

Abstract
Objective
Isopropylamine NONOate (IPA/NO) is a nitroxyl (HNO) donor at physiological pH. HNO is a positive inotrope and vasodilator, but little is known about its effect on neointimal hyperplasia. The aims of this study are to determine the effect of IPA/NO on endothelial and vascular smooth muscle cells (VSMC) in vitro, and to determine if IPA/NO inhibits neointimal hyperplasia in vivo.
Methods
VSMC were harvested from the abdominal aortas of male Sprague Dawley rats, and human umbilical vein endothelial cells were purchased from ATCC. In vitro, cellular proliferation was assessed by 3H-thymidine incorporation, cell migration was assessed using the scrape assay, and cell death was assessed using Guava Personal Cell Analysis (PCA). Cell cycle analysis was performed using propidium iodide staining and FACS analysis. Protein expression was assessed using Western blot analysis. Phosphorylated proteins were assessed using immunoprecipitation and Western blot analysis. In vivo, the carotid artery injury model was performed on male Sprague Dawley rats treated with (n=12) or without (n=6) periadventitial IPA/NO (10 mg). Arteries harvested at 2 weeks were assessed for morphometrics using ImageJ. Inflammation was assessed using immunohistochemistry. Endothelialization was assessed by Evans blue staining of carotid arteries harvested 7 days after balloon injury from rats treated with (n=6) or without (n=3) periadventitial IPA/NO (10 mg).
Results
In vitro, 1000 μmol/L IPA/NO inhibited both VSMC (38.7±4.5% inhibition vs. control, P=0.003) and endothelial cell proliferation (54.0±2.9% inhibition vs. control, P=<0.001) without inducing cell death or inhibiting migration. In VSMC, this inhibition was associated with an S-phase cell cycle arrest and increased expression of cyclin A, cyclin D1 and the cyclin-dependent kinase inhibitor p21. No change was noted in the phosphorylation status of cdk2, cdk4, or cdk6 by IPA/NO. In rodents subjected to the carotid artery balloon injury model, IPA/NO caused significant reductions in neointimal area (298±20 vs. 422±30, P=<0.001) and medial area (311±14 vs. 449±16, P=<0.001) compared to injury alone, and reduced macrophage infiltration to 1.7±0.8 from 16.1±3.5 cells per high power field (P=<0.001). IPA/NO also prevented re-endothelialization compared to injury alone (55.9±0.5% non-endothelialized vs. 21±4.4%, respectively, P=0.001). Lastly, a 50% mortality rate was observed in the IPA/NO-treated groups.
Conclusions
In summary, while IPA/NO modestly inhibited neointimal hyperplasia by inhibiting VSMC proliferation and macrophage infiltration, it also inhibited endothelial cell proliferation and induced significant mortality in our animal model. Since HNO is being investigated as a treatment for congestive heart failure, our results raise some concerns about the use of IPA/NO in the vasculature and suggest that further studies be conducted on the safety of HNO donors in the cardiovascular system.
INTRODUCTION
Isopropylamine NONOate (IPA/NO) was originally synthesized as a diazeniumdiolate by Drago and Karstetter in 1961.1 However, it was mostly disregarded as a nitric oxide (NO) donor because it was relatively unstable and its conversion to NO occurred in relatively low yield.2 Later, interest in the compound grew when it was realized that IPA/NO released nitroxyl (HNO) at predictable rates. IPA/NO has a short half-life in solution (t1/2 ≈ 2 minutes), and HNO is not the only nitrogen species released from its decomposition:3 IPA/NO releases HNO and NO, the levels of which depend on reaction conditions such as buffer pH and initial concentration of IPA/NO.3 At physiologic pH, the major pathway of IPA/NO breakdown yields HNO and isopropanol, and the minor pathway yields NO and the parent compound, isopropylamine (IPA). The higher the initial concentration of IPA/NO, and the higher the pH of the initial solution, the more likely IPA/NO is to break down to HNO and isopropanol.
Another HNO donor, Angeli’s salt, has been used for years for in vivo and in vitro studies because it is stable and decomposes with known kinetics in physiological buffers.4, 5 Like IPA/NO, Angeli’s salt has a short half-life and releases multiple nitrogen oxides—HNO and nitrite (NO2−)—upon decomposition in solution.4 Unlike Angeli’s salt, IPA/NO is organic and thus can be further modified with protective groups to extend the short half-life in physiological conditions and mitigate any potential side effects from unwanted breakdown products.
Several recent studies have shown that HNO is a positive inotrope,6 a vasodilator,7 and subject to redox chemistry regulation.8, 9 Since the positive inotropic action of HNO is unaffected by beta-blockers—indeed, it acts synergistically with them 6—and, since the vasodilatory effects of HNO are not subject to development of tolerance,10 nitroxyl donors are being investigated as agents for treatment of congestive heart failure 11 and replacements for nitroglycerin or dobutamine.6, 12 These data are the impetus for the renewed interest in understanding the biochemical and biological activity of HNO, especially in its capacity to act both similar to and opposite of NO in the vasculature.
With the growing amount of evidence suggesting a therapeutic potential for HNO in the cardiovascular system, we sought to determine if the organic HNO donor IPA/NO would have an effect on the development of neointimal hyperplasia following arterial injury in the vasculature, since this was unknown. Therefore, the aim of this study was two-fold: 1) to determine the effect of IPA/NO on endothelial and vascular smooth muscle cell (VSMC) proliferation and death in vitro; and, 2) to determine if IPA/NO inhibits neointimal hyperplasia in vivo. Our hypothesis is that IPA/NO will inhibit neointimal hyperplasia following vascular injury.
MATERIALS AND METHODS
Cell culture
VSMC were harvested and cultured from the abdominal aorta of 10–12 week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN), and maintained as previously described.13, 14 Human umbilical vein endothelial cells (HUVEC, line CRL-1730) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained according to ATCC protocols. Cells were used for in vitro experimentation between passages 3–9.
IPA/NO preparation
IPA/NO was synthesized by L.K.K. and J.E.S. as previously described.15 Stock solutions (10 mmol/L) were prepared by dissolving solid IPA/NO in the appropriate complete culture medium just before use. The final pH during treatment was 7.8.
Griess reaction
To compare the amount of NO released by IPA/NO and the NO donor diethylenetriamine NONOate (DETA/NO), a Greiss reaction was conducted. Briefly, after samples were maintained at 37°C and 5% CO2 for 24 hours, duplicate 100 μL aliquots were combined with an equal volume of the Griess reagents (1% sulfanilamide/0.1% naphthylethylenediamine dihydrochloride/2.5% H3PO4). Nitrite, which is a stable breakdown product of NO metabolism and serves as a proxy for NO production, was measured in the samples by determining their absorbance at 550 nm in a microplate reader.
Cell proliferation
VSMC or HUVEC plated in 12-well plates (4×104 cells/well) were growth-arrested for 24 hours in medium without fetal bovine serum (FBS). Cells were then incubated in complete medium containing tritiated [3H]-thymidine (5 μCi/mL, PerkinElmer, Wellesley, MA) and IPA/NO (100–1000 μmol/L) for 24 hours. [3H]-thymidine incorporated into trichloroacetic-acid-precipitated DNA was measured with a Wallac WinSpectral 1414 liquid scintillation counter (Wallac, Turku, Finland).
Cell death
VSMC or HUVEC were plated and growth-arrested as above, followed by incubation in complete medium containing IPA/NO (100–1000 μmol/L) for 24 hours. Cells were collected, pelleted, resuspended in 1X Hank’s Balanced Salt Solution (HBSS) (Invitrogen, Carlsbad, CA), diluted five-fold (VSMC) or two-fold (HUVEC) in Guava ViaCount Reagent (Guava Technologies, Hayward, CA), and cell death assessed by Guava PCA as previously described. 16–18
Cell migration assay
VSMC and HUVEC plated in 6-well plates (1×105 and 2×105 cells/well, respectively) were growth-arrested as above. Monolayers were injured by a single scrape with a 1000 μL pipet tip, followed by treatment with IPA/NO as above, then immediately photographed, and photographed again 24 hours after treatment. Blinded counting of nuclei of cells that migrated into the empty space created by the scrape was performed at both time points using Adobe Photoshop 8.0 (Adobe Systems Inc., San Jose, CA), and quantitation was performed using ImageJ (National Institutes of Health, Bethesda, MD).
Flow cytometry
VSMC plated in 10-cm plates (9×105 cells/plate) were growth-arrested as above, then exposed to complete medium containing IPA/NO (100–1000 μmol/L) for 24 hours. Cells were trypsinized, pelleted, resuspended in 1X phosphate-buffered saline (PBS), and then fixed with ice-cold 70% ethanol. After ethanol fixation, cells were resuspended in a propidium iodide (PI) (Invitrogen, Carlsbad, CA) staining solution [1X PBS (pH 7.4), 50 g/mL PI, 204 μg/mL RNase A (Sigma, St. Louis, MO), 0.1% Triton X-100 (Fischer Biotech, Fair Lakes, NJ)], incubated for 15 minutes at 37°C, then analyzed on a Coulter Epic XL flow cytometer (Coulter, Hialeah, FL). Data were analyzed using ModFit 3.1 LT (Verity, Topsham, ME).
Western blot analysis
Whole-cell suspensions of IPA/NO-treated and untreated VSMC were prepared as previously described 19 and protein concentrations determined by bicinchoninic acid assays performed according to the manufacturer’s instructions (Pierce, Rockford, IL). Whole-cell suspensions were subjected to acrylamide gel electrophoresis on 8–13% gels, after which proteins were transferred to a nitrocellulose membrane. Protein expression was determined using antibodies to p21 (1:500), p27 (1:1000), cdc2 (1:1000), cyclin-dependent kinase (cdk) 2 (1:1000), cdk4 (1:500), cdk6 (1:1000), cyclin A (1:1000) and cyclin D1 (1:1000), all from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The antibody to cyclin B1 (1:250) was from BD Pharmingen (San Jose, CA).
Immunoprecipitation
For immunoprecipitations, IPA/NO-treated and untreated VSMC were lysed using lysis buffer A (20 mmol/L Tris-HCl [pH 7.5], 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L ethyleneglycoltetraacetic acid (EGTA), 1% (v/v) Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerol phosphate, 1 mmol/L sodium orthovanadate, 1 μg/mL leupeptin, and 1 mmol/L PMSF), and 300 μg of lysate was incubated with 1 μg of rabbit IgG plus 20 μL of protein A/G sepharose (both from Santa Cruz) for 1 hour at 4°C on a rotating wheel. The beads were then pelleted by centrifugation for 5 minutes at 1500 rpm and 4°C in an Eppendorf 5417R centrifuge (Westbury, NY), and the supernatant transferred to a fresh tube, to which was added 20 μL of protein A/G sepharose and 2.5 μg of either cdc2, cdk2, cdk4 or cdk6 antibody. After overnight incubation at 4°C on a rotating wheel, the beads were pelleted at 2500 rpm for 15 minutes at 4°C and the supernatants carefully removed. The beads were then subjected to acrylamide gel electrophoresis, transferred to nitrocellulose membranes, and protein levels were detected using the antibody concentrations listed above. Phosphorylated proteins were detected by phosphothreonine antibody (1:750, Santa Cruz). Levels of phosphorylated and non-phosphorylated protein were quantified using ImageJ software.
Animal surgery
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication 85-23, 1996) and approved by the Northwestern University Animal Care and Use Committee. Eleven-week-old male Sprague-Dawley rats weighing between 350–400 g were anesthetized with inhaled isofluorane (0.5–3%). Atropine was administered subcutaneously (0.1 mg/kg) to decrease airway secretions. After a midline neck incision, the left common carotid artery (CCA), external carotid artery (ECA), and the internal carotid artery (ICA) were dissected and proximal and distal control obtained with microclips. A transverse arteriotomy was created on the ECA. A 2 F Fogarty catheter (generously provided by Edward Life Sciences) was inserted into the CCA through the ECA, and the CCA injured by inflating the balloon to 5 atmospheres of pressure for 5 minutes. Following injury, the catheter was removed, the ECA ligated, and flow restored to the CCA and ICA, as previously described.16, 17, 20 After injury and restoration of flow, 10 mg of IPA/NO powder was applied evenly to the periadventitial surface of the injured CCA of rats in the treatment group. The neck incision was closed, and carotid arteries were harvested at 14 days post-injury. In order to obtain the 6 rats required for statistical significance in each treatment group, we operated on 12 animals in the IPA/NO group, due to a 50% mortality rate. Control groups included no injury and injury alone (n=6/group).
Tissue processing
Carotid arteries were harvested following in situ perfusion-fixation with cold 1X PBS (250 mL) and 2% paraformaldehyde (500 mL). Vessels were frozen in TissueTek O.C.T. compound (Sakura Finetek USA, Torrance, CA) and cut into 5-micron sections throughout the entire injured segment, as previously described.21
Morphometric analysis
Carotid arteries harvested at 14 days (n=6/group) were examined histologically for evidence of neointimal hyperplasia using routine hematoxylin-eosin (H&E) staining. Six equally-spaced sections from the area of injury were stained from each animal. Digital images of stained sections were collected with light microscopy using an Olympus BHT microscope (Melville, NY) with 4X and 10X objectives. For morphometric analysis, intimal area, medial area and arterial circumference were measured (arbitrary units) using ImageJ software. For cell density assessment, cell nuclei were counted on 3 equally-spaced sections from each animal in each treatment group by a blinded observer using the 40X objective. Nuclei were counted in the intima, media, and adventitia from four high power fields for each section.
Immunohistochemistry
To assess inflammation, carotid artery sections were also stained for presence of leukocytes and macrophages as follows. After fixation for 5 minutes in cold acetone, sections were permeabilized with 0.3% Trition X-100 for 20 minutes and incubated in 0.3% hydrogen peroxide in methanol for 15 minutes to block endogenous peroxidases. After blocking with horse serum for 30 minutes, primary antibody diluted in 0.5% BSA was added for 1 hour as follows: anti-CD45 (leukocyte, 1:500), anti-CD68 (macrophage, 1:500; both from AbD Serotec, Raleigh, NC). Next, biotinylated secondary antibody diluted 1:5000 in 0.5% BSA was added for 1 hour, followed by addition of streptavidin-bound horseradish peroxidase for 30 minutes. The stain was developed by incubation in the chromogen 3,3′-diaminobenzidine tetrahydrochloride (DAB) for 90 seconds, and a counterstain of hematoxylin was applied. Coverslips were affixed with gelvatol, and allowed to dry overnight. For each type of staining, six equally-spaced sections from the area of injury were stained from each animal. Digital images of stained sections were collected with light microscopy using an Olympus BHT microscope (Melville, NY) with 4X, 10X and 40X objectives. For inflammation staining, blinded counting of positive cells per high powered field was performed using ImageJ.
TUNEL staining
To assess the effect of IPA/NO on cell death in the balloon injury model, rat carotid artery sections were stained using the DeadEnd Colorimetric Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) system (Promega, Madison, WI), per the manufacturer’s instructions. Briefly, sections were washed in 0.85% NaCl, washed in 1X PBS, and fixed in 4% paraformaldehyde. After rinsing with 1X PBS, sections were treated with 20 μg/ml proteinase K, and then washed in 1X PBS and fixed in 4% paraformaldehyde again. After another wash in 1X PBS, sections were covered with Equilibration buffer, covered with biotinylated nucleotide mix and a plastic coverslip, and then incubated for 60 minutes at 37°C. The plastic coverslips were removed and the end-labeling reaction terminated by incubation in 2X SSC for 15 minutes at room temperature. After 3 washes in 1X PBS, the sections were blocked for endogenous peroxidases and developed with DAB chromogen as described above. The sections were coverslipped with gelvatol and sections were digitally imaged as described above.
Endothelialization
Rat carotid arteries harvested at 7 days post-injury (n = 3/group) were examined for endothelialization. In order to obtain the 3 rats for each treatment group, we operated on 6 animals in the IPA/NO treatment group due to a 50% mortality rate. Thirty minutes prior to sacrifice, 11-week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing between 395–495 g received an intravenous injection of Evans blue dye (0.5 mL of 0.5%, Sigma, St. Louis, MO). Following in situ perfusion with 500 mL of 1X PBS, carotid arteries were removed and photographed. Areas of denuded endothelium were identified by blue staining, and the portion of the injured area stained blue was quantitated using ImageJ.
Statistical analysis
To determine the number of animals required for each experimental group, a power analysis was performed. In order to detect a difference of 20% between the treatment groups with a power of 0.8, a standard deviation of 10%, and an alpha of 0.05, an n=6 per treatment group was required. Results are expressed as mean ± standard error of the mean (SEM). Differences between multiple groups were analyzed using one-way analysis of variance (ANOVA) with the Student-Newman-Keuls post hoc test for all pairwise comparisons (SigmaStat; SPSS, Chicago, IL). Statistical significance was assumed when P<0.05.
RESULTS
IPA/NO inhibits VSMC and HUVEC proliferation, but does not cause cell death or inhibit migration in vitro
To determine the effect of IPA/NO on VSMC and HUVEC proliferation, 3H-thymidine incorporation was assessed over a range of IPA/NO concentrations (100–1000 μmol/L). After 24 hours of treatment, IPA/NO inhibited proliferation in VSMC in a concentration-dependent manner, with the highest concentration of IPA/NO (1000 μmol/L) inducing a 38.7±4.5% inhibition of VSMC proliferation relative to control (P=0.003, Figure 1A), similar to the NO donor S-nitroso-N-acetylpenicillamine (SNAP, 1000 μmol/L). A 54.0±2.9% inhibition of HUVEC proliferation relative to control was also observed (P=<0.001, Figure 2A). Separate experiments in our laboratory showed that the NO donor DETA/NO increased endothelial cell proliferation at concentrations below 62.5 μM, but inhibited proliferation at higher concentrations, up to 1000 μM (data not shown). Proliferation assays were also performed with the parent compound, isopropylamine (IPA), and breakdown product, isopropanol, of IPA/NO. In VSMC, a statistically significant effect on proliferation was observed with IPA and isopropanol (P<0.001, Figure 1A), while the effect of IPA and isopropanol in HUVEC was negligible (Figure 2A). Of note, Griess reactions revealed that IPA/NO released significantly less NO than the NO donor DETA/NO at the same concentration (1000 μM). IPA/NO released 491.8±1.0 μM nitrite after 24 hours, while DETA/NO released 853.4±71.6 μM nitrite.
Figure 1.

IPA/NO prevents VSMC proliferation in vitro, but does not cause cell death or affect migration. (A) Proliferation of VSMC was assessed using 3H-thymidine incorporation after 24 hours of treatment with IPA/NO. (B) VSMC death was assessed via Guava PCA after 24 hours of treatment with IPA/NO. (C) The effect of IPA/NO (1000 μmol/L) on VSMC migration was assessed by blinded counting of nuclei migrating into the scraped area. The effects of the parent compound, IPA, and breakdown product of IPA/NO, isopropanol, on proliferation, death and migration were also assessed at 1000 μmol/L. The effects of the NO donor S-nitroso-N-acetylpenicillamine (SNAP) on proliferation and cell death were assessed at 1000 μmol/L. *P<0.005 vs. control. †P<0.05 vs. 1000 μmol/L IPA/NO. n = 3/treatment group. Data shown are representative of three separate experiments.
Figure 2.

IPA/NO prevents HUVEC proliferation in vitro, but does not cause cell death or affect migration. (A) Proliferation of HUVEC was assessed using 3H-thymidine incorporation. (B) HUVEC death was assessed via Guava PCA after 24 hours of treatment with IPA/NO. (C) The effect of IPA/NO (1000 μmol/L) on HUVEC migration was assessed by blinded counting of nuclei migrating into the scraped area. The effects of the parent compound, IPA, and breakdown product, isopropanol, of IPA/NO on proliferation, death and migration were also assessed at 1000 μmol/L. *P<0.005 vs. control. n = 3/treatment group. Data shown are representative of three separate experiments.
VSMC and HUVEC treated with IPA/NO were assessed for cell death using Guava PCA. No statistically significant increase in cell death between control and IPA/NO-treated groups was observed (Figure 1B, 2B). IPA, isopropanol, SNAP (VSMC), and DETA/NO (endothelial cells, data not shown) also did not induce significant cell death in either cell type. These results were consistent across all experimental replicates. Migration assays revealed that IPA/NO, IPA, or isopropanol had no statistically significant effect on VSMC or HUVEC migration at 24 hours (Figure 1C, 2C).
IPA/NO induces S-phase arrest
In order to determine the effect of IPA/NO on cell cycle progression, treated and untreated VSMC were subjected to flow cytometry analysis. As seen in Figure 3, treatment with IPA/NO for 24 hours induced a 49% increase in the S-phase population and a concomitant 20% decrease in the G0/G1 population, compared to control VSMC. These changes were statistically significant (P<0.05 vs. control).
Figure 3.

IPA/NO causes S-phase cell cycle arrest. (A) FACS analysis of untreated and IPA/NO-treated (1000 μmol/L) VSMC. (B) Graphical representation of the flow cytometry results after 24 hours of IPA/NO treatment. *P<0.05 vs. control. n = 3/treatment group. Data shown are representative of three separate experiments.
IPA/NO affects cell cycle protein expression and activity
Since IPA/NO inhibited VSMC proliferation in association with an S-phase cell cycle arrest, Western blot analysis of IPA/NO-treated VSMC was performed to assess the expression of cell cycle proteins. IPA/NO induced a marked increase in the expression of the cyclin-dependent kinase inhibitor (CDKI) p21, relative to untreated cells (Figure 4A). Interestingly, IPA/NO lowered the expression of the CDKI p27 in a concentration-dependent manner, and caused a large increase in cyclin D1 expression (Figure 4A). The cdk4-cyclin D1 complex is known to be inhibited by p27 and p21, but cdk4 expression did not appear to be affected. IPA/NO increased the expression of cyclin A, but had no appreciable effect on cyclin B1, cdc2, cdk2 or cdk6 (Figure 4A). With respect to the activity of the cdks, IPA/NO had no effect on the level of phosphorylated cdc2, cdk2, cdk4, or cdk6 (Figure 4B). Taken together, these data suggest that while IPA/NO affects the expression of the cyclins, as well as the CDKIs p21 and p27, it has a minimal effect on the expression and phosphorylation status of the cdks.
Figure 4.

IPA/NO affects cell cycle protein expression, but not the phosphorylation state of cdks. (A) The expression of cyclins, cdks, and CDKIs were assessed in VSMC by Western blot analysis in the presence or absence of IPA/NO for 24 hours. Data shown are representative of three separate experiments. (B) IPA/NO-treated and untreated VSMC lysates were immunoprecipitated (IP) with the indicated antibody, then analyzed for expression of phosphorylated (P) and non-phosphorylated (non-P) cdks. Graphs show the ratio of phosphorylated to non-phosphorylated protein as determined by densitometry. S: starved, C: control, and I: IPA/NO.
IPA/NO moderates neointimal hyperplasia in vivo
Since IPA/NO was shown to inhibit VSMC proliferation in vitro (Figure 1), we sought to determine whether it would prevent neointimal hyperplasia following balloon arterial injury in the rat carotid artery in vivo. As can be seen qualitatively in Figure 5A, external application of 10 mg of IPA/NO powder to the periadventitial surface of the carotid artery immediately after balloon injury reduced neointimal area by 29% (Figure 5B, P=<0.001), medial area by 31% (P=<0.001), and had no effect on arterial circumference, relative to injury alone. The approximately 30% reduction in both intimal and medial area rendered the traditionally-used metric of intima to media area ratio useless, as it was approximately 1 for both treated and untreated rats, despite the clearly observed statistically significant differences. Cell density analysis revealed that IPA/NO reduced the number of nuclei in the intima (97.6±8.8 vs. 152.8±8.0; P<0.001) and media (7.5±0.8 vs. 49.3±2.6; P<0.001) relative to injury alone, while increasing the number of nuclei in the adventitia (141.2±8.1 vs. 99.5±8.3; P<0.001).
Figure 5.
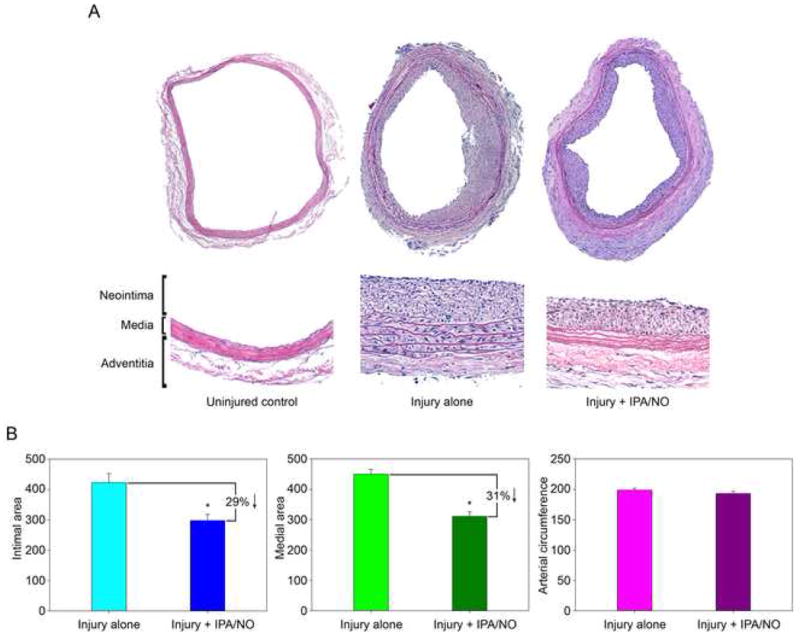
IPA/NO moderates neointimal hyperplasia in vivo. (A) Balloon-injured rat carotid arteries treated with or without exogenous periadventitial administration of IPA/NO powder (10 mg) were harvested 14 days after injury and stained by H&E. Whole mount and close-up sections are shown. (B) Quantitation of intimal area, medial area and arterial circumference using ImageJ software (n = 6/group). *P<0.001 vs. injury alone. Units are arbitrary.
IPA/NO decreases apoptosis in the neointima and media following balloon injury
Since IPA/NO was shown to inhibit VSMC proliferation without causing cell death in vitro, and since IPA/NO moderately inhibited neointimal hyperplasia in vivo, TUNEL staining was performed to assess the effects of IPA/NO on cell death in vivo. As seen in Figure 6, positive staining for apoptosis is visible in each layer of the injured artery at 2 weeks, but is markedly decreased in the neointima and media after administration of IPA/NO, with only the adventitia showing positive staining.
Figure 6.

IPA/NO decreases apoptosis in the neointima and media of balloon-injured arteries. Balloon-injured rat carotid arteries treated with or without exogenous periadventitial administration of IPA/NO powder (10 mg) and harvested 14 days after injury (n = 4/group) were subjected to TUNEL staining for apoptosis. Arrows indicate positive staining.
IPA/NO decreases inflammation caused by balloon injury
To ascertain the effect of IPA/NO on inflammation in vivo, the presence of leukocytes (CD45) and macrophages (CD68) in sections of balloon-injured carotid arteries from IPA/NO-treated and untreated animals was assessed via immunoperoxidase staining. As seen in Figure 7A, no positive staining was observed in uninjured vessels, while injured vessels showed a marked increase in both leukocyte and macrophage staining. IPA/NO treatment resulted in a 90% decrease in macrophage infiltration following balloon injury (Figure 7B, P<0.001), but had no significant effect on leukocyte infiltration.
Figure 7.

IPA/NO decreases inflammation in balloon-injured arteries. (A) Balloon-injured rat carotid arteries treated with or without exogenous periadventitial administration of IPA/NO powder (10 mg) and harvested 14 days after injury (n = 6/group) were subjected to immunoperoxidase staining using antibodies against leukocytes (CD45) and macrophages (CD68). Arrows indicate positive staining. (B) Blinded quantitation of positive CD45 and CD68 staining in untreated and IPA/NO-treated injured vessels was performed using ImageJ (n = 6/group). *P<0.001 vs. injury alone.
IPA/NO inhibits re-endothelialization
To further elucidate the effects of IPA/NO in vivo and confirm the in vitro HUVEC results, Evans blue staining of treated and untreated injured carotid arteries was performed. As seen in Figure 8A and B, the injured group treated with IPA/NO had larger areas of positive blue staining in the balloon-injured area relative to injured rats that received no IPA/NO (55.9±0.5% vs. 21.0±4.4%, P=0.001). This was consistently observed in all IPA/NO-treated rats, and represents active inhibition of reformation of an intact endothelial cell layer following arterial injury. Untreated injured rats were virtually negative for Evans blue staining, indicating the endothelial cell layer was reformed in these rats.
Figure 8.

IPA/NO prevents re-endothelialization. (A) Balloon-injured rats were administered Evans blue dye intravenously 7 days post-injury, and the harvested carotid arteries were photographed to assess the extent of endothelial regeneration. Blue staining indicates a lack of endothelium (n = 3/group). (B) Quantitation of photographs from panel (A) using ImageJ software. *P=0.001, n = 3/group.
DISCUSSION
In this manuscript, we demonstrate that IPA/NO inhibits VSMC and endothelial cell proliferation without inhibiting migration or inducing cell death. Central to our hypothesis, periadventitial administration of IPA/NO modestly reduced the formation of neointimal hyperplasia and prevented inflammation in an in vivo rodent model of arterial injury; however, IPA/NO also prevented the reformation of an intact endothelial cell layer in the injured area, caused significant mortality in the rat carotid artery balloon injury model, and depleted the medial layer of cells, raising concerns over the use of HNO donors in vivo.
The main metabolic breakdown product of IPA/NO is HNO, a highly reactive compound that rapidly and irreversibly dimerizes and dehydrates to form nitrous oxide (N2O).22 Because this dimerization is so rapid and irreversible, it is very difficult to measure HNO release in vitro via a colorimetric assay, like the Griess reaction used to measure nitrite. While the release profile of IPA/NO has been described in the chemistry literature,3, 23 there have been relatively few studies of the actions of HNO in the vasculature. However, some of the basic effects of this molecule have been described. Irvine et al. showed that pretreatment of isolated pre-contracted rat aorta with HNO from Angeli’s salt did not induce tolerance to subsequent doses of Angeli’s salt;10 however, pretreatment with nitroglycerin led to induction of tolerance to subsequent nitroglycerin treatments.10 HNO has also been shown to be a positive inotrope. Paolocci et al. showed that HNO from Angeli’s salt acted as an inotrope in conscious healthy dogs and dogs with failing hearts.6, 24 The inotropy induced by HNO was not affected by the β-blocker propranolol, and HNO administered with dobutamine nearly doubled the inotropic effect of dobutamine alone.6 Thus, HNO clearly induces effects in the cardiovascular system, making it a molecule of interest clinically.
Our work demonstrates that IPA/NO affects VSMC and endothelial cell proliferation, yet highlights the differences by which NO and IPA/NO mediate their effects in the vasculature. NO inhibits VSMC proliferation,14, 25, 26 and we have shown that IPA/NO also inhibits VSMC proliferation. NO stimulates endothelial cell proliferation,26–28 while we have shown that IPA/NO inhibits endothelial cell proliferation. NO induces VSMC death,27, 28 but IPA/NO had no effect on VSMC death. NO and IPA/NO both induce cell cycle arrest, but where NO is known to induce G0/G1 cell cycle arrest in VSMC,19, 29 IPA/NO induced S-phase arrest in VSMC. Consistent with the inhibition of proliferation, IPA/NO caused increased expression of the CDKI p21 in VSMC, similar to the actions of NO; however, whereas NO increases the expression of both p21 and p27, IPA/NO decreased p27 expression.14, 30 Additionally, IPA/NO increased expression of both cyclin A and cyclin D1 in VSMC, probably as a means to overcome cell cycle inhibition. Despite this increase in cyclin D1 expression, IPA/NO affected neither the expression of cdk4 or cdk6, nor the level of the active, phosphorylated versions of these proteins. Lastly, despite the observed S-phase arrest, IPA/NO had no effect on the level of phosphorylated cdk2 in VSMC. More work is required to determine the reasons for the differences between the effects of NO and IPA/NO. Taken together, however, these data paint a picture of HNO from IPA/NO as an important biological molecule in the vasculature which can act both similarly to, yet distinct from, NO.
While our study clearly demonstrates that IPA/NO induces effects in the vasculature that are distinct from those induced by NO, limitations to this work exist. First, while we have demonstrated short- and medium-term effects of IPA/NO in the vasculature of rats,21 these data will need to be validated in a large animal model. Furthermore, the durability of this therapy will require examination over much longer time points. Second, while we evaluated the effects of the parent compound, IPA, and the breakdown product, isopropanol, of IPA/NO, in vitro, we did not study the effects of these compounds in the rat carotid artery injury model in vivo. Furthermore, while the effects we observed on VSMC and HUVEC were distinct from NO, we cannot be certain that NO release did not make a small contribution to our results. Though characterizations of IPA/NO have shown it to behave very similarly to Angeli’s salt, and distinct from NO,23 and IPA/NO is known to decompose primarily to HNO and isopropanol at a pH and concentration similar to those used in this work (L. K. Keefer, unpublished observations), the release pattern of IPA/NO is complex. Therefore, though HNO is the main molecule released from IPA/NO, the effects of the other compounds released (i.e., NO, isopropanol, IPA) cannot be discounted. Indeed, isopropanol and isopropylamine are known irritants, and reduced VSMC proliferation in vivo, so it is possible they are contributing to the effects we observed in vivo.
Despite extensive experiments to determine the signaling pathway by which IPA/NO exerted its effects on VSMC proliferation, we were unable to identify one that mediated the effects of IPA/NO on VSMC proliferation, including cGMP and cAMP (data not shown). While it is clear that IPA/NO acts to lessen the formation of neointimal hyperplasia, the mechanism of this action is not clear. The results of our immunoperoxidase staining showed that IPA/NO reduced macrophage infiltration in the neointima, media, and adventitia of injured vessels. We also observed fewer nuclei in the media of IPA/NO-treated arteries, and a paucity of TUNEL positive staining in the neointima and media of IPA/NO-treated arteries. Interestingly, work by other groups has shown that: 1) macrophages act as a source of migratory SMC;31 2) the level of reactive oxygen species in balloon-injured vessels is increased, leading to a loss of medial cells due to apoptosis;32 and, 3) adventitial fibroblasts convert to myofibroblasts and contribute to the neointima following arterial injury.33 Given all these data, we speculate that periadventitial application of IPA/NO reduces the formation of neointimal hyperplasia and repopulation of the media by preventing the migration and differentiation of macrophages and adventitial fibroblasts, as well as by inhibiting VSMC proliferation. This hypothesis is indirectly supported by our cell density data, which demonstrates a stark lack of nuclei in the media of IPA/NO-treated arteries, as well as an increase in the number of adventitial nuclei, and may also explain the lack of apoptosis observed in the media and neointima of IPA/NO-treated arteries.
Another important limitation to the use of HNO in the vasculature deserves mention. Though HNO donors seem promising due to their specificity and chemistry distinct from that of NO, the question of toxicity remains an important one. In the course of this study, we operated on 12 rats to obtain the group of 6 required for statistical significance. The rate of this mortality, which we believe was secondary to airway edema, was much higher than that observed using diazeniumdiolates that donate primarily NO, so this effect may be related to the HNO released by IPA/NO. Consistent with this IPA/NO-induced increase in animal mortality, early studies did show that the HNO donor Angeli’s salt was cytotoxic.34 Furthermore, it is known that breakdown products of some of the diazeniumdiolates are potentially toxic amines.35 Thus, toxicity of any HNO donor must be addressed prior to administration of these compounds to patients.
Lastly, since the endothelial cell layer produces many vasoprotective molecules and serves as a natural barrier of the vascular wall against platelets and leukocytes, the observation that IPA/NO prevents reformation of the endothelial layer is highly problematic in terms of its use as a therapeutic agent in the cardiovascular system. In fact, the effect of IPA/NO delivery in vivo with respect to its antiproliferative effects on endothelial cells calls to mind the effects of the FDA-approved coronary artery drug-eluting stents which release sirolimus and paclitaxel. Several pooled or meta-analysis studies have revealed either similar mortality or worse mortality of these drug-eluting stents compared to bare-metal stents.36, 37 Thus, it stands to reason that IPA/NO may be subject to the same potentially deleterious effects in the vasculature if used to prevent the development of neointimal hyperplasia in vivo.
In summary, we found that IPA/NO, an organic HNO donor, inhibited VSMC and endothelial cell proliferation in vitro without inhibiting migration or inducing cell death. This inhibition of VSMC proliferation was mediated through S-phase cell cycle arrest. IPA/NO also modestly inhibited the development of neointimal hyperplasia following arterial injury in vivo and reduced inflammation. However, IPA/NO prevented reformation of an intact endothelial cell layer and caused significant mortality in vivo. Thus, although HNO donors may be useful in treating a variety of cardiovascular diseases, the concerns raised by our data, and those of others, must be fully addressed before these compounds should be used clinically.
Acknowledgments
The authors would like to express their thanks to Edwards Life Sciences for generously providing the Fogarty catheters, and to Qun Jiang, MD, Bo Fu, and Janet Martinez, AAS, for their assistance. This work was supported by the National Institutes of Health (1K08HL084203 to M.R.K.), the American Vascular Association (to M.R.K.), Department of Veterans Affairs (Merit Review Grant to M.R.K.), Mrs. Hilda Rosenbloom (to M.R.K.), Ms. Eleanor Baldwin (to M.R.K.), the National Cancer Institute, National Institutes of Health with SAIC-Frederick, Inc. (Contract NO1-CO-12400 to J.E.S.), and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (to L.K.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drago RS, Karstetter BR. The reaction of nitrogen(II) oxide with various primary and secondary amines. J Am Chem Soc. 1961;83:1819–22. [Google Scholar]
- 2.Fukuto JM, Bartberger MD, Dutton AS, Paolocci N, Wink DA, Houk KN. The physiological chemistry and biological activity of nitroxyl (HNO): the neglected, misunderstood, and enigmatic nitrogen oxide. Chem Res Toxicol. 2005 May;18(5):790–801. doi: 10.1021/tx0496800. [DOI] [PubMed] [Google Scholar]
- 3.Dutton AS, Suhrada CP, Miranda KM, Wink DA, Fukuto JM, Houk KN. Mechanism of pH-dependent decomposition of monoalkylamine diazeniumdiolates to form HNO and NO, deduced from the model compound methylamine diazeniumdiolate, density functional theory, and CBS-QB3 calculations. Inorg Chem. 2006 Mar 20;45(6):2448–56. doi: 10.1021/ic051505z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonner FT, Ravid B. Thermal decomposition of oxyhyponitrite (sodium trioxodinitrate(II)) in aqueous solution. Inorg Chem. 1975;14:558–63. [Google Scholar]
- 5.Hughes MN, Wimbledon PE. The chemistry of trioxodinitrates. 1. Decomposition of sodium trioxdinitrate (Angeli’s salt) in aqueous solution. J Chem Soc Dalton Trans. 1976;8:703–7. [Google Scholar]
- 6.Paolocci N, Katori T, Champion HC, St JM, Miranda KM, Fukuto JM, et al. Positive inotropic and lusitropic effects of HNO/NO- in failing hearts: independence from beta-adrenergic signaling. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5537–42. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukuto JM, Chiang K, Hszieh R, Wong P, Chaudhuri G. The pharmacological activity of nitroxyl: a potent vasodilator with activity similar to nitric oxide and/or endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1992 Nov;263(2):546–51. [PubMed] [Google Scholar]
- 8.Arnelle DR, Stamler JS. NO+, NO, and NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995 Apr 20;318(2):279–85. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 9.Wong PS, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, et al. Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998 Apr 21;37(16):5362–71. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 10.Irvine JC, Favaloro JL, Widdop RE, Kemp-Harper BK. Nitroxyl anion donor, Angeli’s salt, does not develop tolerance in rat isolated aortae. Hypertension. 2007 Apr;49(4):885–92. doi: 10.1161/01.HYP.0000259328.04159.90. [DOI] [PubMed] [Google Scholar]
- 11.Tocchetti CG, Wang W, Froehlich JP, Huke S, Aon MA, Wilson GM, et al. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007 Jan 5;100(1):96–104. doi: 10.1161/01.RES.0000253904.53601.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feelisch M. Nitroxyl gets to the heart of the matter. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):4978–80. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu SM, Hung LM, Lin CC. cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway. Circulation. 1997 Mar 4;95(5):1269–77. doi: 10.1161/01.cir.95.5.1269. [DOI] [PubMed] [Google Scholar]
- 14.Kibbe MR, Li J, Nie S, Watkins SC, Lizonova A, Kovesdi I, et al. Inducible nitric oxide synthase (iNOS) expression upregulates p21 and inhibits vascular smooth muscle cell proliferation through p42/44 mitogen-activated protein kinase activation and independent of p53 and cyclic guanosine monophosphate. J Vasc Surg. 2000 Jun;31(6):1214–28. doi: 10.1067/mva.2000.105006. [DOI] [PubMed] [Google Scholar]
- 15.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, et al. Complexes of .NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J Med Chem. 1991 Nov;34(11):3242–7. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 16.Ahanchi SS, Varu VN, Tsihlis ND, Martinez J, Pearce CG, Kapadia MR, et al. Heightened efficacy of nitric oxide-based therapies in type II diabetes mellitus and metabolic syndrome. Am J Physiol Heart Circ Physiol. 2008 Dec;295(6):H2388–H2398. doi: 10.1152/ajpheart.00185.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapadia MR, Chow LW, Tsihlis ND, Ahanchi SS, Eng JW, Murar J, et al. Nitric oxide and nanotechnology: a novel approach to inhibit neointimal hyperplasia. J Vasc Surg. 2008 Jan;47(1):173–82. doi: 10.1016/j.jvs.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han J, Talorete TP, Yamada P, Isoda H. Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology. 2009 Jan;59(1):45–53. doi: 10.1007/s10616-009-9191-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kibbe MR, Nie S, Seol DW, Kovesdi I, Lizonova A, Makaroun M, et al. Nitric oxide prevents p21 degradation with the ubiquitin-proteasome pathway in vascular smooth muscle cells. J Vasc Surg. 2000 Feb;31(2):364–74. doi: 10.1016/s0741-5214(00)90166-6. [DOI] [PubMed] [Google Scholar]
- 20.Pearce CG, Najjar SF, Kapadia MR, Murar J, Eng J, Lyle B, et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free Radic Biol Med. 2008 Jan 1;44(1):73–81. doi: 10.1016/j.freeradbiomed.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shears LL, Kibbe MR, Murdock AD, Billiar TR, Lizonova A, Kovesdi I, et al. Efficient inhibition of intimal hyperplasia by adenovirus-mediated inducible nitric oxide synthase gene transfer to rats and pigs in vivo. J Am Coll Surg. 1998 Sep;187(3):295–306. doi: 10.1016/s1072-7515(98)00163-x. [DOI] [PubMed] [Google Scholar]
- 22.Shafirovich V, Lymar SV. Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc Natl Acad Sci U S A. 2002 May 28;99(11):7340–5. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, et al. Comparison of the NO and HNO donating properties of diazeniumdiolates: primary amine adducts release HNO in vivo. J Med Chem. 2005 Dec 29;48(26):8220–8. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- 24.Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, et al. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc Natl Acad Sci U S A. 2001 Aug 28;98(18):10463–8. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989 May;83(5):1774–7. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo JP, Panday MM, Consigny PM, Lefer AM. Mechanisms of vascular preservation by a novel NO donor following rat carotid artery intimal injury. Am J Physiol. 1995 Sep;269(3 Pt 2):H1122–H1131. doi: 10.1152/ajpheart.1995.269.3.H1122. [DOI] [PubMed] [Google Scholar]
- 27.Nishio E, Fukushima K, Shiozaki M, Watanabe Y. Nitric oxide donor SNAP induces apoptosis in smooth muscle cells through cGMP-independent mechanism. Biochem Biophys Res Commun. 1996 Apr 5;221(1):163–8. doi: 10.1006/bbrc.1996.0563. [DOI] [PubMed] [Google Scholar]
- 28.Iwashina M, Shichiri M, Marumo F, Hirata Y. Transfection of inducible nitric oxide synthase gene causes apoptosis in vascular smooth muscle cells. Circulation. 1998 Sep 22;98(12):1212–8. doi: 10.1161/01.cir.98.12.1212. [DOI] [PubMed] [Google Scholar]
- 29.Sarkar R, Gordon D, Stanley JC, Webb RC. Cell cycle effects of nitric oxide on vascular smooth muscle cells. Am J Physiol. 1997 Apr;272(4 Pt 2):H1810–H1818. doi: 10.1152/ajpheart.1997.272.4.H1810. [DOI] [PubMed] [Google Scholar]
- 30.Sato J, Nair K, Hiddinga J, Eberhardt NL, Fitzpatrick LA, Katusic ZS, et al. eNOS gene transfer to vascular smooth muscle cells inhibits cell proliferation via upregulation of p27 and p21 and not apoptosis. Cardiovasc Res. 2000 Sep;47(4):697–706. doi: 10.1016/s0008-6363(00)00137-1. [DOI] [PubMed] [Google Scholar]
- 31.Bayes-Genis A, Campbell JH, Carlson PJ, Holmes DR, Jr, Schwartz RS. Macrophages, myofibroblasts and neointimal hyperplasia after coronary artery injury and repair. Atherosclerosis. 2002 Jul;163(1):89–98. doi: 10.1016/s0021-9150(01)00771-7. [DOI] [PubMed] [Google Scholar]
- 32.Pollman MJ, Hall JL, Gibbons GH. Determinants of vascular smooth muscle cell apoptosis after balloon angioplasty injury. Influence of redox state and cell phenotype. Circ Res. 1999 Jan 8;84(1):113–21. doi: 10.1161/01.res.84.1.113. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, O’Brien JE, Fard A, Mannion JD, Wang D, Zalewski A. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996 Oct 1;94(7):1655–64. doi: 10.1161/01.cir.94.7.1655. [DOI] [PubMed] [Google Scholar]
- 34.Wink DA, Feelisch M, Fukuto J, Chistodoulou D, Jourd’heuil D, Grisham MB, et al. The cytotoxicity of nitroxyl: possible implications for the pathophysiological role of NO. Arch Biochem Biophys. 1998 Mar 1;351(1):66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- 35.Ragsdale RO, Karstetter BR, Drago RS. Decomposition of the Adducts of Diethylamine and Isopropylamine with Nitrogen(II) Oxide. Inorg Chem. 1965;4:420–2. [Google Scholar]
- 36.Spaulding C, Daemen J, Boersma E, Cutlip DE, Serruys PW. A pooled analysis of data comparing sirolimus-eluting stents with bare-metal stents. N Engl J Med. 2007 Mar 8;356(10):989–97. doi: 10.1056/NEJMoa066633. [DOI] [PubMed] [Google Scholar]
- 37.Lagerqvist B, James SK, Stenestrand U, Lindback J, Nilsson T, Wallentin L. Long-term outcomes with drug-eluting stents versus bare-metal stents in Sweden. N Engl J Med. 2007 Mar 8;356(10):1009–19. doi: 10.1056/NEJMoa067722. [DOI] [PubMed] [Google Scholar]