and calmodulin-independent fragments. J Biol Chem. 1987;262:13828–13834. [PubMed] [Google Scholar]
- 21.Ikebe M, Maruta S, Reardon S. Location of the inhibitory region of smooth muscle myosin light chain kinase. J Biol Chem. 1989;264:6967–6971. [PubMed] [Google Scholar]
- 22.Stull JT, Krueger JK, Kamm KE, Gao Z-H, Zhi G, Padre R. Myosin light chain kinase. In: Barany M, editor. Biochemistry of smooth muscle contraction. Academic Press; San Diego, New York, Boston, London, Sydney, Toronto: 1996. pp. 119–130. [Google Scholar]
- 23.Ito M, Dabrowska R, Guerriero R, Hartshorne DJ. Identification in turkey gizzard of an acidic protein related to the C-terminal portion of smooth muscle myosin light chain kinase. J Biol Chem. 1989;264:13971–13974. [PubMed] [Google Scholar]
- 24.Collinge M, Matrisian PE, Zimmer WE, Shattuck RL, Lukas TJ, Van EL, Watterson DM. Structure and expression of a calcium-binding protein gene contained within a calmodulin-regulated protein kinase gene. Molecular & Cellular Biology. 1992;12:2359–2371. doi: 10.1128/mcb.12.5.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silver DL, Vorotnikov AV, Watterson DM, Shirinsky VP, Sellers JR. Sites of interaction between kinase-related protein and smooth muscle myosin. J Biol Chem. 1997;272:25353–25359. doi: 10.1074/jbc.272.40.25353. [DOI] [PubMed] [Google Scholar]
- 26.Holden HM, Ito M, Hartshorne DJ, Rayment I. X-ray structure determination of telokin, the C-terminal domain of myosin light chain kinase, at 2.8 A resolution. J Mol Biol. 1992;227:840–851. doi: 10.1016/0022-2836(92)90226-a. [DOI] [PubMed] [Google Scholar]
- 27.Stull JT, Lin PJ, Krueger JK, Trewhella J, Zhi G. Myosin light chain kinase: functional domains and structural motifs. Acta Physiol Scand. 1998;164:471–482. doi: 10.1111/j.1365-201x.1998.tb10699.x. [DOI] [PubMed] [Google Scholar]
- 28.Mayr GW, Heilmeyer LMGJ. Shape and substructure of skeletal muscle myosin light chain kinase. Biochemistry. 1983;22:4316–4326. doi: 10.1021/bi00287a024. [DOI] [PubMed] [Google Scholar]
- 29.Ausio J, Malencik DA, Anderson SR. Analytical sedimentation studies of turkey gizzard myosin light chain kinase and telokin. Biophys J. 1992;61:1656–1663. doi: 10.1016/S0006-3495(92)81969-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin P, Luby-Phelps K, Stull JT. Properties of filament-bound myosin light chain kinase. J Biol Chem. 1999;274:5987–5994. doi: 10.1074/jbc.274.9.5987. [DOI] [PubMed] [Google Scholar]
- 31.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 32.Tan RY, Mabuchi Y, Grabarek Z. Blocking the Ca2+-induced conformational transitions in calmodulin with disulfide bonds. J Biol Chem. 1996;271:7479–7483. doi: 10.1074/jbc.271.13.7479. [DOI] [PubMed] [Google Scholar]
- 33.Stafford WF. Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 34.Stafford WF. Boundary analysis in sedimentation velocity experiments. Methods Enzymol. 1994;240:478–501. doi: 10.1016/s0076-6879(94)40061-x. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Stafford WF. An optical thermometer for direct measurement of cell temperature in the Beckman instruments XL-A analytical ultracentrifuge. Anal Biochem. 1995;224:199–202. doi: 10.1006/abio.1995.1030. [DOI] [PubMed] [Google Scholar]
- 36.Stafford WF, Sherwood PJ. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem. 2004;108:231–243. doi: 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Perkins SJ. Protein volumes and hydration effects. Eur J Biochem. 1986;157:169–180. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- 38.Tanford C. Physical Chemistry of Macromolecules. John Wiley and Sons; New York: 1961. [Google Scholar]
- 39.Kuntz ID, Kauzman W. Hydration of Proteins and Polypeptides. Adv Prot Chem. 1974;28:239–345. doi: 10.1016/s0065-3233(08)60232-6. [DOI] [PubMed] [Google Scholar]
- 40.Mabuchi K. Heavy-meromyosin-decorated actin filaments: a simple method to preserve actin filaments for rotary shadowing. Journal of Structural Biology. 1991;107:22–28. doi: 10.1016/1047-8477(91)90027-t. [DOI] [PubMed] [Google Scholar]
- 41.Mabuchi K. Melting of myosin and tropomyosin: electron microscopic observations. Journal of Structural Biology. 1990;103:249–256. doi: 10.1016/1047-8477(90)90043-c. [DOI] [PubMed] [Google Scholar]
- 42.Grabarek Z, Gergely J. Zero-length crosslinking procedure with the use of active esters. Anal Biochem. 1990;185:131–135. doi: 10.1016/0003-2697(90)90267-d. [DOI] [PubMed] [Google Scholar]
- 43.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 44.Kopp J, Schwede T. The SWISS-MODEL Repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res. 2004;32:D230–234. doi: 10.1093/nar/gkh008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 47.Peitsch MC. Protein modeling by E-mail. Bio/Technology. 1995;13:658–660. [Google Scholar]
- 48.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein molecules in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 49.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 50.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pearson RB, Ito M, Morrice NA, Smith AJ, Condron R, Wettenhall RE, Kemp BE, Hartshorne DJ. Proteolytic cleavage sites in smooth muscle myosin-light-chain kinase and their relation to structural and regulatory domains. Eur J Biochem. 1991;200:723–730. doi: 10.1111/j.1432-1033.1991.tb16237.x. [DOI] [PubMed] [Google Scholar]
- 52.Padre RC, Stull JT. Functional assembly of fragments from bisected smooth muscle myosin light chain kinase. J Biol Chem. 2000;275:26665–26673. doi: 10.1074/jbc.M001769200. [DOI] [PubMed] [Google Scholar]
- 53.Numata T, Katoh T, Yazawa M. Functional Role of the C-Terminal Domain of Smooth Muscle Myosin Light Chain Kinase on the Phosphorylation of Smooth Muscle Myosin. J Biochem (Tokyo) 2001;129:437–444. doi: 10.1093/oxfordjournals.jbchem.a002875. [DOI] [PubMed] [Google Scholar]
- 54.Sobieszek A. Regulation of smooth muscle myosin light chain kinase. Allosteric effects and co-operative activation by calmodulin. J Mol Biol. 1991;220:947–957. doi: 10.1016/0022-2836(91)90365-d. published erratum appears in J Mol Biol 1991 Dec 20;222(4):1173. [DOI] [PubMed] [Google Scholar]
- 55.Sobieszek A, Strobl A, Ortner B, Babiychuk EB. Ca2+ calmodulin-dependent modification of smooth-muscle myosin light-chain kinase leading to its co-operative activation by calmodulin. Biochem J. 1993;295:405–411. doi: 10.1042/bj2950405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Babiychuk EB, Babiychuk VS, Sobieszek A. Modulation of smooth muscle myosin light chain kinase activity by Ca2+/calmodulin-dependent, oligomeric-type modifications. Biochemistry. 1995;34:6366–6372. doi: 10.1021/bi00019a015. [DOI] [PubMed] [Google Scholar]
- 57.Krueger JK, Olah GA, Rokop SE, Zhi G, Stull JT, Trewhella J. Structures Of Calmodulin and a Functional Myosin Light Chain Kinase In the Activated Complex - a Neutron Scattering Study. Biochemistry. 1997;36:6017–6023. doi: 10.1021/bi9702703. [DOI] [PubMed] [Google Scholar]
- 58.Politou AS, Gautel M, Improta S, Vangelista L, Pastore A. The elastic I-band region of titin is assembled in a “modular” fashion by weakly interacting Ig-like domains. J Mol Biol. 1996;255:604–616. doi: 10.1006/jmbi.1996.0050. [DOI] [PubMed] [Google Scholar]
- 59.Improta S, Krueger JK, Gautel M, Atkinson RA, Lefevre JF, Moulton S, Trewhella J, Pastore A. The assembly of immunoglobulin-like modules in titin: implications for muscle elasticity. J Mol Biol. 1998;284:761–777. doi: 10.1006/jmbi.1998.2028. [DOI] [PubMed] [Google Scholar]
- 60.Knighton DR, Pearson RB, Sowadski JM, Means AR, Ten EL, Taylor SS, Kemp BE. Structural basis of the intrasteric regulation of myosin light chain kinases. Science. 1992;258:130–135. doi: 10.1126/science.1439761. [DOI] [PubMed] [Google Scholar]
- 61.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 62.Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 63.Hu SH, Parker MW, Lei JY, Wilce M, Benian GM, Kemp BE. Insights into autoregulation from the crystal structure of twitchin kinase. Nature. 1994;369:581–584. doi: 10.1038/369581a0. [DOI] [PubMed] [Google Scholar]
- 64.Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 65.Mrosek M, Labeit D, Witt S, Heerklotz H, von Castelmur E, Labeit S, Mayans O. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. Faseb J. 2007;21:1383–1392. doi: 10.1096/fj.06-7644com. [DOI] [PubMed] [Google Scholar]
- 66.Areschoug T, Linse S, Stalhammar-Carlemalm M, Heden LO, Lindahl G. A proline-rich region with a highly periodic sequence in Streptococcal beta protein adopts the polyproline II structure and is exposed on the bacterial surface. J Bacteriol. 2002;184:6376–6383. doi: 10.1128/JB.184.22.6376-6383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Forbes JG, Jin AJ, Ma K, Gutierrez-Cruz G, Tsai WL, Wang K. Titin PEVK segment: charge-driven elasticity of the open and flexible polyampholyte. J Muscle Res Cell Motil. 2005;26:291–301. doi: 10.1007/s10974-005-9035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Granzier HL, Labeit S. Titin and its associated proteins: the third myofilament system of the sarcomere. Adv Protein Chem. 2005;71:89–119. doi: 10.1016/S0065-3233(04)71003-7. [DOI] [PubMed] [Google Scholar]
- 69.Nagy A, Grama L, Huber T, Bianco P, Trombitas K, Granzier HL, Kellermayer MS. Hierarchical extensibility in the PEVK domain of skeletal-muscle titin. Biophys J. 2005;89:329–336. doi: 10.1529/biophysj.104.057737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma K, Kan L, Wang K. Polyproline II helix is a key structural motif of the elastic PEVK segment of titin. Biochemistry. 2001;40:3427–3438. doi: 10.1021/bi0022792. [DOI] [PubMed] [Google Scholar]
- 71.Ma K, Wang K. Malleable conformation of the elastic PEVK segment of titin: non-co-operative interconversion of polyproline II helix, beta-turn and unordered structures. Biochem J. 2003;374:687–695. doi: 10.1042/BJ20030702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carrasco B, Garcia de la Torre J. Hydrodynamic properties of rigid particles: comparison of different modeling and computational procedures. Biophys J. 1999;76:3044–3057. doi: 10.1016/S0006-3495(99)77457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Somlyo AP, Devine CE, Somlyo AV, Rice RV. Filament organization in vertebrate smooth muscle. Philos Trans R Soc Lond B Biol Sci. 1973;265:223–229. doi: 10.1098/rstb.1973.0027. [DOI] [PubMed] [Google Scholar]
- 74.Gallagher PJ, Stull JT. Localization of an actin binding domain in smooth muscle myosin light chain kinase. Mol Cell Biochem. 1997;173:51–57. doi: 10.1023/a:1006876318155. [DOI] [PubMed] [Google Scholar]
- 75.Ye LH, Hayakawa K, Kishi H, Imamura M, Nakamura A, Okagaki T, Takagi T, Iwata A, Tanaka T, Kohama K. The structure and function of the actin-binding domain of myosin light chain kinase of smooth muscle. J Biol Chem. 1997;272:32182–32189. doi: 10.1074/jbc.272.51.32182. [DOI] [PubMed] [Google Scholar]
- 76.Cross RA, Sobieszek A. Influence of smooth muscle myosin conformation on myosin light chain kinase binding and on phosphorylation. FEBS Lett. 1985;188:367–374. doi: 10.1016/0014-5793(85)80404-x. [DOI] [PubMed] [Google Scholar]
- 77.Sobieszek A. Phosphorylation reaction of vertebrate smooth muscle myosin: an enzyme kinetic analysis. Biochemistry. 1985;24:1266–1274. doi: 10.1021/bi00326a032. [DOI] [PubMed] [Google Scholar]
- 78.Ngai PK, Walsh MP. Purification of smooth-muscle myosin free of calmodulin and myosin light-chain kinase. Susceptibility to oxidation. Biochem J. 1987;246:205–211. doi: 10.1042/bj2460205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hong F, Haldeman BD, John OA, Brewer PD, Wu YY, Ni S, Wilson DP, Walsh MP, Baker JE, Cremo CR. Characterization of tightly associated smooth muscle myosin-myosin light-chain kinase-calmodulin complexes. J Mol Biol. 2009;390:879–892. doi: 10.1016/j.jmb.2009.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 81.Fukuda N, Granzier HL. Titin/connectin-based modulation of the Frank-Starling mechanism of the heart. J Muscle Res Cell Motil. 2005;26:319–323. doi: 10.1007/s10974-005-9038-1. [DOI] [PubMed] [Google Scholar]
- 82.Qin XR, Kurosaki C, Yoshida M, Hayashi F, Yokoyama S. Solution structure of the fourth Ig-like domain from myosin light chain kinase, smooth muscle. To be Published. [Google Scholar]
- 83.Kobe B, Heierhorst J, Feil SC, Parker MW, Benian GM, Weiss KR, Kemp BE. Giant Protein Kinases - Domain Interactions and Structural Basis Of Autoregulation. EMBO Journal. 1996;15:6810–6821. [PMC free article] [PubMed] [Google Scholar]
- 84.Mayans O, van der Ven PF, Wilm M, Mues A, Young P, Furst DO, Wilmanns M, Gautel M. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395:863–869. doi: 10.1038/27603. [DOI] [PubMed] [Google Scholar]
- 85.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]