

Abstract
Background
Silent cerebral infarct (SCI) is the most common cause of serious neurological disease in sickle cell anemia (SCA), affecting approximately 22% of children. The goal of this trial is to determine whether blood transfusion therapy will reduce further neurological morbidity in children with SCI, and if so, the magnitude of this benefit.
Procedure
The Silent Cerebral Infarct Transfusion (SIT) Trial includes 29 clinical sites and 3 subsites, a Clinical Coordinating Center, and a Statistical and Data Coordinating Center, to test the following hypothesis: prophylactic blood transfusion therapy in children with SCI will result in at least an 86% reduction in the rate of subsequent overt strokes or new or progressive cerebral infarcts as defined by magnetic resonance imaging (MRI) of the brain. The intervention is blood transfusion versus observation. Two hundred and four participants (102 in each treatment assignment) will ensure 85% power to detect the effect necessary to recommend transfusion therapy (86% reduction), after accounting for 10% drop out and 19% crossover rates. MRI examination of the brain is done at screening, immediately before randomization and study exit. Each randomly assigned participant receives a cognitive test battery at study entry, 12–18 months later, and study exit and an annual neurological examination. Blood is obtained from all screened participants for a biologic repository containing serum and a renewable source of DNA.
Conclusion
The SIT Trial could lead to a change in standard care practices for children affected with SCA and SCI, with a consequent reduction in neurological morbidity.
Keywords: blood transfusion therapy, clinical trials, hemoglobinopathies, sickle cell anemia, silent cerebral infarcts
BACKGROUND AND RATIONALE
Introduction
Sickle cell anemia (SCA) is a recessive genetic disorder caused by a point mutation that results in the substitution of valine for glutamic acid at the sixth position in the beta chain of hemoglobin. This homozygous gene disorder affects one of every 400 African–American newborns and approximately 70,000 persons in the United States [1]. The major cause of morbidity and mortality in SCA is vaso-occlusion. Although sequelae of SCA can occur in any organ, one of the most devastating results is arguably cerebral injury. Stroke in children has not been as extensively studied as it has in adults. In a study of children aged 1–14 years with stroke in the Baltimore-Washington Cooperative Young Stroke Study, SCA was the most common cause of ischemic stroke [2]. The study described in this manuscript, the Silent Cerebral Infarct Transfusion (SIT) Trial, represents the largest intervention trial of ischemic neurological injury in children, and will provide a model for subsequent studies of strokes in children.
Overt Strokes and Silent Cerebral Infarction (SCI)
Cerebral infarcts in children with SCA may impair motor and cognitive function. Overt strokes in patients with SCA are characterized by the sudden and persistent onset of neurological deficits, usually resulting in clear recognition and prompt treatment. In the period before transcranial Doppler (TCD) screening, these overt strokes occurred in approximately 9% of patients with SCA before their 14th birthday [3]. SCI is defined as an area of abnormal hyperintensity on T2-weighted magnetic resonance imaging (MRI) of the brain in a patient with no history or physical findings of a focal neurological deficit in a corresponding localizing vascular distribution [4]. For the purposes of this trial, we have included in the definition that the lesion must be ≥ 3 mm in diameter and visible in at least two planes of T2-weighted images (axial and coronal). Absence of neurological symptoms or exam abnormalities corresponding to the MRI lesions must also be confirmed by a neurologist.
SCI is the most commonly recognized form of neurological injury in children with SCA, reported to occur in approximately 22% to 27% of these children who have not suffered a clinically evident (overt) stroke [5, 6]. SCI is characterized by an increased risk for further progression of neurological disease, including overt stroke [5], new or progressive brain lesions (SCI) on MRI [5], poor academic attainment [7], and lower I.Q. when compared with children with SCA who have normal MRI of the brain [8] or siblings without SCA [9].
The cause of SCI is not clear. The majority of SCI occur in the deep white matter border zones of the carotid circulation, affecting predominantly frontal and parietal lobe subcortical white matter [10]. Preliminary data suggest small precapillary arteriole vessel obstruction [11]. Unfortunately, there are no imaging data to confirm that these microinfarcts are associated with SCI lesions seen on brain MRI.
Feasibility of Blood Transfusion to Prevent Further Neurological Injury Among Children with SCA
On the basis of evidence that SCI are morbid and progressive and to provide support for a multicenter clinical trial, we conducted a single arm feasibility intervention study, in which children with SCA and SCI received blood transfusion therapy [12]. Seven subjects had evaluable baseline and repeat MRIs. Of these seven, there were a total of 18 infarcts. One infarct increased in size; all other lesions were stable or became smaller in diameter while on transfusion therapy during the time of follow-up (mean 3.9 years, range 2.1–5.7) and no new SCI was detected. Seven children in the study completed transfusion therapy for over 2 years, with an average duration of therapy of 2.7 years (range 1–6). On the basis of these data, we concluded that blood transfusion therapy is feasible for the prevention of progression of SCI and parents are willing to accept blood transfusion therapy to prevent further neurological injury. While the optimal duration of transfusion therapy has not been determined systematically for abnormal TCD velocity or overt strokes, evidence from both primary and secondary prevention stroke studies suggests that the risk of primary or secondary strokes continues at least throughout childhood. In the SIT Trial, participants are transfused or observed for 36 months, on the basis of the incidence of new SCI among individuals with existing SCI [5], results of a feasibility study with a mean follow-up of approximately 32 months of transfusions [12] and expert opinion of the hematologists from the participating pediatric hematology centers.
METHODS: STUDY OBJECTIVES AND ORGANIZATION
Main Hypothesis
The overall goal of this trial is to determine whether blood transfusion therapy will limit overt clinical strokes, or new or progressive SCI in children with SCA. Our primary hypothesis is that prophylactic blood transfusion therapy in children with SCI will result in a reduction of at least 86% in the proportion of participants with clinically evident strokes or new or progressive SCI.
Secondary Hypotheses
A secondary, but nonetheless important, hypothesis is that blood transfusion therapy will preserve cognition when compared with the observation arm (Secondary Hypothesis 1). In addition, we test the hypothesis that the overall benefits of transfusion will outweigh the associated risks (Secondary Hypothesis 2) and justify transfusion in the eyes of the parents and health care providers.
Study Organization
The study group for the SIT Trial consists of four central units: a clinical coordinating center and central imaging center (Electronic Radiology Laboratory) located in St. Louis, Missouri, and a statistical and data coordinating center and biologic repository located in Baltimore, MD, as well as 29 clinical centers and 3 subsites in the United States, Canada, France, and the United Kingdom. (See Supplementary Figure 1, supplemental online material: http://sitstudy.wustl.edu).
STUDY DESIGN AND CONDUCT OF THE TRIAL
Overall Design
The SIT Trial is a multicenter intervention trial, in which participants with SCI are randomized to receive blood transfusion therapy or observation for 36 months. Given the nature of the intervention (transfusion), masking of the site investigators and personnel to the intervention is not feasible. The adjudications of the primary and secondary outcome measures are masked in terms of the participant’s identity and intervention. The study utilizes an intention-to-treat analysis, allowing a conservative estimate of the differences between groups and a better estimate of the effectiveness and potential public health implications of the decision to treat, rather than a pure estimate of efficacy alone [13].
Primary Endpoint
The primary outcome measure is the presence of new or progressive cerebral infarction as assessed by MRI of the brain or the occurrence of overt stroke. Alternative endpoints such as cognition were considered, as preservation of neurological function is our ultimate goal; however, new or progressive infarcts or stroke was chosen for the following reasons: (1) the lack of specificity of change in cognitive test scores over time, (2) the objective and quantifiable nature of lesions assessed by MRI, (3) the close association of cognitive impairment with the presence of SCI, and (4) the likelihood that prevention of new or progressive MRI findings would provide more convincing evidence to change clinical practice than change in cognitive test scores.
As the primary outcome measure for the trial is the presence of new or progressive cerebral infarction on MRI, precise methods are used to ensure accuracy and reproducibility of the endpoint determination. A qualitative assessment is based on the presence or absence of new or progressive cerebral infarcts on a 3-point confidence scale (1 = definitely not present, 2 = indeterminate, 3 = definitely present), the same definition used by the Comprehensive Study of Sickle Cell Disease (CSSCD) to demonstrate a 24% increase in the incidence of SCI in children with SCI over a 5.2-year mean follow-up period [5]. An enlarging SCI-like lesion is defined as a qualifying lesion that increases in size along any linear dimension in the axial plane. The quantitative assessment by the neuroradiologists involves independent identification and measurement of each SCI-like lesion, with subsequent joint review to establish the consensus measurement. When all three neuroradiologists performed independent measurements of SCI-like lesions from a subset of the screening MRIs (N = 30), kappa statistics for each two-way comparison of neuroradiologist’s readings ranged from 0.68 to 0.77, all within the “substantial” range for agreement. On the basis of these assessments, a minimum threshold of at least 3 mm linear enlargement is the definition of enlargement of a SCI-like lesion in the SIT Trial.
Additional Design Considerations
A large effect size of 86% was chosen to assess the efficacy of blood transfusion therapy to reduce the incidence of the primary outcome. Several factors were considered: (1) the demonstrated efficacy of transfusions in preventing both primary (89% relative risk reduction for stroke in the Stroke Prevention Trial in Sickle Cell Anemia [STOP]) [14] and secondary (84% relative risk reduction in a retrospective cohort) [15] stroke; (2) the absence of recurrences of SCI in the 18 participants followed in the STOP trials who had SCI at baseline and received blood transfusion therapy [16], suggesting complete suppression of new lesions by transfusions in this limited cohort; and (3) the known toxicities and high family burden associated with chronic blood transfusion therapy.
Figure 1 shows the assumptions used to estimate the number of participants needed for screening.
FIGURE 1.
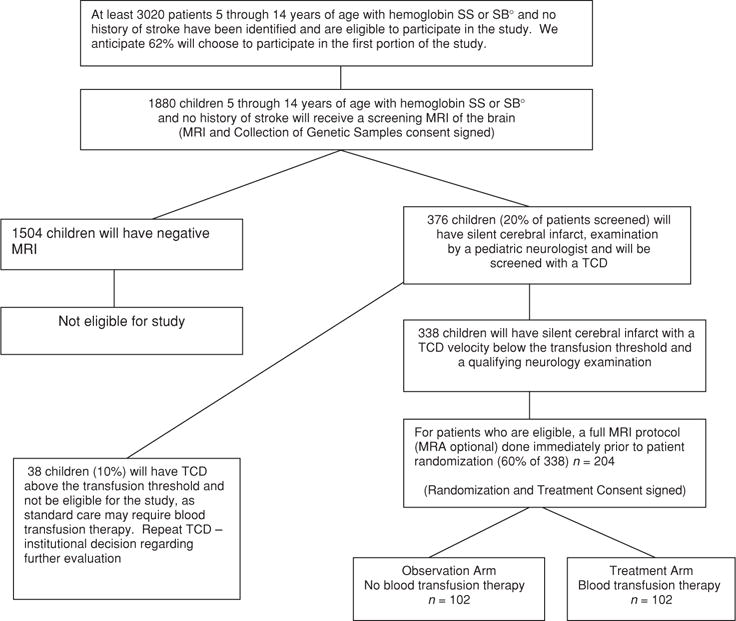
Flow diagram showing progress of the postulated number of participants from screening to randomization.
We also anticipated that, given the length of the trial, changes in technology would occur during the course of the trial, for example, some sites would upgrade from 1.5 to 3.0 Tesla MRI. The accommodations for these changes are described in the full protocol in the supplemental online material.
Conduct of the Trial
Overview
We initially anticipated that the trial would be conducted over a six-and-half-year period, divided into four phases: (1) Standardization; (2) Enrollment and Active Clinical Trial; (3) Close Out; and (4) Data Analysis. This was subsequently changed to a period of eight and a half years. All participants are to be treated or followed for 3 years.
The SIT Trial is a two-stage study. During the first stage, eligible participants are screened for SCI. During the second stage, participants with documented, stable SCI are randomly allocated to receive either blood transfusion therapy or observation (Figure 1).
Subject Eligibility
Inclusion and exclusion criteria for screening and randomly allocating participants to receive either blood transfusion therapy or observation for 36 months are described in Tables 1 and 2, respectively. Inclusion criteria for random allocation of eligible participants with SCI include all of the criteria for the screening phase of the trial. On screening MRIs, suspected SCI are classified only as SCI-like lesions, based on consensus of opinion of two of three members of the neuroradiology committee; to be eligible for randomization, the participant must have a normal neurological examination or a neurological abnormality that is inconsistent with the neuroanatomical location of the lesion on MRI. A standardized neurological examination is performed by the site neurologist and adjudicated by the neurology committee; only after this adjudication is the lesion determined to be silent (SCI).
TABLE 1.
Inclusion and Exclusion Criteria for Screening
| Inclusion criteria for screening | Exclusion criteria for screening at initial screening evaluation and after informed consent has been signed, as well as during the interval up to randomization. |
|---|---|
|
|
TABLE 2.
Inclusion and Exclusion Criteria for Randomization
| Inclusion criteria for randomization | Exclusion criteria for randomization |
|---|---|
|
|
Participant Assessment
Screening for SCI
Screening visits to determine eligibility for the trial include informed consent, a comprehensive medical and educational history, a neurological examination by a hematologist, a screening MRI of the brain, a blood specimen for the genetic repository, and the first educational session. If there are no infarct-like lesions on the MRI, the participant is not eligible for the randomized component of the trial (see Supplemental Table I, supplemental online material).
Postscreening educational visits are held to inform the participant/caregiver of the MRI results and to discuss the next phase of the trial, to allow the parent/caregiver and participant to determine if they are truly willing to be allocated by chance to either the observation or blood transfusion arm of the trial. Education sessions are a mainstay of the trial, and are intended to improve the quality of participant experience, as well as adherence to the concepts and principles of the trial.
Prerandomization Evaluations for All Participants with MRIs of the Brain Positive for Infarct-Like Lesions
Participants with infarct-like lesions willing to accept random assignment receive a standardized neurological examination (see below) by a study neurologist and a TCD examination. To be eligible for randomization, the participant must have TCD velocities that are below the transfusion threshold (nonimaging method ≥ 200 cm/sec or imaging method ≥ 185 cm/sec), as well as no neurological deficit corresponding to the findings on MRI of the brain. Participants with a high conditional TCD measurement (nonimaging method 180–199 cm/sec or imaging method 170–184 cm/sec) undergo repeat TCD within one month and prior to randomization. If the prerandomization TCD remains in the high conditional range and the participant meets all other inclusion criteria, the participant may be randomly allocated to receive blood transfusion therapy or observation. The schedules of evaluation for participants in the treatment and observation arms are shown in Supplemental Tables II and III, respectively, in the supplemental online material.
The TCD criteria were established for safety, to avoid enrolling participants who are at a level of risk for stroke for which treatment recommendations have been established in a controlled clinical trial [14]. This also reduces the likelihood that participants in the nontreatment arm will cross over to the treatment arm because of the detection of an elevated TCD.
Extensive procedures are in place to maintain blinding of the local neurologist to the location of MRI lesions. The central neurology committee reviews the MRI and the neurological examination, to adjudicate whether the infarction is silent and the child is eligible for randomization. A second MRI of the brain is obtained within six months of the initial screening MRI of the brain, to ensure that the participant has not developed a new or enlarging SCI prior to randomization.
Neurological Evaluation
In addition to the neurological evaluation performed to establish the presence of a SCI prior to randomization, the standardized neurological examination is performed at months 12, 24, and at study exit by the site pediatric neurologist.
Randomization
Participants are randomly assigned to either the treatment or observation arm after the second MRI of the brain is adjudicated by the neuroradiology committee.
For participants randomly assigned to the treatment arm, transfusions are begun within 2 weeks of random assignment (preferably within 4 weeks of the prerandomization MRI of the brain), to reduce the likelihood of reaching a neurological or neuroimaging endpoint before receiving the treatment.
Transfusion Management, Efficacy, and Safety
For participants randomized to receive transfusion therapy, automated exchange transfusion (erythrocytapheresis) is encouraged; simple transfusion or partial exchange transfusion are acceptable. Participants are transfused with leukocyte-poor, hemoglobin S-negative, packed red blood cells, 10 to 15 ml per kg per transfusion. Blood is matched for ABO, Rh (C, D, E), and Kell antigens, with additional matching for other minor blood group antigens by the institutional physician, as clinically indicated. The intent is to maintain the hemoglobin above 9–9.5 gm/dl and hemoglobin S ≤ 30%.
Participants are transfused initially at 2-week intervals, until the hemoglobin S level is reduced to ≤ 30%, with the intention of meeting this goal within 4 weeks after randomization. In the event that the hemoglobin S level cannot be consistently kept ≤ 30% and transfusions have been performed at a reasonable period of every 3 to 4 weeks, these participants will be considered effectively transfused.
Chelation therapy using deferoxamine or an oral chelator is expected for all participants on the transfusion arm, using the study recommendations as detailed in the full protocol in the supplemental online material.
Child Health Questionnaire (CHQ) and Utility Assessment
As various factors are considered when parents/caregivers make health care decisions for their children, a measure of health-related quality of life and overall utility is part of the study. The CHQ [17] has been validated in a wide range of pediatric populations, and has been used by researchers on cancer [18]. A Utility Assessment is also completed on parents/caregivers and health care providers. The Utility Assessment is designed to determine whether, in the eyes of the investigators and parents, the overall benefit of transfusion therapy outweighs the disadvantages. In the event that the study outcome is favorable with regard to the primary outcome, this assessment will assure that there is overall benefit to the participant and provide an indication of how participants and caregivers balance the overall benefits and disadvantages of transfusion therapy.
Cognitive Studies
The primary index of cognitive change is general intellectual ability (I.Q.), as assessed by the Wechsler Abbreviated Scale of Intelligence (WASI) [19] for children 6 years of age and older, or the Wechsler Preschool and Primary Scale of Intelligence-III (WPPSIII) [20] for children 5 years of age. A secondary index is executive behavior, as assessed by the Behavior Rating Inventory of Executive Function (BRIEF) [21]. The BRIEF emphasizes frontally mediated executive abilities and is used because most SCI associated with SCA affect the frontal lobes [10]. Masked cognitive test results are reviewed remotely by the psychology committee, using procedures similar to those of the Pediatric AIDS Clinical Trial Group [22].
The committee develops clinical consensus ratings similar to those reported by Ryan et al. [23] to classify cognitive change from baseline to study exit. A list of measures and procedural details are shown in supplemental online material (see Supplemental Table IV and the full protocol).
Postrandomization Evaluation
The Utility Assessment, CHQ, and cognitive testing are administered within the first three months of randomization for participants in the observation arm. Given that the baseline hemoglobin level may affect test results, the Utility Assessment and CHQ are administered before the participant’s first blood transfusion in the treatment arm, allowing for comparability between the treatment and observation arm at baseline. For cognition, the analysis will be change in I.Q. over time within groups; thus, cognitive testing is done when the maximum hemoglobin S level is less than 30% (after the third, but prior to the fifth transfusion). These choices were made to avoid false attribution of improvements to prevention of SCI, when in fact they might be due to the acute effects of transfusion.
Interim Evaluations (12 to 18 Months After Study Entry)
Cognitive and behavioral assessment on all participants and TCD measurements will be done at 12–18 months after study entry for participants who have a normal range TCD and are in the observation arm. Participants in the observation arm with a high conditional TCD measurement will have TCDs at 6, 12, and 24 months after study entry. Interim TCDs are not a study requirement in the treatment arm.
Annual Evaluations and Study Exit
All participants will have an annual neurological exam with the study neurologist. Upon study exit, all participants will have an MRI of the brain, cognitive, behavioral, and neurological assessment.
Evaluation and Adjudication of Suspected Strokes
Special effort is made within the study to identify acute or subacute events that could represent an overt stroke, as this is a study endpoint and important for participant safety. Participants who have acute neurological events that are suggestive of, but not clearly indicative of, an overt stroke receive an examination by the study neurologist and an MRI of the brain (within 24 h of the neurological event), using the SIT Trial imaging protocol and including diffusion weighted sequences if possible, as findings on MRI may influence the decision about whether a study endpoint has been reached (stroke or new or progressive SCI). Neurological evaluation by the study site neurologist includes a pediatric adaptation of the NIH Stroke Scale (PedNIHSS) [24] (see Supplemental Table V, supplemental online material).
Official determination of an overt stroke is adjudicated by three members of the neurology committee, based on historical information, the neurological examination, and a MRI of the brain. Criteria for establishing the diagnosis of stroke and transient ischemic attack (TIA) are shown in Supplemental Table VI of the supplemental online material.
Participants who are designated as having had overt strokes or new or progressive SCI are considered to have reached the primary outcome of the study and are offered the options to be treated with blood transfusion therapy, stop blood transfusion or consider some other therapeutic choice, on the basis of the decision of the local study site investigator and family.
Further details of the evaluation and adjudication of suspected strokes are included in the full protocol in the supplemental online material.
Banking of DNA and Plasma Specimens
The SIT Trial represents a unique opportunity to study longitudinally a large, well-defined cohort of patients with and without SCI. Provision of a blood sample for genetic studies is part of the initial consent and screening procedures; therefore, the genetics samples will be obtained on all participants who receive MRIs of the brain. Lymphocytes are harvested and transformed using Epstein-Barr Virus, to allow for a renewable source of DNA and RNA for this study and a repository is maintained. Consent for the use of DNA, RNA, and plasma for anonymous studies is obtained as part of the main consent form.
Statistical Considerations
Sample Size and Statistical Analysis
Eligible children are allocated to transfusion and observation arms of the study by the Statistical and Data Coordinating Center, with randomization to each study arm balanced within each clinical center. Adjustments will be made to control for age and gender in the comparisons of the transfusion and observation arms, as age may be related to the risk of overt stroke [3] and gender is associated with the secondary outcome measurement of I.Q. [25]. Adams et al. demonstrated that patients with TCD measurements less than 200 cm/sec and greater than 170 cm/sec are at risk for overt strokes, but suggest the risk is not sufficiently high to justify blood transfusion therapy [14]. High normal TCD velocities in the presence of SCI may further increase the risk of future cerebral infarcts or new strokes as assessed by MRI; therefore, the analysis will be adjusted for this factor as well. Block randomization will be used, with a unique randomization schedule for each study site. All study participants will be randomly allocated to receive a maximum of 3 years of observation or blood transfusion therapy. Each participant will be followed through study exit.
Primary Hypothesis
We anticipate the study will have 85% power to detect a difference of at least 86% in the primary study outcome (i.e., the incidence of overt stroke and new or progressive SCI) in children who receive blood transfusion therapy, compared with children who do not receive this therapy. The sample size assumes a 22% recurrence rate in the observation arm and a 3% recurrence rate in the transfused group for a two-tailed alpha = 0.05. To allow for an expected 16% crossover rate from transfusion to observation, an expected 3% crossover rate from observation to transfusion, and an expected 10% dropout rate, 102 children will be randomly assigned to each arm. For the primary hypothesis, a p value of <0.05 will be considered statistically significant.
Reasons for the high projected crossover rate include: (1) some children may develop complications due to transfusion and will be switched to observation after randomization and (2) some children may have: (a) elevated TCD measurements after randomization and will be placed on transfusion; (b) repetitive acute chest syndrome that is not responsive to hydroxyurea; or (c) other complications of SCA.
Secondary Hypotheses
For the analysis of Secondary Hypothesis 1 (transfusion therapy limits the further decline in intellectual abilities compared to observation), all participants will be assessed with a cognitive battery at randomization, 12 to 18 months, and at study exit. The major indicator of cognitive loss will be a decline in I.Q., as assessed by the WASI [19] (children 6–14 years of age) or WPPSIII [20] (children 5 years of age). The BRIEF will be used similarly to assess behavioral decline [21].
Using data for participants from English-speaking countries, a two-group t-test will be used to assess changes in I.Q. and BRIEF T scores from baseline to study exit between the two treatment groups. With 84 participants in each arm (total sample of 168, assuming that approximately 10% of 184 participants are lost-to-follow-up), we will have 80% power (two-tailed alpha = 0.01) to detect an 8-point difference in I.Q. between the study arms (on a scale with M = 100 and SD = 15) and 82% power (2-tailed alpha = 0.01) to detect a 5.5-point difference in BRIEF T scores (on a scale with M = 50 and SD = 10) between study arms. In addition to using statistical techniques to evaluate change, clinical significance of cognitive change will be evaluated on the basis of clinical consensus ratings from the psychology committee. An intention-to-treat analysis will be used [26].
After completion of the study, the critical question will be as follows: What level of efficacy makes blood transfusion therapy worthwhile? For analysis of Secondary Hypothesis 2 (the overall benefit of blood transfusion therapy for SCI outweighs the associated risks), parents and health care providers may have different perceptions of the risk/benefit balance (both within and between groups). Utility Assessment will be performed through ranking, rating scale, and standard gamble for the following outcomes: SCA without major problems, painful event, acute chest crisis, SCI, overt stroke, blood transfusion therapy, blood transfusion therapy and viral infection, and blood transfusion therapy and chelation.
We will elicit assessments from one parent/caregiver for each participant, and health care providers at participating centers. Parents will provide assessments within 3 months of randomization (Observation Arm) or before the first transfusion (Transfusion Arm). Assessments will be repeated at exit from the study.
For the two-planned secondary analyses and for all other secondary analyses, a p value of ≤0.01 will be considered strong evidence against the null hypothesis; a p value of >0.01 and ≤0.05 will be considered some evidence against the null hypothesis; and a p value >0.05 will be considered no evidence against the null hypothesis.
Data Analysis
Details of the data analysis plan are included in the full protocol in the supplemental online material.
Interim Data and Stopping Rules and Data and Safety Monitoring
An independent Data and Safety Monitoring Board (DSMB), appointed by NINDS, reviews study progress and safety results every six months. In addition, an independent Medical Safety Monitor reviews all serious adverse events, including overt strokes, in real time. Local site Principal Investigators are encouraged to contact the Medical Safety Monitor, to avoid investigator bias in treating these events. The local investigators are discouraged from seeking advice from study leadership regarding treatment of overt neurological events in study participants.
As no MRI of the brain is performed between the baseline assessment and the final assessment at 36 months, formal monitoring of the primary outcome will be to consider the overt stroke rate between the two study arms. If there is a significant difference in the overt stroke rates between the two study arms, the DSMB will decide to stop or continue the study. As a reduction in stroke risk in the treatment group compared to the control group is expected, a difference in stroke rate significantly greater than expected would be needed to halt the study. The DSMB will need to take into account the lack of an accepted treatment for SCI. Similarly, as there is a known association of SCI and lower I.Q., it is unlikely that the study would be halted because of a difference in I.Q. at an interim analysis, unless the difference greatly exceeded that expected.
SIGNIFICANCE OF THE SIT TRIAL
SCI remain the most frequent morbid neurological condition in patients with SCA; on the basis of previous studies, approximately 22% to 27% of all school-age children will have a SCI on MRI of the brain [5, 6, 27], lower cognitive scores [8, 9, 28], and poor academic attainment [7], when compared with children without SCI or siblings. Presently, there is no systematic strategy to identify or treat this population, despite the fact that they have significant neurological morbidity, with an increased risk of further neurological compromise. At the completion of this study, we will be able to determine whether blood transfusion therapy will prevent further cerebral ischemic injury and whether the measured benefits outweigh the risks. Advances in our understanding and treatment of SCI will likely lead to a decrease in the burden associated with ischemic injury in children with SCA.
This study will provide valuable longitudinal information on the cognition and behavior of children with SCI. Additionally, after completion of this study, we will understand more about the relationship between the burden and the location of the cerebral infarcts and the magnitude and nature of cognitive deficits. These data are important for establishing effective cognitive rehabilitation strategies for children with strokes or other brain injuries, (e.g., trauma or treatment such as radiation therapy or neurosurgery for children with brain tumors). We are hopeful that insights gained from this population, whose SCI arise because of a single gene mutation, undoubtedly modified by other accessory genetic and environmental factors, will provide insights into the etiology and treatment of SCI in other patient groups. The availability of an extensive archive of MRI data and additional disease-related information gained throughout this study will enhance future investigations. Finally, the availability of a biologic repository with an infinite supply of DNA, in association with a well-defined phenotype for SCI, should provide the opportunity and impetus for continued study of this and other sickle cell-related problems for the indefinite future.
Supplementary Material
Acknowledgments
This study is supported by cooperative agreements U01-NS042804 and U01-NS042940 from the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health. The authors would like to acknowledge the extraordinary efforts of the study coordinators for the SIT Trial and the families and children with SCA who were participants in the trial.
Coordinators
Coordinators for the SIT Trial at the time of this submission include Liz Dackiw from Johns Hopkins University School of Medicine, Anjum Zaki (Subsite coordinator) from the Georgetown University Hospital, Joan Marasciulo (Subsite coordinator) from the Sinai Hospital, Chrissy Rhoads, (Subsite coordinator) from University of Maryland, Barbara Speller-Brown from Children’s National Medical Center, Ruth Baldwin and Fran Wright from the University of North Carolina, Mary DeBarr from the Rainbow Babies and Children’s Hospital, Kami Perdue from The Research Center at Nationwide Children’s Hospital, Marlene Eaton from the Cincinnati Comprehensive Sickle Cell Center, Cincinnati Children’s Hospital Medical Center, Cindy Davis from Indiana University Purdue University Indianapolis, Cynthia Burnett from Wayne State University, Janice R. Beatty from Northwestern University, Danielle Jirovec from Medical College of Wisconsin, Susan Sarcone from the Children’s Mercy Hospital and Clinics, Cindy Terrill and Terianne Lindsey from Washington University School of Medicine, Angela Mull from the Arkansas Children’s Hospital Research Institute, Mary D. Jones from University of Alabama at Birmingham, Kim Gross from University of Mississippi Medical Center, Bogdan Dinu from Baylor College of Medicine, Michael Henson and Brad Cook from University of Texas Southwestern Medical Center, Jacqueline Davis from Wake Forest University Health Sciences, Jeffrey E. Olson from Children’s Hospital of Philadelphia, Giselle Padmore-Pennington from Guy’s and Saint Thomas’ Hospital NHS Foundation Trust, Heather Newell from The Royal London NHS Trust, Annette Gilmore from Central Middlesex Hospital North West London Hospitals NHS Trust, Annie Kamdem from Hopital Intercommual de Créteil, and Nagina Parmar from University of Toronto.
Active Clinical Centers
James F. Casella, (Principal Investigator), Harold Lehman and John J. Strouse (Coinvestigators) from Johns Hopkins University School of Medicine, Corina Gonzalez (Subsite Investigator) from Georgetown University Hospital, Jason Fixler and Joseph M. Wiley (Subsite Investigators) from Sinai Hospital, Neil Grossman (Subsite Investigator) from the University of Maryland, Helge Hartung (Principal Investigator) (previously Caterina P. Minniti and Ana Burgos) from Children’s National Medical Center, Rupa Redding-Lallinger, MD (Principal Investigator) from University of North Carolina, Beng Fuh (Principal Investigator) (previously Charles Daeschner and Mario Grossi) from East Carolina University, Brian Berman (Principal Investigator) (previously Anthony Villella) from the Rainbow Babies and Children’s Hospital, Melissa M. Rhodes (Principal Investigator) (previously Mark A. Ranalli) from Ohio State University, Karen Kalinyak (Principal Investigator) from the Cincinnati Children’s Hospital Medical Center, Mark Heiny (Principal Investigator) (previously Kathleen Neville) from Indiana University Purdue University Indianapolis, Sharada A. Sarnaik (Principal Investigator) from Wayne State University, Alexis A. Thompson (Principal Investigator) from Northwestern University, Julie A. Panepinto (Principal Investigator) from Medical College of Wisconsin, Gerald M. Woods (Principal Investigator) from Children’s Mercy Hospital and Clinics, Allison King (Principal Investigator) from Washington University School of Medicine, Thomas H. Howard (Principal Investigator) from University of Alabama at Birmingham, Rathi V. Iyer (Principal Investigator) from University of Mississippi Medical Center, Gladstone Airewele (Principal Investigator) from Baylor College of Medicine, Charles T. Quinn (Principal Investigator) from University of Texas Southwestern Medical Center, Hernan Sabio (Principal Investigator) from Wake Forest University Health Sciences, Janet L. Kwiatkowski (Principal Investigator) from The Children’s Hospital of Philadelphia, Melanie Kirby-Allen (Principal Investigator) from University of Toronto, Fenella Kirkham (Principal Investigator) from University College London Institute of Child Health, Baba Inusa (Principal Investigator) from Guy’s and Saint Thomas’ Hospital NHS Foundation Trust, Paul Telfer (Principal Investigator) from The Royal London NHS Trust, and Francoise Bernaudin (Principal Investigator) from Hopital Intercommual de Creteil.
Inactive Clinical Centers
Scott T. Miller (Principal Investigator) from the State University of New York - Downstate, Suzanne L. Saccente (Principal Investigator) from Arkansas Children’s Hospital Research Institute, Charles Scher (Principal Investigator) from Tulane University Health Sciences Center, Thomas Coates (Principal Investigator) from Children’s Hospital of Los Angeles, Michelle Afif (Principal Investigator) (previouslyJoanna Howard) from Central Middlesex Hospital North West London Hospitals NHS Trust, and David Rees from King’s College Hospital NHS Foundation Trust.
Resource Centers
Project Office: Deborah Hirtz (Project Scientist) and Claudia S. Moy (Program Director) from the National Institutes of Health - National Institute of Neurological Disorders and Stroke.
Clinical Coordinating Center: Michael R. DeBaun (Principal Investigator for the Clinical Coordinating Center and SIT Trial), Teresa Roediger (Project Manager), (previously Colleen Kilbourne-Glynn) and Cindy Terrill (Project Manager) from Washington University School of Medicine.
Statistical and Data Coordinating Center: Bruce Barton (Principal Investigator) and Adrienne Brandon (Project Manager) from Maryland Medical Research Institute.
Electronic Radiology Laboratory (ERL): Fred Prior (Director), Stephen M. Moore (Systems Administrator), and Bruce A. Vendt (Project Manager) from the Mallinckrodt Institute of Radiology.
Biologic Repository: James F. Casella, (Director), Kimberly Jones, John J. Strouse, and Emily Barron-Casella from Johns Hopkins University School of Medicine. Margaret Penno and Tanya Ray from the Genetic Resources Core Facility Cell Center (GRCF).
Committees
Data and Safety Monitoring Board: David Schoenfeld (Chair), Massachusetts General Hospital,; Thomas Adamkiewicz, Morehouse School of Medicine; Stephen Ashwal, Loma Linda University; H. Stacy Nicholson, Oregon Health & Science University; Guillaume Sebire, Universite De Sherbrooke; Janice Cordell, National Institutes of Health.
Advisory Committee: William Powers (Chair), University of North Carolina, Robert J. Adams, Medical University of South Carolina; Jeffrey Schatz, University of South Carolina; Yuko Palesch, Medical University of South Carolina.
Executive Committee: Michael R, DeBaun (Chair and Principal Investigator for the SIT Trial), Robert C. McKinstry, III, and Michael J. Noetzel, Washington University School of Medicine; James F. Casella (Vice Chair and Co-principal Investigator for the SIT Trial), Johns Hopkins University School of Medicine; Bruce Barton, Maryland Medical Research Institute; Rebecca N. Ichord, Children’s Hospital of Philadelphia; Fred Prior, Mallinckrodt Institute of Radiology, Washington University School of Medicine; Deborah Hirtz, National Institutes of Health - National Institute of Neurological Disorders and Stroke; and Desiree White, Washington University Department of Psychology. 2004–2005 Site Investigators: Caterina Minniti, Children’s National Medical Center; Thomas Coates, Children’s Hospital of Los Angeles; Thomas H. Howard, University of Alabama at Birmingham. 2005–2006 Site Investigators: Gladstone Airewele, Baylor College of Medicine; Karen Kalinyak, Cincinnati Comprehensive Sickle Cell Center; Anthony Villella, Rainbow Babies and Children’s Hospital. 2006–2007 Site Investigators: Charles T. Quinn, University of Texas Southwestern; Julie A. Panepinto, Medical College of Wisconsin; Rupa Redding-Lallinger, University of North Carolina-Chapel Hill,. 2007–2008 Site Investigators: Thomas H. Howard, University of Alabama at Birmingham; Sharada Sarnaik, Wayne State University. 2008–2009 Site Investigators: Charles T. Quinn, University of Texas Southwestern; Sharada Sarnaik, Wayne State University; Beng Fuh, East Carolina University.
Neurology Committee: Michael J. Noetzel (Chair), Washington University School of Medicine; Michael Dowling, University of Texas Southwestern Medical Center; Deborah Hirtz, National Institutes of Health – National Institute of Neurological Disorders and Stroke; Rebecca N. Ichord, Children’s Hospital of Philadelphia; E. Steve Roach, Nationwide Children’s Hospital.
Psychology Committee: Desiree White (Chair), Washington University Department of Psychology,; T. David Elkin, University of Mississippi Medical Center; Mary M. George, Baylor College of Medicine; H. Gerry Taylor, Rainbow Babies and Children’s Hospital.
Neuroradiology Committee: Robert C. McKinstry, III (Chair), Washington University School of Medicine; William S. Ball, Jr., University of Cincinnati; Michael A. Kraut, Johns Hopkins Hospital; Marilyn Siegel, Washington University School of Medicine.
Publications and Presentation Committee: Comprised of members of the SIT Trial executive committee. Additional members of the Publications Committee may be appointed by the chair of the executive committee on an ad hoc basis.
Protocol Review Committee: Comprised of members of the SIT Trial executive committee. Additional members of the Protocol Review Committee may be appointed by the chair of the executive committee on an ad hoc basis.
Definitions
Silent cerebral infarct-like lesions: An MRI signal abnormality visible on two views on the T-2 weighted images (axial and coronal). The signal abnormality must measure at least 3 mm in one dimension, based on consensus of two of three neuroradiologists.
Silent cerebral infarct: A silent cerebral infarct is a silent cerebral infarct-like lesion as reported by the neuroradiology committee that has been adjudicated as silent by the neurology committee. A lesion is adjudicated as silent only if the neurological examination is normal or there is an abnormality on the neurological exam that cannot be explained by the location of the MRI lesion.
Transient Ischemic Attack (TIA): A neurological deficit lasting less than 24 h with a negative MRI of the brain
Screening: Patient visits, testing to determine eligibility and patient/parent willingness to participate in the trial.
Stroke: A neurological deficit lasting more than or less than 24 h with a positive MRI (new vascular lesion which could explain deficit) or a neurological deficit lasting more than 24 h with a negative MRI or vascular lesion that does not explain deficit.
Footnotes
Declaration of Interest: The authors report no conflict of interest. The authors alone are responsible for the content and writing of this paper.
NOTE
Additional Materials
Additional figures and tables as well as a copy of the full protocol for the SIT Trial are included as supplemental online material (http://sitstudy.wustl.edu).
Contributor Information
James F. Casella, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Allison A. King, Washington University School of Medicine, St. Louis, Missouri, USA.
Bruce Barton, Maryland Medical Research Institute, Baltimore, Maryland, USA.
Desiree A. White, Washington University Department of Psychology, St. Louis, Missouri, USA.
Michael J. Noetzel, Washington University School of Medicine, St. Louis, Missouri, USA.
Rebecca N. Ichord, Children’s Hospital of Philadelphia, Philadelphia, USA.
Cindy Terrill, Washington University School of Medicine, St. Louis, Missouri, USA.
Deborah Hirtz, National Institutes of Health, National Institute of Neurological Disorders and Stroke, Bethesda, Maryland, USA.
Robert C. McKinstry, Washington University School of Medicine, St. Louis, Missouri, USA.
John J. Strouse, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Thomas H. Howard, University of Alabama Medical Center, Birmingham, Alabama, USA.
Thomas D. Coates, Children’s Hospital, Los Angeles, California, USA.
Caterina P Minniti, Children’s National Medical Center, Washington, DC, USA.
Andrew D. Campbell, University of Michigan, Ann Arbor Michigan, USA.
Bruce A. Vendt, Washington University School of Medicine, St. Louis, Missouri, USA.
Harold Lehmann, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Michael R. DeBaun, Washington University School of Medicine, St. Louis, Missouri, USA.
References
- 1.Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151:839–845. doi: 10.1093/oxfordjournals.aje.a010288. [DOI] [PubMed] [Google Scholar]
- 2.Earley CJ, Kittner SJ, Feeser BR, et al. Stroke in children and sickle-cell disease: Baltimore-Washington Cooperative Young Stroke Study. Neurology. 1998;51:169–176. doi: 10.1212/wnl.51.1.169. [DOI] [PubMed] [Google Scholar]
- 3.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–294. [PubMed] [Google Scholar]
- 4.Glauser TA, Siegel MJ, Lee BC, DeBaun MR. Accuracy of neurologic examination and history in detecting evidence of MRI-diagnosed cerebral infarctions in children with sickle cell hemoglobinopathy. J Child Neurol. 1995;10:88–92. doi: 10.1177/088307389501000203. [DOI] [PubMed] [Google Scholar]
- 5.Pegelow CH, Macklin EA, Moser FG, et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. 2002;99:3014–3018. doi: 10.1182/blood.v99.8.3014. [DOI] [PubMed] [Google Scholar]
- 6.Kwiatkowski JL, Zimmerman RA, Pollock AN, et al. Silent infarcts in young children with sickle cell disease. Br J Haematol. 2009;146:300–305. doi: 10.1111/j.1365-2141.2009.07753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schatz J, Brown RT, Pascual JM, et al. Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease. Neurology. 2001;56:1109–1111. doi: 10.1212/wnl.56.8.1109. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong FD, Thompson RJ, Jr, Wang W, et al. Cognitive functioning and brain magnetic resonance imaging in children with sickle cell disease. Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Pediatrics. 1996;97:864–870. [PubMed] [Google Scholar]
- 9.Moran CJ, Siegel MJ, DeBaun MR. Sickle cell disease: imaging of cerebrovascular complications. Radiology. 1998;206:311–321. doi: 10.1148/radiology.206.2.9457180. [DOI] [PubMed] [Google Scholar]
- 10.Moser FG, Miller ST, Bello JA, et al. The spectrum of brain MR abnormalities in sickle-cell disease: A report from the Cooperative Study of Sickle Cell Disease. AJNR Am J Neuroradiol. 1996;17:965–972. [PMC free article] [PubMed] [Google Scholar]
- 11.Baird RL, Weiss DL, Ferguson AD, et al. Studies in sickle cell anemia: XXI. Clinico-pathological aspects of neurological manifestations. Pediatrics. 1964;34:92–100. [PubMed] [Google Scholar]
- 12.King AA, Noetzel M, White DA, et al. Blood transfusion therapy is feasible in a clinical trial setting in children with sickle cell disease and silent cerebral infarcts. Pediatr Blood Cancer. 2008;50:599–602. doi: 10.1002/pbc.21338. [DOI] [PubMed] [Google Scholar]
- 13.Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ. 1999;319:670–674. doi: 10.1136/bmj.319.7211.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. New Engl J Med. 1998;339:5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 15.Scothorn DJ, Price C, Schwartz D, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapyfor at least five years after initial stroke. J Pediatr. 2002;140:348–354. doi: 10.1067/mpd.2002.122498. [DOI] [PubMed] [Google Scholar]
- 16.Pegelow CH, Wang W, Granger S, et al. Silent infarcts in children with sickle cell anemia and abnormal cerebral arteryvelocity. Arch Neurol. 2001;58:2017–2021. doi: 10.1001/archneur.58.12.2017. [DOI] [PubMed] [Google Scholar]
- 17.Landgraf JM. CHQ Manual. Boston: Health Act; 2000. [Google Scholar]
- 18.Nixon SK, Maunsell E, Desmeules M, et al. Mutual concurrent validity of the child health questionnaire and the health utilities index: an exploratory analysis using survivors of childhood cancer. Int J Cancer Suppl. 1999;12:95–105. doi: 10.1002/(sici)1097-0215(1999)83:12+<95::aid-ijc18>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 19.Wechsler D. Wechsler Abbreviated Scale of Intelligence Manual. San Antonio: The Psychological Corporation; 1999. [Google Scholar]
- 20.Wechsler D. Wechsler Pre-School and Primary Scale of Intelligence, WPPSI. New York: The Psychological Corporation; 1967. [Google Scholar]
- 21.Gioia GA, Isquith PK, Guy SC, Kenworthy L. Behavior Rating Inventory of Executive Function. Lutz: Psychological Assessment Resources; 2000. [Google Scholar]
- 22.Raskino C, Pearson DA, Baker CJ, et al. Neurologic, neurocognitive, and brain growth outcomes in human immunodeficiency virus-infected children receiving different nucleoside antiretroviral regimens. Pediatric AIDS Clinical Trials Group 152 Study Team. Pediatrics. 1999;104:e32. doi: 10.1542/peds.104.3.e32. [DOI] [PubMed] [Google Scholar]
- 23.Ryan C, Adams KM, Heaton RK, Grant I. Neurobehavioral assessment of medical patients in clinical trials: the DCCT experience. In: Mohr EBP, editor. Handbook of Clinical Trials: The Neurobehavioral Approach. Amsterdam/Lisse: Swets & Zeitlinger BV; 1991. pp. 215–242. [Google Scholar]
- 24.Ichord R, Smith S, Garcia-Espana F, et al. Pediatric adaptation of NIH stroke scale predicts outcome after arterial ischemic stroke in children. Stroke. 2005;36:480–481. Abstract. [Google Scholar]
- 25.Hurtig AL, White LS. Psychosocial adjustment in children and adolescents with sickle cell disease. J Pediatr Psychol. 1986;11:411–427. doi: 10.1093/jpepsy/11.3.411. [DOI] [PubMed] [Google Scholar]
- 26.Armitage P. Exclusions, losses to follow-Up, and withdrawals in clinical trials. In: Shapiro SLT, editor. Clinical Trials: Issues and Approaches. New York: Marcel Dekker; 1983. pp. 99–113. [Google Scholar]
- 27.Buchanan GR, DeBaun MR, Quinn CT, Steinberg MH. Sickle cell disease. Hematology Am Soc Hematol Educ Program. 2004:35–47. doi: 10.1182/asheducation-2004.1.35. [DOI] [PubMed] [Google Scholar]
- 28.Craft S, Schatz J, Glauser TA, et al. Neuropsychologic effects of stroke in children with sickle cell anemia. J Pediatr. 1993;123:712–717. doi: 10.1016/s0022-3476(05)80844-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.