

Abstract
It has recently been reported that relatively short-term inhibition of vascular endothelial growth factor (VEGF) signaling can cause photoreceptor cell death, a potentially clinically important finding since VEGF blockade has become an important modality of treatment of ocular neovascularization and macular edema. However, in a set of studies in which we achieved extended and complete blockage of VEGF-induced vascular leakage through retinal expression of a VEGF binding protein, we did not observe any toxicity to retinal neurons. To follow-up on these apparently discrepant findings, we designed a set of experiments with the kinase inhibitor SU4312, which blocks phosphorylation of VEGF receptors, to look directly for evidence of VEGF inhibition-related retinal toxicity. Using transgenic mice with sustained expression of VEGF in photoreceptors, we determined that periocular injection of 3 μg of SU4312 every 5 days markedly suppressed subretinal neovascularization, indicating effective blockade of VEGF signaling. Wild-type mice given periocular injections of 5 μg of SU4312 every 5 days for up to 12 weeks showed normal scotopic and photopic electroretinograms (ERGs), no TUNEL stained cells in the retina, and no reduction in outer nuclear layer thickness. Incubation of cultured ganglion cells or retinal cultures containing photoreceptors with high doses of SU4312 did not reduce cell viability. These data suggest that blocking VEGF signaling in the retina for up to 12 weeks does not damage photoreceptors nor alter ERG function and should reassure patients who are receiving frequent injections of VEGF antagonists for choroidal and retinal vascular diseases.
Vascular endothelial growth factor (VEGF) is an important stimulator of retinal and choroidal neovascularization and excessive retinal vascular permeability (Campochiaro, 2004). While it seems likely that other proteins also contribute to these processes, it is remarkable what can be achieved with selective antagonists of VEGF in various retinal and choroidal vascular diseases. Monthly injections of ranibizumab, an Fab that binds all isoforms of VEGF-A, causes significant improvement of 3 or more lines of visual acuity in 34–40% of patients with neovascular age-related macular degeneration (AMD), with a mean improvement of eight letters (Brown et al., 2006; Rosenfeld et al., 2006). Ranibizumab represents the first treatment to reverse severe vision loss in neovascular AMD and has revolutionized its treatment. In patients with diabetic macular edema, 5 intraocular injections of ranibizumab over a span of 6 months resulted in a mean improvement of 12 letters at month 7 (Nguyen et al., 2006). In patients with macular edema due to central retinal vein occlusion (CRVO) or branch retinal vein occlusion (BRVO), monthly injections of ranibizumab for 3 months resulted in marked improvement in vision; in a group of 40 patients (20 with CRVO and 20 with BRVO) treated with 0.3 or 0.5 mg of ranibizumab the mean improvement was approximately 15 letters (Campochiaro et al., 2008). These results have been confirmed in two phase III clinical trials in which patients with macular edema due to BRVO showed mean improvements of 16 and 18 letters with monthly injections of 0.3 or 0.5 mg of ranibizumab for 6 months and patients with CRVO showed mean improvements of 13 and 15 letters with monthly injections of 0.3 or 0.5 mg of ranibizumab for 6 months.
Despite the undeniable short-term benefits seen in patients with a variety of retinal and choroidal diseases treated with VEGF antagonists, some investigators have sounded a note of caution. Fenestrated vascular beds are more dependent upon VEGF, raising concern that the choriocapillaris, which is fenestrated, may be damaged over time. VEGF type 2 receptors have been identified on neurons, including retinal neurons, and it has been suggested that their activation may promote cell survival, while their inhibition could lead to cell death (Jin et al., 2000; Kilic et al., 2006).
To explore the effects of long term blockade of VEGF on the retina and choroid, we expressed high levels of a potent VEGF binding protein in the retinas of mice for up to 7 months (Ueno et al., 2008). These mice had complete suppression of the pro-permeability effects of exogenous VEGF, confirming good VEGF blockade, but they showed no abnormalities of the choriocapillaris and no evidence of any damage to photoreceptors, ganglion cells, or other retinal cells. Electroretinograms (ERGs), a widely used test of retinal function, were completely normal throughout 7 months of VEGF blockade.
Contrary to our findings, other laboratories have suggested that local or systemic treatment of mice with agents that bind VEGF can cause severe damage to retinal ganglion cells (RGCs) (Nishijima et al., 2007) or photoreceptors (Saint-Geniez et al., 2008). Another report has suggested that three intraocular injections of an antibody directed against VEGF over the span of a week caused pigmentary changes in the retina observed with fundus photography and alterations in ERGs (Takeda et al., 2009). These studies have potentially significant clinical implications and have caused considerable concern for patients who are receiving and for physicians who are administering intraocular injections of VEGF antagonists. In order to address the important issues involved, and to try to reconcile the apparent differences between these published studies and our own results, we have performed additional experiments to carefully look for any evidence of retinal toxicity from VEGF blockade. A particularly efficient way to antagonize VEGF is by administration of tyrosine kinase inhibitors that block phosphorylation of VEGF receptors. 3-(4-Dimethylaminobenzylidene)-1,3-dihydroindol-2-one (SU4312) is a relatively selective inhibitor of VEGF receptors 1 and 2 (Sun et al., 1998; Kendall et al., 1999). While VEGF binding proteins are competitive antagonists, SU4312 is a noncompetitive inhibitor that is capable of completely silencing VEGF signaling regardless of the level or source (intracellular or extracellular) of VEGF. In fact, cultured endothelial cells that are healthy in the presence of VEGF binding proteins, cannot tolerate SU4312, because it blocks autocrine stimulation of VEGF receptors (Lee et al., 2007). In this study, we identified a treatment regimen with SU4312 that strongly suppressed the angiogenic effects of overexpression of VEGF in the retina and used it to investigate the effects of blocking VEGF signaling in normal mouse retina for up to 12 weeks. We also explored the effect of SU4312 in vitro upon cultured retinal cells.
Materials and Methods
Drugs and reagents
The VEGF receptor tyrosine kinase inhibitor SU4312 was purchased from Sigma–Aldrich (St. Louis, MO). A stock solution of 25 mg/ml of SU4312 was prepared in DMSO and then diluted in phosphate-buffered saline (PBS) to a concentration of 1 mg/ml for periocular injections or diluted in growth media (GM; Neurobasal media, B27 supplement, 2 mM glutamine, and 1 U/ml Penicillin/Streptomycin) for in vitro cell culture experiments. Human recombinant VEGF was obtained from R&D Systems, Inc. (Minneapolis, MN). An equivalent solution of DMSO in PBS or GM was used for vehicle controls.
Animal use
Mice and rats were treated in accordance with the recommendations of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Assessment of effects of SU4312 on VEGF signaling in the retina
The activity of SU4312 was assessed by Western blots to measure phosphorylation of Akt, which is a downstream molecule in the VEGF signaling pathway and whose phosphorylation is required for VEGF survival-promoting activity (Gerber et al., 1998). C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME; n =3 in each group) were given an intraocular injection of 1 μl of PBS containing 1 μM VEGF with or without 1 μM SU4312. Mice that did not receive any injections were used as negative controls. Intravitreous injections were given with a Harvard pump apparatus (Harvard Apparatus, Holliston, MA) and pulled-glass micropipettes, as previously described (Mori et al., 2001). Six hours after intraocular injections, the mice were euthanized and retinas were dissected. Retinal homogenates were run in immunoblots as previously described (Saishin et al., 2003). The primary antibody was rabbit anti-Phospho Akt (#9271 Cell Signaling, Danvers, MA) and rabbit anti-beta actin (Cell Signaling).
Assessment of effects of SU4312 on VEGF-induced retinal neovascularization
Hemizygous rhodopsin/VEGF transgenic mice that express VEGF in photoreceptors (Okamoto et al., 1997; Tobe et al., 1998) were given a periocular injection of 3 μg of SU4312 every 4, 5, or 7 days starting at post natal day (P) 14. At P28, the mice were anesthetized, perfused with fluorescein-labeled dextran (2 × 106 average molecular weight, Sigma–Aldrich), and retinal flat mounts were examined by fluorescence microscopy (Axioskop2 plus; Zeiss, Thornwood, NY) at 400× magnification, which provides a narrow depth of field, so that when neovascularization along the outer edge of the retina is brought into focus, the remainder of the retinal vessels are out of focus, allowing easy delineation and quantification of the neovascularization. Images were digitized with a three-color charge-coupled device video camera (Cool SNAP™-Pro; Media Cybernetics, Silver Spring, MD) and a frame grabber. Image analysis software (Image-Pro Plus 5.0; Media Cybernetics, Silver Spring, MD) was set to recognize fluorescently stained neovascularization and used to calculate the total area of neovascularization per retina. The investigator performing image analysis was masked with respect to treatment group.
Recording of ERGs
Every 5 days for 12 weeks, adult C57BL/6 mice (4–6 weeks old) were given periocular injections in one eye of 5 μl of vehicle or vehicle containing 5 μg of SU4312. Juvenile mice (P14) were given periocular injections of 3 μl of vehicle or vehicle containing 3 μg of SU4312 every 5 days for 4 weeks. Scotopic and photopic ERGs were recorded at several time points in vehicle-treated and SU4312-treated mice using an Espion ERG machine (Diagnosys LLL, Littleton, MA) as previously described (Okoye et al., 2003; Komeima et al., 2006; Ueno et al., 2008). For scotopic recordings, mice were dark adapted overnight, and for photopic recordings, mice were adapted for 10 min to background white light at an intensity of 30 cd/m2. The mice were anesthetized with an intraperitoneal injection of ketamine hydrochloride (100 mg/kg body weight) and xylazine (5 mg/kg body weight). Pupils were dilated with Midrin P containing 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Santen Pharmaceutical Co., Osaka, Japan). The mice were placed on a pad heated to 39°C and platinum loop electrodes were placed on each cornea after application of Gonioscopic prism solution (Alcon Labs, Fort Worth, TX). A reference electrode was placed subcutaneously in the anterior scalp between the eyes and a ground electrode was inserted into the tail. The head of the mouse was held in a standardized position in a Ganzfeld bowl illuminator that ensured equal illumination of the eyes. Recordings for both eyes were made simultaneously with electrical impedance balanced. Scotopic ERGs were recorded at 11 intensity levels of white light ranging from −3.00 to 1.40 log cd-s/m2 and six measurements were averaged at each flash intensity. Photopic ERGs were recorded at three intensity levels of white light ranging from 0.60 to1.40 log cd-s/m2. Five measurements were averaged at each flash intensity. Intensity response functions were analyzed using unpaired t-test.
Measurement of outer nuclear layer (ONL) thickness
Mice were euthanized, a mark was placed at 12:00 at the corneal limbus, and eyes were removed and frozen in OCT compound. Ten micrometers frozen sections were cut parallel to 12:00 meridian and sections through the optic nerve, superior, and inferior poles were fixed in 4% paraformaldehyde. Sections were stained with hematoxylin and eosin, microscopic images were digitized, and ONL thickness was measured by image at six locations, 25% (S1), 50% (S2), and 75% (S3) of the distance between the superior pole and the optic nerve and 25% (I1), 50% (I2), and 75% (I3) of the distance between the inferior pole the optic nerve (Komeima et al., 2007).
Terminal deoxynucleotidyl transferase-mediated dUTP-biotinide end labeling (TUNEL)
At various time points after initiation of treatment with SU4312, mice were euthanized and 10 μm frozen ocular sections were fixed in 1% paraformaldehyde for 10 min and TUNEL was performed using the Apoptag Red In Situ Apoptosis Detection Kit (Chemicon International, Inc., Billerica, MA) following the manufacturer’s instructions. Slides were counterstained with DAPI (Vectashield; Vector Laboratories, Inc., Burlingame, CA), and viewed with a Nikon Fluorescence Microscope. Images were captured using the same exposure time for each section.
Preparation of RGC cultures
RGCs were cultured as previously described (Kerrison et al., 2005). Briefly, retinas were dissected from P3 to P5 Sprague–Dawley rats and digested with activated papain and DNase prior to dissociation by trituration. The cell suspension was depleted of macrophages by incubation with CD11b/c-coupled (554859, BD Biosciences Pharmigen, San Diego, CA) Dynal magnetic beads (11531D, Invitrogen, Carlsbad, CA). RGCs were then selected from the remaining cells by incubation with magnetic beads coupled to an anti Thy-1 antibody (MAB1406, Millipore, Malborough, MA) via a DNA linker. The beads were washed and the purified Thy-1 immunoreactive cells were removed by cleaving the linker with DNAse. Cells were resuspended in GM supplemented with 5 μM forskolin and seeded at a density of 5,000 cells/well in 96 well culture plates (Falcon, BD Biosciences, Bedford, MA) that had been sequentially coated with poly-D-lysine (0.01 mg/ml, Sigma–Aldrich) and laminin (0.01 mg/ml, Sigma–Aldrich).
Preparation of primary retinal cell cultures
Primary retinal cell cultures were prepared using P3–P4 hRhoGFP/hRhoGFP knock-in mice (Chan et al., 2004). Retinas were dissociated into single cells by papain enzymatic digestion and mechanical trituration using a Pasteur glass pipette as described previously (Bonnet et al., 2004). Cells were resuspended in growth medium and seeded at a density of 32,000 cells/well in 96-well plates that had been coated as described above.
Testing the effects of blocking VEGF signaling in retinal cell cultures
Stock solutions of SU4312 were prepared in DMSO and diluted in growth medium to give five final concentrations ranging from 0 to 6.4 μM. After 72 h (RGCs) or 6 days (primary retinal cell cultures) in culture, with or without various concentrations of SU4312, cells were stained with 5 μM Hoescht 33342 (Invitrogen) and 4 μM ethidium homodimer-1 (Invitrogen), fluorescent dyes that allowed simultaneous counting of nuclei and dead cells. In addition, RGCs were stained with 10 μM calcein-AM (Invitrogen), which stains living cells and provides a means to assess cell bodies and neurites. Rhodopsin expression was monitored by GFP intensity in the retinal cell cultures.
Using a robotic fluorescence-based imaging system (ArrayScan VTI, Thermo Fisher, Cellomics, Pittsburgh, PA) images of up to 40 fields per well were acquired and stored on a Dell Xenon-based 6-terabyte server. RGC cells were identified as Hoechst-positive, ethidium homodimer-negative cells that surpassed an empirically determined threshold level of calcein AM staining. Neurites were identified as calcein-stained extensions of cells and their length and number were measured by image analysis. Rod photoreceptors were identified as Hoechst-positive, hRhoGFP-positive and ethidium homodimer-negative cells that surpassed a threshold level of empirically determined hRhoGFP expression. To provide references for comparison, primary retinal cell cultures were treated with 20 nM staurosporin (Sigma–Aldrich) which increases expression of Rho or 1 ng/ml of ciliary neurotrophic factor (CNTF, Prepro Tech, Rocky Hill, NJ) which decreases expression of Rho (Bradford et al., 2005). Using custom algorithms created using the Cellomics Neuronal Profiling and Target Activation image-analysis software packages, relative cell death, normalized rod photoreceptor viability and relative levels of hRhoGFP expression in live rod photoreceptor cells were determined. These data were normalized to values obtained from scanning cells in wells containing DMSO vehicle. Data are reported as the mean (±SEM) and statistical comparisons were made by one-way ANOVA.
Detection of VEGFR2 mRNAs by PCR
Mouse RGCs were isolated according to a published procedure (Barres and Chun, 1993). Mouse retinas were dissociated as described above and seeded at 1.5 million cells per well. Total RNA was isolated from three wells at 1, 2, 3, 4, and 7 days using the RNeasy mini kit (QIAGEN, Valencia, CA) with genomic DNA digestion. After quantification of RNA, cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen) and diluted for PCR. PCR was performed using the following primers: for mouse VEGFR2, Forward CACCTGCCAGGCCTGCAA, Reverse GCTTGGTGCAGGCACCTA; for rhodopsin, Forward TGCCCTCAGGGATGTACC, Reverse ACCTGGATCATGGCGTTG; and for GAPDH, Forward TCAACAGCAACTCCCACTCTTCCA, Reverse ACCCTGTTGCTGTAGCCGTATTCA. Quantitative PCR was performed with Eva Green fluorescent dye (Biotium, Hayward, CA) on a real time thermocycler (MyiQ; Bio-Rad, Hercules, CA). Relative starting quantities of template cDNA were normalized to GAPDH levels.
Results
SU4312 blocks VEGF-induced phosphorylation of Akt in mouse retina
Akt is a key second messenger that mediates the survival-promoting activity of VEGF (Gerber et al., 1998). Injection of VEGF into the vitreous cavity resulted in phosphorylation of Akt in the retina and intraocular injection of SU4312 blocked VEGF-induced phosphorylation of Akt (Fig. 1).
Fig. 1.
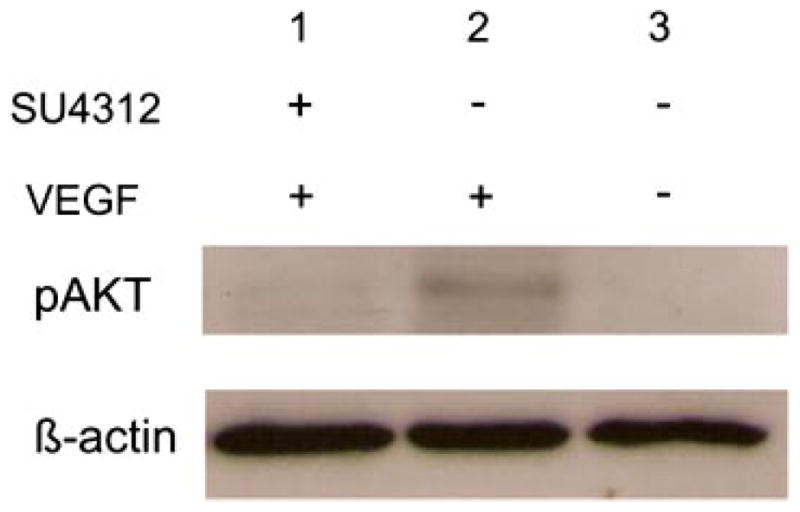
SU4312 blocks vascular endothelial growth factor (VEGF)-induced phosphorylation of Akt in mouse retina. Retinas were dissected from C57BL/6 mice that were uninjected or 6 h after intraocular injection of 1 μl of 10−6 M μg of VEGF or coinjection of 1 μl containing 10−6 M VEGF and 3 × 10−6 M SU4312. Retinal homogenates (80 μg of protein) were run in immunoblots using an antibody specific for phosphorylated AKT, stripped and reblotted with an antibody for β-actin. Retinas from eyes injected with VEGF showed a signal for phosphorylated Akt (lane 2) which was not present in the retinas of uninjected eyes (lane 3) and was blocked by co-injection of SU4312 with the VEGF (lane 1).
Identification of a treatment regimen with SU4312 that completely blocks the effects of VEGF in the retina
Transgenic mice in which the rhodopsin promoter drives expression of VEGF in the retina (rho/VEGF mice) have sustained expression of VEGF and progressive subretinal neovascularization (NV) starting at postnatal day (P) 7 (Okamoto et al., 1997; Tobe et al., 1998). At P14, there are normally a few tufts of subretinal NV. Starting at P14, hemizygous rho/VEGF mice were given a periocular injection of 3 μl of vehicle or 3 μl containing 3 μg of SU4312 every 4, 5, or 7 days. After 2 weeks during which these three groups of mice received a total of 4, 3, or 2 injections, the mice were perfused with fluorescein-labeled dextran and retinal flat mounts were examined by fluorescence microscopy. The eyes of all mice injected with vehicle, regardless of the frequency of injections, showed numerous tufts of NV on the outer surface of the retina (Fig. 2A–C). Mice given two injections of SU4312 seven days apart also showed many tufts of NV on the outer surface of the retina (Fig. 2D), but mice given injections of SU4312 every 5 days (Fig. 2E) or every 4 days (Fig. 2F) showed few tufts of NV. Image analysis with the investigator masked with respect to treatment group was used to quantify these results. Eyes of mice given two injections of SU4312 over the span of 2 weeks had no significant difference in mean (±SEM) area of NV on the outer surface of the retina compared to eyes of mice that received two injections of vehicle (Fig. 2G, first column). However, eyes given injections of SU4312 every 5 days (second column) or 4 days (third column) had a significant reduction in the area of NV on the outer surface of the retina compared to their corresponding vehicle-injected control group. Fellow eyes of mice given injections of SU4312 every 5 or 4 days also showed significant reductions in the area of NV compared to eyes from vehicle-injected controls indicating that the amount of SU4312 getting into the systemic circulation was sufficient to block VEGF signaling in the fellow eye. Since there was similar strong blockade of VEGF effects with injections every 5 days or every 4 days, in subsequent experiments SU4312 was injected every 5 days but the dose was increased to 5 μg.
Fig. 2.
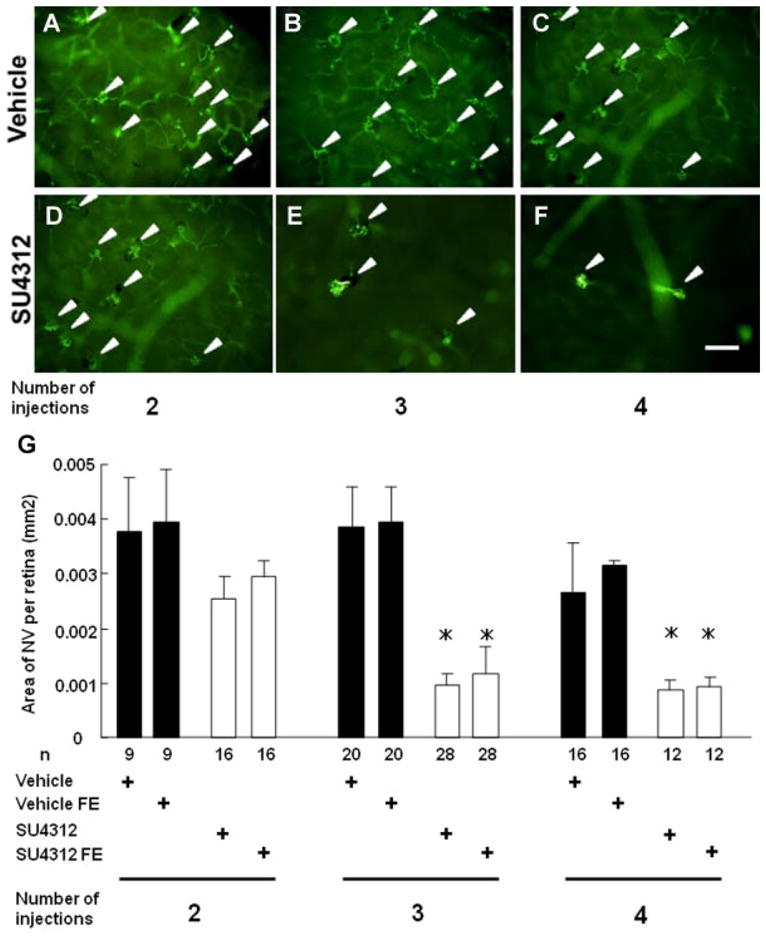
Effect of periocular injections of SU4312 in transgenic mice in which the rhodopsin promoter drives expression of VEGF in photoreceptors (rho/VEGF mice). Starting at postnatal day (P) 14, hemizygous rho/VEGF mice were given a periocular injection of 3 μl of vehicle or 3 μl containing 3 μg of SU4312 every 4, 5, or 7 days. At P28, these three groups of mice which had had a total of 4, 3, or 2 injections were perfused with fluorescein-labeled dextran and retinal flat mounts were examined by fluorescence microscopy. The eyes of all mice injected with vehicle, regardless of the frequency of injections, showed numerous tufts of neovascularization (NV) on the outer surface of the retina (A–C). Mice given two injections of SU4312 every 7 days a part also showed many tufts of NV on the outer surface of the retina (D), but mice given injections of SU4312 every 5 days (E) or every 4 days (F) showed few tufts of NV. Image analysis with the investigator masked with respect to treatment group showed that eyes of mice given two injections of SU4312 over the span of 2 weeks had no significant difference in mean (±SEM) area of NV on the outer surface of the retina compared to eyes of mice that received two injections of vehicle (G, first column). However, eyes given injections of SU4312 every 5 days (second column) or 4 days (third column) had a significant reduction in the area of NV on the outer surface of the retina than their corresponding vehicle-injected control group. Fellow eyes (FE) of mice given injections of SU4312 every 5 or 4 days also showed significant reductions in the area of NV compared to eyes from vehicle-injected controls indicating that the amount of SU4312 getting into the systemic circulation was sufficient to block VEGF signaling in the fellow eye. *P < 0.001 by ANOVA with Bonferroni/Dunn’s correction for multiple comparisons. Scale bar =100 μm.
Retinal function as assessed by electroretinograms (ERGs) is not reduced by blockade of VEGF signaling in the retina
Adult C57BL/6 mice were given a periocular injection of 5 μg of SU4312 in one eye every 5 days and a separate control group of mice was given periocular injections of vehicle every 5 days. Scotopic and photopic ERGs were done 4, 6, 8, 10, and 12 weeks after initiating injections. Scotopic ERG wave forms at 12 weeks after initiating injections appeared very similar in vehicle-treated mice and SU4312-treated mice (Fig. 3A,B). The mean a-wave and b-wave amplitudes at each of the stimulus intensities in the eyes of mice treated for 12 weeks with SU4312 were not significantly different from those in mice treated with vehicle (Fig. 3C,D). They also were not different from those seen in fellow eyes of mice treated with vehicle. Photopic ERG wave forms appeared similar in eyes of mice treated for 12 weeks with vehicle or SU4312 (Fig. 3E,F). Measurement of mean photopic b-wave amplitudes at each of 3 stimulus intensities confirmed that there was no difference between eyes treated with SU4312 and those treated with vehicle (Fig. 3G). The ERG wave forms obtained at 4, 6, 8, and 10 weeks after initiation of treatment were very similar to those seen at the 12-week time point and there were no differences between treatment groups (data not shown).
Fig. 3.

Prolonged blockade of VEGF signaling in the retina did not reduce electroretinogram (ERG) a- or b-wave amplitudes. Adult (4–6weeks old) C57BL/6 mice were given a periocular injection of 5 μl of vehicle or 5 μl of vehicle containing 5 μg of SU4312 every 5 days. Scotopic and photopic ERGs were done 4, 6, 8, 10, and 12 weeks after initiating injections. The scotopic ERG wave forms at 12 weeks (12w) after initiating injections appeared very similar in vehicle-treated mice (A) and SU4312-treated mice (B). The mean (±SEM) a-wave (C) and b-wave amplitudes (D) are shown for the 12-week time point. There was no significant difference in amplitude at any flash intensity between SU4312-treated eyes and vehicle-treated eyes and the curves for the fellow eyes (FE) for each group were superimposable on the curves for SU4312-treated and vehicle-treated eyes. Photopic ERG waveforms also appeared similar in eyes of mice treated for 12 weeks with vehicle (E) or SU4312 (F). The mean (±SEM) photopic b-wave amplitudes (G) showed no significant difference between eyes treated with SU4312 or vehicle for 12 weeks, or for FE for each group at three different flash intensities. The data from earlier time points are identical to those shown here for the 12-week time point.
Inhibition of VEGF signaling in the retina for up to 12 weeks did not cause apoptosis
Adult C57BL/6 mice were given a periocular injection of 5 μg of SU4312 every 5 days and TUNEL staining for apoptosis was done on ocular sections 4, 6, 8, 10, and 12 weeks after starting treatment. There were no TUNEL-positive cells in the retina of any mice treated with SU4312 for any of these time periods (Fig. 4A–D). Rd10 mice are homozygous for a missense mutation in exon 13 of the β subunit of the Pde6b gene, which results in rod cell death starting by P18 (Chang et al., 2002; Gargini et al., 2007). Ocular sections from P20 rd10 mice were used as a positive control and they showed numerous TUNEL-positive cells in the ONL (Fig. 4E).
Fig. 4.
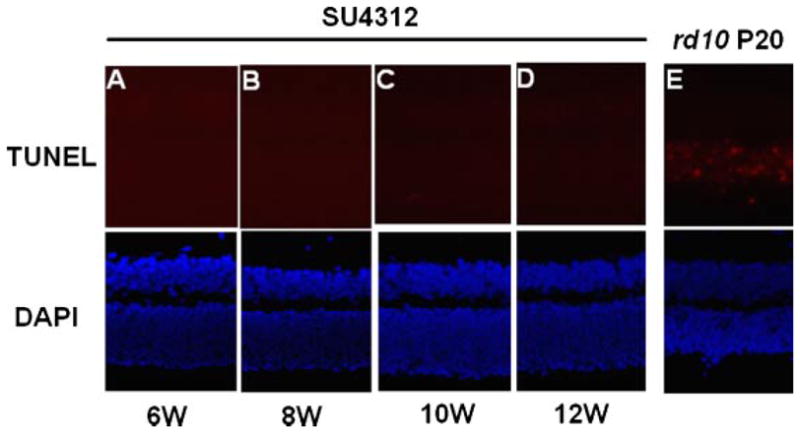
Apoptosis was not induced in the retinas of mice treated with SU4312. Adult C57BL/6 mice were given a periocular injection of 5 μg of SU4312 every5 days and were euthanized after 4,6, 8, 10, or12 weeks. TUNEL staining of ocular frozen sections alsostained with DAPI to visualize nuclei showed no labeled cells in the retinas of mice treated with SU4312 for 6 (A), 8 (B), 10 (C), or 12 (D) weeks, but there were numerous labeled cells in the retina of postnatal day (P) 20 rd10 mice (E).
No reduction of outer nuclear layer (ONL) thickness after blockade of VEGF signaling for up to 12 weeks
Death of photoreceptors results in loss of photoreceptor nuclei which occupy the ONL and this causes thinning of the ONL. However, the ONL thickness differs in different locations of the retina and thus comparisons between groups must be made with great care to insure that all comparisons are made at equivalent retinal locations. Using an approach that compares the same six locations in different eyes, it was found that at each of the locations tested there was no difference in mean ONL thickness between eyes of mice treated with SU4312 for 6 weeks compared to eyes of mice treated with vehicle for 6 weeks (Fig. 5A). Furthermore, fellow eyes of the mice in these two groups showed no differences from the two primary groups of eyes in mean ONL at corresponding locations. Treatment with SU4312 for 8 weeks (Fig. 5B) and 12 weeks (Fig. 5C) also did not lead to any significant changes in ONL thickness. Representative images of retinas from the four groups at the 12-week time point illustrate that at the same location retinas from eyes treated with SU4312 had an ONL and INL that appeared very similar to those seen in fellow eyes or in other mice treated with vehicle or their fellow eyes (Fig. 5D–G).
Fig. 5.
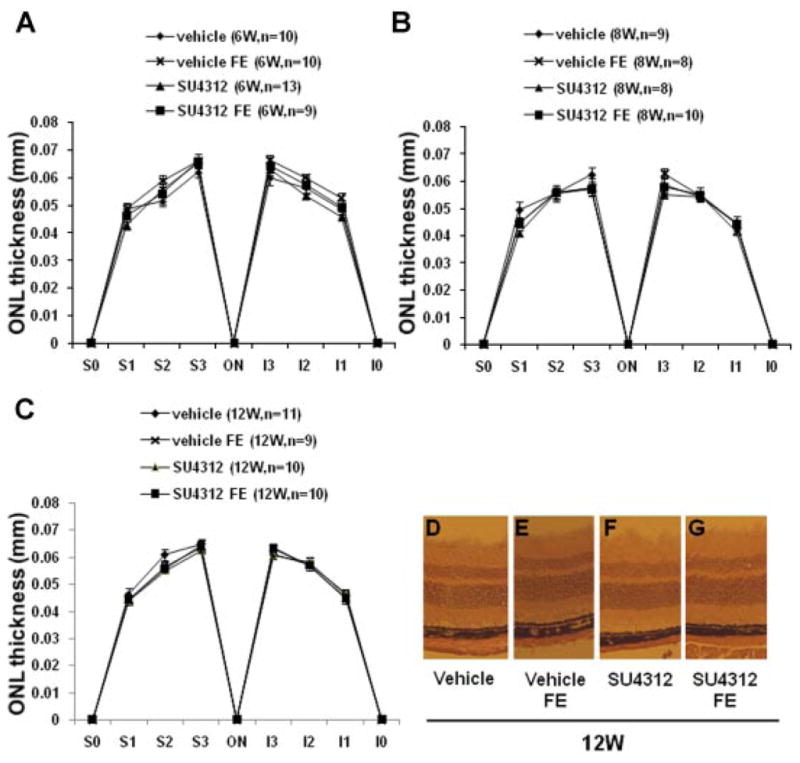
Treatment with SU4312 for upto 12 weeks did not cause reduction in thickness of the outer nuclear layer (ONL). Adult C57BL/6 mice were given periocular injections of 5 μl of vehicle or vehicle containing 5 μg of SU4312 every 5 days in one eye and after 4, 6, 8, 10, or 12 weeks, ONL thickness was measured by image analysis at six locations in each eye; 25% (S1), 50% (S2), and 75% (S3) of the distance between the superior pole and the optic nerve and 25% (I1), 50% (I2), and 75% (I3) of the distance between the inferior pole and the optic nerve. Each point represents the mean (±SEM) for the designated number of mice in each group. Compared to eyes injected with vehicle, eyes injected with SU4312 for 6 weeks (A), 8 weeks (B), or 12 weeks (C) showed no difference in mean ONL thickness at each of the corresponding six locations. The mean ONL thickness values for both of these groups were also not different from values in contralateral fellow eyes (FE) of eyes injected with SU4312 or FE of eyes injected with vehicle. Representative images are shown only for the 12-week time point—the retinas from SU4312-injected eyes, their contralateral fellow eyes (FE), vehicle-injected eyes from separate mice and their FE all looked similar.
Blockade of VEGF signaling in young mice does not cause retinal damage
Newly developed neuronal cells are often more susceptible to damage than mature neuronal cells. To maximize the chance of identifying VEGF toxicity in the retina we sought to block VEGF signaling in young mice. However, VEGF is critical for retinal vascular development and administration of VEGF antagonists during the first 2 weeks after birth causes retinal damage indirectly by perturbing the immature retinal vasculature. The earliest time point that strong antagonists of VEGF can be given without causing retinal damage from reduced perfusion is P14 (Seo et al., 1999; Ozaki et al., 2000). Starting at P14, mice were given periocular injections of 3 μg of SU4312 every 5 days. Three micrograms were used instead of 5 μg, because 3 μl is the maximum volume that can be given by periocular injection in baby mice and 1 mg/ml is the limit of SU4312 solubility. As shown in Figure 1, this regimen is able to completely block the effects of high level expression of VEGF in the retina. After 4 weeks of treatment, there were no TUNEL-positive cells in the retinas of mice treated with SU4312 (Fig. 6A) or mice treated with vehicle (Fig. 6B), but numerous TUNEL-positive cells were present in the ONL of P20 rd10 mice (Fig. 6C). Compared to eyes of mice treated with vehicle, eyes of mice treated with SU4312 for 4 weeks starting at P14 showed no difference in ONL thickness at six corresponding locations (Fig. 6D). Likewise, there was no reduction in mean photopic b-wave amplitudes (Fig. 6E), or scotopic a- and b-wave amplitudes (Fig. 6F,G).
Fig. 6.

SU4312 did not cause apoptosis, reduction of outer nuclear layer thickness, or reduction of ERG amplitudes in young mice. Starting at postnatal day (P) 14, C57BL/6 mice were given periocular injections of 3 μl ofvehicleor 3 μl of vehicle containing 3 μg of SU4312 every 5 days. After 4 weeks (4w) of injections, there were no TUNEL-positive cells in the retinas of mice treated with SU4312 (A) or vehicle (B), but numerous TUNEL-positive cells in the retinas of P20 rd10 mice (C). The sections were also stained with DAPI to visualize the nuclei (shown beneath each fluorescent image). There were no significant differences in mean (±SEM) outer nuclear layer (ONL) thickness at six corresponding locations in eyes injected with SU4312 for 4 weeks, their contralateral fellow eyes (FE), eyes of mice injected with vehicle for 4 weeks or their FE (D). There was no reduction in mean photopic b-wave amplitudes (E), or scotopic a- (F) and b-wave amplitudes (G) in eyes of mice treated with SU4312 for 4 weeks compared to vehicle-treated mice, or FE of each type of mice.
Treatment of cultured retinal ganglion cells with SU4312 does not reduce viability or axon outgrowth
Our in vivo data indicate that prolonged blockade of VEGF signaling does not damage RGCs or photoreceptors in the retina. Perhaps surrounding cells and extracellular matrix provide survival signals that compensate for blockade of VEGF signaling. To test for this possibility, we investigated the effects of VEGF receptor blockade in cultured retinal cells. We first tested cultures of isolated rat RGCs. The mRNA for VEGF receptor 2 is expressed in RGCs in vivo (Bocker-Meffert et al., 2002) and this expression is maintained in cultured RGCs (Fig. 7, left panel). Unlike whole retina, in which there are high levels of mRNA for rhodopsin, there was no detectable rhodopsin mRNA in the RGC cultures, indicating that there are minimal if any contaminating photoreceptor cells (Fig. 7, right panel).
Fig. 7.
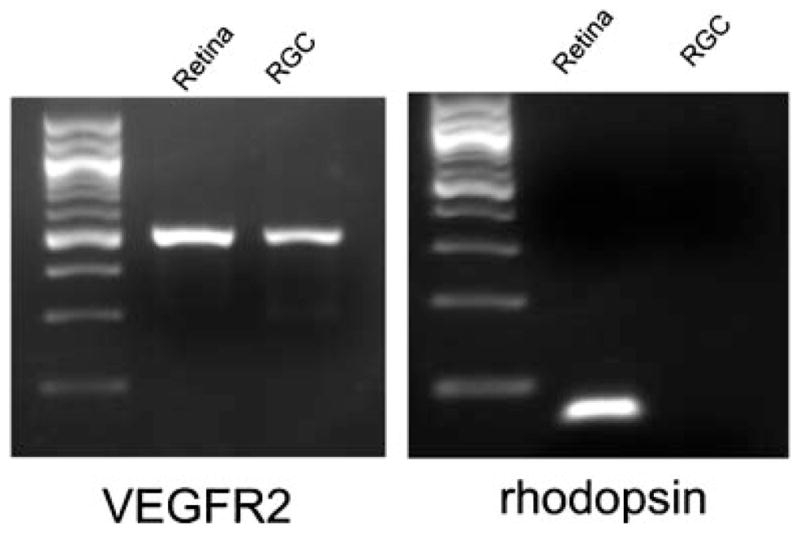
Expression of mRNA for vascular endothelial cell growth factor receptor 2 (VEGFR2) in retinal ganglion cell (RGC) cultures. Total RNA was prepared from immunopurified murine RGCs and from total mouse retina (as positive control), cDNA was synthesized, and 1 μg was used for RT-PCR using primers specific for VEGFR2 or rhodopsin. Both RGC cultures and total retina showed clear expression of VEGFR2 mRNA. The mRNA for rhodopsin was present in total retina, but not in the immunopurified RGCs.
RGC cultures were maintained in normal growth media or growth media supplemented with 0.1, 0.4, 1.6, or 6.4 μM SU4312. After 72 h, there was no difference in mean cell number of cultures exposed to any of the concentrations of SU4312 compared to unexposed cultures, indicating that blockade of VEGF signaling did not reduce viability of RGCs in vitro (Fig. 8A). Compared to ganglion cells maintained in normal growth medium, those exposed to various concentrations of SU4312 showed no significant difference in mean neurite length (Fig. 8B).
Fig. 8.
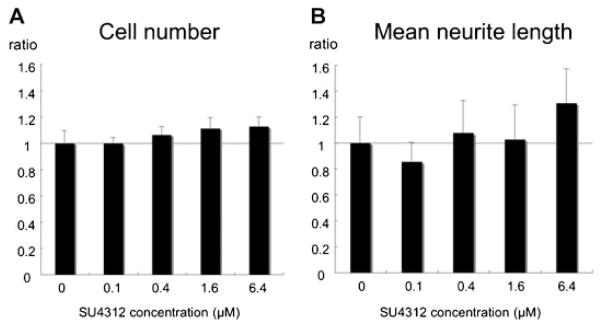
Blockade of vascular endothelial growth factor (VEGF) signaling does not alter viability or mean neurite length of cultured retinal ganglion cells. Rat RGC cultures were maintained in normal growth media or growth media supplemented with 0.1, 0.4, 1.6, or 6.4 μM SU4312 (A,B). After 72 h cell number (A) and mean neurite length (B) were analyzed by image analysis. The bars represent the mean (±SEM) cell number or neurite length value for each concentration of SU4312 normalized to control cultures without SU4312, which are defined to have a value of 1. There were no significant differences between any of the treatment groups and the controls in cell number or mean neurite length.
Treatment of dissociated retinal cell cultures with SU4312 does not reduce photoreceptor viability
Knock-in mice in which the murine rhodopsin gene was replaced by a human rhodopsin/green fluorescent protein (GFP) fusion gene (Chan et al., 2004) were used to generate dissociated retinal cell cultures. Since the rhodopsin/GFP expressed by the rod photoreceptors in these cultures is fluorescently labeled, it is easy to track photoreceptor development and survival. Consistent with prior experiments, the control molecules ciliary neurotrophic factor (CNTF) and staurosporin, a neurotrophic factor and a non-specific tyrosine kinase inhibitor, respectively, both moderately reduced total retinal cell survival under our culture conditions (Fig. 9). In terms of rhodopsin expression, as expected, staurosporin increased the percent of live rods and the relative level of rhodopsin/GFP expression, whereas CNTF reduced both of these parameters. Treatment with SU4312 did not have a consistent effect on any of the survival or rhodopsin expression parameters measured. To rule out the possibility that the lack of an observed consistent and significant effect with SU4312 treatment could have been due to the absence of VEGFR2 expression, we performed QPCR analysis and found that the mixed retinal cultures had easily detectable VEGFR2 mRNA expression throughout the culture period (data not shown).
Fig. 9.
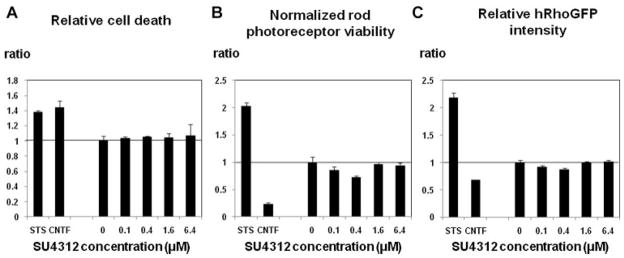
Blockade of vascular endothelial growth factor (VEGF) signaling does not alter viability or rhodopsin expression of dissociated mixed retinal cell cultures. Mixed retinal murine cell cultures were maintained in normal growth media or growth media supplemented with 0.1, 0.4, 1.6, or 6.4 μM SU4312 (all cultures contained 0.1% DMSO). Staurosporine (20 nM) was used as a positive control, and CNTF (1 ng/ml) was used as a negative control. After 6 days in vitro, cultures were imaged on an ArrayScan VTI and cell number and fluorescence intensity (relative rhodopsin level) per cell were quantified by image analysis. Each bar represents the mean value (±SEM) of the indicated parameter for the given SU4312 concentration normalized to the mean of the negative control cultures (three cultures for each experimental group). One-way ANOVA analysis did not show any significant effect of SU4312 treatment on the measured cell parameters.
Discussion
In a previous study, we demonstrated that expression of a VEGF binding protein in the retina completely suppressed VEGF-induced leakage, but even 7 months of continuous expression of the binding protein failed to cause any reduction in retinal function as assessed by ERGs or cause any morphologic evidence of retinal cell death (Ueno et al., 2008). In the midst of our study, it was reported that systemic injections of soluble VEGFR1 three times a week in mice for 8 weeks or intraocular injections of an anti-VEGF antibody for 6 weeks caused loss of 50% of RGCs (Nishijima et al., 2007). This caused us to perform a quantitative analysis of RGC survival by counting RGC axons in the optic nerve of mice in which a VEGF binding protein had been expressed in the retina for 7 months; there was no difference in RGC axons and hence RGCs in these mice compared to corresponding controls (Ueno et al., 2008).
Soon after our study was published, a study by Saint-Geniez et al. (2008) suggested that systemic injection of 2.5 ×109 Ad-CMV-sFlt1 caused massive cell death in the inner nuclear layer (INL) and outer nuclear layer (ONL) of the retina. Subsequently, a study by Takeda et al. (2009) suggested that intraocular injections of 3 μg of an anti-VEGF antibody on days 0, 3, and 6 in mice caused pigmentary changes in the retina and some alterations in ERGs. In view of these reports, we sought additional ways to rigorously test the effects of blocking VEGF in the eye.
One of the most efficient ways to block the effects of VEGF is with small molecules that selectively inhibit autophosphorylation of VEGF receptors, because such antagonists tend to penetrate tissues better than protein antagonists and thus provide pharmacokinetic advantages. In addition, since they penetrate cell membranes, they block the effects of intracellular as well as extracellular VEGF, and therefore can eliminate autocrine as well as paracrine effects of VEGF (Lee et al., 2007). We first showed that intraocular injection of the commercially available VEGF receptor kinase inhibitor SU4312 blocked the ability of VEGF to phosphorylate Akt, which is necessary for VEGF-mediated survival signaling. We then sought to identify a regimen of administration of SU4312 that provided long term suppression of VEGF-stimulated subretinal neovascularization. We utilized rho/VEGF transgenic mice in which the rhodopsin promoter drives sustained expression of VEGF in photoreceptors starting at P7, which results in a few buds of subretinal neovascularization at P14 that increase and coalesce into an extensive network of neovascularization over time (Okamoto et al., 1997; Tobe et al., 1998). Periocular injections of 3 μg of SU4312 every 5 days starting at P14 completely suppressed the growth of subretinal neovascularization. The ability to block the effects of high levels of VEGF in the retina indicates that this regimen would certainly maintain sufficient levels of SU4312 in the eye to block all effects of endogenous levels of VEGF. We then tested the effect of a higher dose of SU4312, periocular injection of 5 μg every 5 days, in adult wild-type mice and found that complete blockade of VEGF signaling in the retina for up to 12 weeks did not result in any reduction in scotopic ERG a- or b-wave amplitudes or photopic b-wave amplitudes, indicating that rod, cone, and second order neuron functioning in the retina was normal. This lack of observable functional changes after 12 weeks of blockade of VEGF signaling was accompanied by a lack of any evidence of photoreceptor cell death; there was no apoptosis and no reduction in thickness of the ONL, which contains the photoreceptor nuclei. Since juvenile mice are more susceptible to neurotoxic effects than adult mice, we tested whether we could identify some subtle toxicity from VEGF inhibition by starting injections of 3 μg of SU4312 at P14 and continuing every 5 days for 4 weeks. Like adult mice, these young mice showed no evidence of cell death or reduced function in the retina. Since surrounding cells and extracellular matrix can be a major source of survival signals and might mask potential toxic effects, we also tested isolated cells in vitro because they might provide a more sensitive assay for potential negative effects of VEGF signaling blockade on retinal cell function and survival. Treatment of isolated retinal ganglion cells or dissociated retinal cells containing photoreceptors with a wide range of doses of SU4312, including doses shown to cause damage to isolated capillary endothelial cells (Lee et al., 2007), failed to cause death of retinal cells. Thus, even with this most stringent test, we failed to identify any deleterious effect of blocking VEGF signaling in retinal cells. These data and our previously published data (Ueno et al., 2008) indicate that blockade of the effects of VEGF in the retina for up to 7 months does not cause detectable retinal damage or dysfunction.
When studies in the literature arrive at opposite conclusions, it is important to closely examine the design of the studies and quality of data that lead to the differing conclusions. Figure 4 in Saint-Geniez et al. shows widespread TUNEL positive cells in the INL and ONL 14 days after systemic injection of Ad-sFlt1. The number of reported apoptotic cells is difficult to reconcile with our results. We did not see any increase in TUNEL positive cells with prolonged blockade of VEGF signaling, and in fact the number of apoptotic cells described by Saint-Geniez et al. far exceeded that which we observed at the peak of photoreceptor death in the rd10 mouse, in which there is extensive mutation-induced photoreceptor loss (see Fig. 4, this manuscript). Such extensive apoptosis would likely lead to blindness, as is the case in rd10 mice, and yet patients that receive intraocular injections of ranibizumab or VEGF Trap-Eye (which is more similar to sFlt1) have complete blockade of VEGF for much longer than 14 days and show no recognizable retinal dysfunction. A metanalysis taking into account all of the published literature in which intraocular injections of VEGF antagonists were given for neovascular AMD failed to identify any evidence of retinal toxicity from blocking VEGF and concluded that anti-VEGF treatments are safe and effective (Ip et al., 2008). The reason for the difference between the results reported by Saint-Geniez et al. and our results is unclear, and requires further study, but we feel that our data clearly demonstrate that VEGF signaling in the retina can be significantly blocked for prolonged periods of time without detectable evidence of retinal cell damage. Although Saint-Geniez et al. reported that their null adenoviral vector did not elicit a significant effect, perhaps the damage they observed with their Ad-sFlt1 construct somehow reflected an off-target effect or some synergy related to the adenoviral delivery system employed. The explanation for our divergent cell culture results is also unclear, but could potentially reflect the different cell culture systems employed—for our photoreceptor studies we used total retinal cultures while Saint-Geniez et al. used photoreceptor enriched cultures.
The paper by Takeda et al. states that intraocular injections of 2 ng or 3 μg of an anti-VEGF antibody, as compared to mice injected with vehicle or antagonists of CCR3, can cause pigmentary changes on fundus photographs and ERG alterations. Pigmentary changes are generally indicative of reactive changes in the retinal pigmented epithelium. In our experience, even after a single intraocular injection of any solution using pulled glass micropipettes and a foot-activated injector, there are usually pigmentary changes. Takeda et al. performed three intraocular injections 3 days apart with a 33-gauge needle and a hand-activated syringe, a technique that one would expect to be more damaging than the use of thinner glass micropipettes. Thus, it is not surprising that the mice injected with anti-VEGF antibody showed some pigmentary changes. From our experience, what is more surprising is the pristine appearance of the retinas of the mice given three intraocular injections of PBS or CCR3 antagonists. Objective analysis of pigmentary changes would require quantitative and masked analysis of fundus photos following a systematic protocol. In addition, to assess the possible meaning of pigmentary changes they should be combined with histopathologic studies of retinal structure. To assess possible ERG alterations, Takeda et al. showed a series of selected ERG waveforms and stated that they showed reduced retinal function in mice injected with anti-VEGF antibody. Again, due to inherent variability in ERG measurements, objective assessment of possible electrophysiological changes from administration of an anti-VEGF antibody requires a systematic, quantitative, and statistically rigorous analysis of multiple ERG recordings.
There are many patients with neovascular AMD who depend upon frequent injections of a VEGF antagonist to maintain useful vision. It would clearly be unfortunate if the vision promoting effects of VEGF antagonism are in fact countered by effects that promote retinal cell dysfunction and death, as has been suggested by the several recent studies discussed above. However, it would be even more unfortunate if patients are denied or forgo potentially vision saving treatments because of safety concerns that, with further investigation, turn out to be incorrect. Although no single mouse study is going to fully prove or disprove whether humans undergoing anti-VEGF treatment are at risk for retinal toxicity, we feel that the data presented in this manuscript should be reassuring to patients and physicians alike in that it demonstrates that long-term and high-level blockade of VEGF signaling can be achieved without detectable toxic effects on retinal cells. Given the continuing controversy, we strongly encourage other investigators and clinicians in the field to continue to closely scrutinize and analyze the effects of VEGF antagonism in the retina.
Acknowledgments
Contract grant sponsor: National Eye Institute;
Contract grant numbers: EY12609, EY009769, P30EY1765.
The authors wish to thank John Wilson and Theodore Wensel (Baylor College of Medicine) for generously providing the rhodopsin/GFP knock-in mice. This is study was supported by National Eye Institute (EY12609, EY009769, and core grant P30EY1765). P.A.C. is the George S. and Dolores Dore Eccles Professor of Ophthalmology and Neuroscience. D.J.Z. is the Guerrieri Family Professor of Ophthalmology, Molecular Biology and Genetics, and Neuroscience. P.A.C. has an institutional consulting agreement with Genentech, Inc. for which payment is made to Johns Hopkins University, has research funding for clinical trials sponsored by Genentech but not for this study, and is on the Data and Safety Monitoring Committee for the View 1 Trial sponsored by Regeneron, Inc.
Literature Cited
- Barres BA, Chun Linda LY. Purification of rat retinal ganglion cells by immunopanning. Neuroprotocols. 1993;2:201–204. [Google Scholar]
- Bocker-Meffert S, Rowenstiel P, Rohl C, Warneke N, Held-Feindt J, Sievers J, Lucius R. Erythropoietin and VEGF promote neural outgrowth from retinal explants in postnatal rats. Invest Ophthalmol Vis Sci. 2002;43:2021–2026. [PubMed] [Google Scholar]
- Bonnet D, Garcia M, Vecino E, Lorentz JG, Sahel J, Hicks D. Brain-derived neurotrophic factor signalling in adult pig retinal ganglion cell neurite regeneration in vitro. Brain Res. 2004;1007:142–151. doi: 10.1016/j.brainres.2004.02.023. [DOI] [PubMed] [Google Scholar]
- Bradford RL, Wang C, Zack DJ, Adler R. Roles of cell-intrinsic and microenvironmental factors in photoreceptor cell differentiation. Dev Biol. 2005;286:31–45. doi: 10.1016/j.ydbio.2005.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S the Anchor Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA. Ocular neovascularisation and excessive vascular permeability. Expert Opin Biol Ther. 2004;4:1395–1402. doi: 10.1517/14712598.4.9.1395. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Hafiz G, Shah SM, Nguyen QD, Ying H, Do DV, Quinlan E, Zimmer-Galler I, Haller JA, Solomon S, Sung JU, Hadi Y, Janjua KA, Choy DF, Arron JR. Ranibizumab for macular edema due to retinal vein occlusions; implication of VEGF as a critical stimulator. Mol Ther. 2008;16:791–799. doi: 10.1038/mt.2008.10. [DOI] [PubMed] [Google Scholar]
- Chan F, Bradley A, Wensel TG, Wilson JH. Knock-in human rhodopsin-GFP fusions as mouse models for human disease and targets for gene therapy. Proc Natl Acad Sci USA. 2004;101:9109–9114. doi: 10.1073/pnas.0403149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurda RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–525. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. J Comp Neurol. 2007;500:222–238. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Ip MS, Scott IU, Brown GC, Brown MM, Ho AC, Huang SS, Recchia FM. Anti-vascular endothelial growth factor pharmacotherapy for age-related macular degeneration. A report of the American Academy of Ophthalmology. Ophthalmology. 2008;115:1837–1846. doi: 10.1016/j.ophtha.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall RL, Rutledge RZ, Mao X, Tebben AJ, Hungate RW, Thomas KA. Vascular endothelial growth factor receptor KDR tyrosine kinase activity is increased by autophosphorylation of two activation loop tyrosine residues. J Biol Chem. 1999;274:6453–6460. doi: 10.1074/jbc.274.10.6453. [DOI] [PubMed] [Google Scholar]
- Kerrison JB, Lewis RN, Otteson DC, Zack DJ. Bone morphogenetic proteins promote neurite outgrowth in retinal ganglion cells. Mol Vis. 2005;11:208–215. [PubMed] [Google Scholar]
- Kilic U, Kilic E, Jarve A, Guo Z, Spudich A, Bieber K, Barzena U, Bassetti CL, Marti HH, Hermann DM. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. J Neuosci. 2006;26:12439–12446. doi: 10.1523/JNEUROSCI.0434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeima K, Rogers BS, Lu L, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natil Acad Sci USA. 2006;103:11300–11305. doi: 10.1073/pnas.0604056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213:809–815. doi: 10.1002/jcp.21152. [DOI] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Duh E, Gehlbach P, Ando A, Takahashi K, Pearlman J, Mori K, Yang HS, Zack DJ, Ettyreddy D, Brough DE, Wei LL, Campochiaro PA. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001;188:253–263. doi: 10.1002/jcp.1114. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Tatlipinar S, Shah SM, Haller JA, Quinlan E, Sung J, Zimmer-Galler I, Do DV, Campochiaro PA. Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am J Ophthalmol. 2006;142:961–969. doi: 10.1016/j.ajo.2006.06.068. [DOI] [PubMed] [Google Scholar]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Tobe T, Hackett SF, Ozaki H, Vinores MA, LaRochelle W, Zack DJ, Campochiaro PA. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: A new model of intraretinal and subretinal neovascularization. Am J Pathol. 1997;151:281–291. [PMC free article] [PubMed] [Google Scholar]
- Okoye G, Zimmer J, Sung JPG, Deering T, Nambu N, Hackett SF, Melia M, Esumi N, Zack DJ, Campochiaro PA. Increased expression of BDNF preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J Neuosci. 2003;23:4164–4172. doi: 10.1523/JNEUROSCI.23-10-04164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki H, Seo M-S, Ozaki K, Yamada H, Yamada E, Hofmann F, Wood J, Campochiaro PA. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol. 2000;156:679–707. doi: 10.1016/S0002-9440(10)64773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY the Marina Study Group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, Raharaj ASR, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, Darland DC, Young MJ, D’Amore PA. Endogenous VEGF is required for visual function: Evidence for a survival role on Muller cells and photoreceptors. Plos One. 2008;3:e3554, 1–13. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saishin Y, Saishin Y, Takahashi K, Melia M, Vinores SA, Campochiaro PA. Inhibition of protein kinase C decreases prostaglandin-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195:210–219. doi: 10.1002/jcp.10238. [DOI] [PubMed] [Google Scholar]
- Seo M-S, Kwak N, Ozaki H, Yamada H, Okamoto N, Fabbro D, Hofmann F, Wood JM, Campochiaro PA. Dramatic inhibition of retinal and choroidal neovascularization by oral administration of a kinase inhibitor. Am J Pathol. 1999;154:1743–1753. doi: 10.1016/S0002-9440(10)65430-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Tran N, Tang F, App H, Hirth P, McMahon G, Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: A novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem. 1998;41:2588–2603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- Takeda A, Baffi JZ, Kleinman ME, Cho WG, Nozaki M, Yamada K, Kaneko H, Albuquerque RJC, Dridi S, Saito K, Raisler BJ, Budd SJ, Geisen P, Munitz A, Ambati BK, Green MG, Ishibasi T, Wright JD, Humbles AA, Gerard CJ, Ogura Y, Pan Y, Smith JR, Grisanti S, Hartnett ME, Rothenberg ME, Ambati J. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460:225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe T, Okamoto N, Vinores MA, Derevjanik NL, Vinores SA, Zack DJ, Campochiaro PA. Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci. 1998;39:180–188. [PubMed] [Google Scholar]
- Ueno S, Pease ME, Wersinger DMB, Masuda T, Vinores SA, Licht T, Zack DJ, Quigley H, Keshet E, Campochiaro PA. Prolonged blockade of VEGF family members does not cause identifiable damage to retinal neurons or vessels. J Cell Physiol. 2008;217:13–22. doi: 10.1002/jcp.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]