

Abstract
The characteristic oxidation or reduction reaction mechanisms of short-chain oxidoreductase (SCOR) enzymes involve a highly conserved Asp-Ser-Tyr-Lys catalytic tetrad. The SCOR enzyme Q9HYA2 from the pathogenic bacterium Pseudomonas aeruginosa was recognized to possess an atypical catalytic tetrad composed of Lys118-Ser146-Thr159-Arg163. Orthologs of Q9HYA2 containing the unusual catalytic tetrad along with conserved substrate and cofactor recognition residues were identified in 27 additional species, the majority of which are bacterial pathogens. However, this atypical catalytic tetrad was not represented within the Protein Data Bank. The crystal structures of unligated and NADPH-complexed Q9HYA2 were determined at 2.3 Å resolution. Structural alignment to a polyketide ketoreductase (KR), a typical SCOR, demonstrated that Q9HYA2's Lys118, Ser146, and Arg163 superimposed upon the KR's catalytic Asp114, Ser144, and Lys161, respectively. However, only the backbone of Q9HYA2's Thr159 overlapped KR's catalytic Tyr157. The Thr159 hydroxyl in apo Q9HYA2 is poorly positioned for participating in catalysis. In the Q9HYA2–NADPH complex, the Thr159 side chain was modeled in two alternate rotamers, one of which is positioned to interact with other members of the tetrad and the bound cofactor. A chloride ion is bound at the position normally occupied by the catalytic tyrosine hydroxyl. The putative active site of Q9HYA2 contains a chemical moiety at each catalytically important position of a typical SCOR enzyme. This is the first observation of a SCOR protein with this alternate catalytic center that includes threonine replacing the catalytic tyrosine and an ion replacing the hydroxyl moiety of the catalytic tyrosine.
Keywords: short-chain oxidoreductase (SCOR), short-chain dehydrogenase/reductase (SDR), NADPH cofactor binding, atypical catalytic tetrad, Q9HYA2, Pseudomonas aeruginosa
Introduction
The largest group (>16,000 members) of proteins in the classical short-chain oxidoreductase (SCOR) family of enzymes is classified by 40 highly conserved residues, including a characteristic N-terminal motif (TGxxxGxG), sequence length of 250–350 amino acids, NAD(H) or NADP(H) cofactor binding, and an Asn-Ser-Tyr-Lys catalytic tetrad.1–3 They are responsible for regulating many vital processes including oxidation/reduction of alcohols, aldehydes, and ketones.1 Optimized sequence alignments identified a probable SCOR protein Q9HYA2 (UniProt reference code) from Pseudomonas aeruginosa PAO1 as possessing the hallmarks of the SCOR family except for atypical catalytic residues (Lys118-Ser146-Thr159-Arg163) present at positions normally associated with the standard catalytic tetrad. P. aeruginosa is an opportunistic pathogen, with cystic fibrosis patients at particular risk. It is the top cause of nosocomial pneumonial infections with a mortality rate of up to 60% associated with multidrug resistant strains.4,5 Although the substrate of this enzyme is unknown, the atypical catalytic site is found in a total of 28 probable SCOR proteins, each from a different species. All of these orthologs exhibit the other classic SCOR motifs and tend to be present within an operon conserved across these species. The crystal structure of Q9HYA2 provides the first insight into this unusual SCOR subfamily.
Results and Discussion
Quality and architecture of the NADPH-bound and free Q9HYA2 structures
The apo and holo crystal structures of the SCOR protein Q9HYA2 from P. aerginousa were solved by molecular replacement (MR). The two forms of the enzyme crystallized in different space groups (apo: C2221; holo: P212121). Data collection and refinement statistics are presented in Table I. Both structures exhibit the typical Rossmann nucleotide-binding fold of alternating β/α secondary structure (a seven parallel β-strand core surrounded by six α-helices) common across this family.2,6
Table I.
Data Collection and Refinement Statistics
| Apo-Q9HYA2 | Holo-Q9HYA2 | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.97946 | 0.97946 |
| Resolution range (Å) | 50.0–2.31 (2.40–2.31) | 50.0–2.30 (2.35–2.30) |
| Number of observed reflections | 74,548 | 215,695 |
| Number of unique reflections | 19,618 | 42,509 |
| Redundancy | 3.8 (2.8) | 5.0 (3.1) |
| Data completeness (%) | 96.7 (83.0) | 92.7 (78.0) |
| Rmerge | 0.065 (0.23) | 0.067 (0.33) |
| I/σ(I) | 15.8 (2.7) | 18.0 (4.0) |
| Space group | C2221 | P212121 |
| Unit cell parameters (Å) | a = 118.2, b= 128.6, c = 59.8 | a = 65.8, b = 112.6, c = 136.6 |
| Refinement | ||
| Resolution used for refinement (Å) | 43.8–2.31 | 32.8–2.30 |
| Rwork/Rfree (%)a | 17.09/23.93 | 17.33/23.79 |
| Number of reflections for refinement | 19,618 | 42,509 |
| Average B factor (Å2)/No. molecules | ||
| Overall protein/residues | 30.2/524 | 42.3/1043 |
| Waters | 32.1/258 | 38.8/457 |
| Cl− ions | 29.5/2 | 87.2/3 |
| Mn2+ ions | 43.5/3 | — |
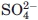 ions ions |
— | 108.8/2 |
| Glycerol | — | 55.4/2 |
| NADPH | — | 65.4/2 |
| RMSD bond angles (°) | 0.924 | 0.990 |
| RMSD bond length (Å) | 0.008 | 0.007 |
| Ramachandran plot | ||
| Residues in most favored regions (%) | 98.5 | 97.9 |
| Additionally allowed regions (%) | 1.5 | 2.1 |
| Generously allowed regions (%) | 0 | 0 |
Values in parenthesis are for high-resolution data shell.
R = Σhkl‖‖Fobs| − |Fcalc‖‖/Σhkl|Fobs|, where Rfree is calculated using a randomly chosen 5% of reflections, which were excluded from structure refinement, and Rwork is calculated using the remaining reflections.
Q9HYA2 exists as a tetramer in solution as judged by size exclusion chromatography (SEC; data not shown), and tetramers were observed in both crystal forms. However, the apo enzyme crystal form contained only a dimer in its asymmetric unit, with the tetramer formed via crystallographic twofold symmetry. The holo enzyme crystal form contained the complete tetramer within its asymmetric unit [Fig. 1(A) and Supporting Information Fig. 1]. The apo enzyme dimer present within the asymmetric unit is formed through interactions between the central β-sheets, similar to the dimer formed between monomers A and B in the holo enzyme structure (Supporting Information Fig. 1). The first three N-terminal residues were disordered in each monomer of the apo structure, and the cofactor and substrate binding sites were unoccupied except for a chloride anion (Cl−) and solvent molecules. For both monomers A and B, a Mn+2 ion was coordinated to His264 and a twofold crystallographic symmetry mate. Mn+2 is thought to only aid crystallization of the apo form because it did not occupy analogous positions in the holo enzyme structure. MolProbity scored the apo structure's geometry in the 98th percentile, and 98.5% of its residues were within Ramachandran plot favorable regions. The monomer A–B interface buries 1460 Å2 per monomer. Superposition of monomers A and B yielded a root-mean-square deviation (RMSD) of 0.42 Å for 261 Cα atoms, with no significant local conformational differences observed.
Figure 1.

Pseudomonas Q9HYA2 structure. A: A surface representation of the holo Q9HYA2 tetramer as present within the asymmetric unit. Each monomer is labeled a–d and individually colored. B: Sequence alignment of the catalytic center of a SCOR enzyme (polyketide ketoreductase, PDB: 1X7H) structurally homologous to Q9HYA2. Catalytic residues of interest have been highlighted with triangles. 1X7H is a typical SCOR enzyme containing the conserved Asn-Ser-Tyr-Lys catalytic tetrad. Q9HYA2 contains a Lys-Ser-Thr-Arg tetrad. The complete alignment is in Supporting Information Figure 3. C: Stereoview of the superposition of apo Q9HYA2 (monomer B; teal) and holo Q9HYA2 (monomer C; green) with NADPH, Arg40, secondary structure elements, and N and C-termini labeled. Regions displaying the greatest differences caused by NADPH binding are displayed in orange on the holo structure. D: Stereoview of superposition of the canonical Asn111-Ser144-Tyr157-Lys161 SCOR catalytic tetrad (1X7H monomer A; teal) with the corresponding residues (Lys118-Ser146-Thr159-Arg163) in holo Q9HYA2 (monomer A; green). Thr159 has two alternate side chain conformations. An interactive view is available in the electronic version of the article.PRO384 Figure 1
In the holo enzyme structure, the individual monomers of the tetramer have been labeled A, B, C, and D, and residues 3–203 and 210–265, 3–265, 5–265, and 4–265, respectively, were built into electron density [Fig. 1(A) and Supporting Information Fig. 1]. The β-sheet interface between monomers A and B, analogous to the asymmetric unit of the apo structure, buries 1320 Å2 per monomer. The dimer interface composed of α-helices 5 and 6 between monomers A and D buries 1580 Å2 per monomer. The analogous interfaces involving the remaining monomers are similar. The complex buries a total of 13,140 Å2 out of a total surface area of 38,100 Å2. Superposition of individual monomers (Cα atoms) produced RMSDs of 0.60–1.32 Å, indicating no large global conformational differences between monomers. Partially disordered NADPH was observed bound to two monomers, A and C, with only the adenine moiety to the PA phosphate group observed. Monomers B and D did not exhibit sufficient FO − FC density to indicate the presence of NADPH. Localized conformational differences between monomers were observed, which correlated to the presence (A and C) or absence (B and D) of bound cofactor. Partial cofactor binding has been observed in other SCOR crystal structures, such as human 17β-hydroxysteroid dehydrogenase (PDB ID: 1QYX) and M. tuberculosis FabG1 (PDB ID: 1UZN).7,8 MolProbity ranked the structure's geometry in the 83th percentile with 97.9% of the residues in a favorable region of the Ramachandran plot.
Structural changes upon cofactor binding
NADP(H) was the predicted cofactor for this protein based upon the presence of a basic residue (Arg40) 19 amino acids from the last glycine in the TGxxxGxG motif.2 The Q9HYA2-NADPH structure confirmed the protein's affinity for this cofactor, with NADP(H) recognition occurring through a salt bridge between Arg40 and the ligand's P2B phosphate group [Fig. 1(C)]. Apo Q9HYA2 monomers A and B were most structurally similar to holo Q9HYA2 monomer D (RMSDs of 0.49 and 0.44 Å, respectively, over 262 Cα atoms). Monomer D was one of the two chains in the holo crystal structure lacking a bound cofactor. Over 262 Cα atoms, the most dissimilar chains were apo monomer B and holo monomer C, with a RMSD of 0.93Å. These superimposed monomers displayed three regions with conformational differences, two of which are involved in cofactor interactions and the third is a loop typically involved in substrate binding.9 The entire α-helix 3 and the preceding loop containing the cofactor recognition residue Arg40 shifts ∼1.4 Å upon NADPH binding, as the Arg40 guanidinium group and the Arg40 and Asp41 main chain amides move to create a binding site for the cofactor's P2B phosphate. The loop region between β4 and α5 contains the conserved NNAG motif. NADPH binding causes main chain rearrangements of Asn94 to Gly98, including a ∼180° flip of Ala95, to accommodate the adenine ribose and phosphate groups (Supporting Information Fig. 2). The third conformational change occurs in a predicted substrate-binding loop, residues Gly195-Trp211 [α−helix 7a, Fig. 1(C)] which shifts by ∼2 Å, reducing the volume of the active site by ∼100 Å3. Binding of cofactor to SCOR enzymes has been observed to cause a conversion from an open to a closed form.10 As only a fragment of the cofactor was stably bound in the holo Q9HAY2 structure, it likely represents an intermediate form of the protein. The presence of both NADPH and a substrate may be required to fully convert the enzyme to the closed form.
Structural comparison of the catalytic tetrad
The SCOR enzyme family contains an Asn-Ser-Tyr-Lys catalytic tetrad. The tetrad amino acids are conserved with 94, 94, 96, and 97% sequence identity, respectively.2 Sequence and structural alignments suggest that Q9HYA2 contains an atypical Lys118-Ser146-Thr159-Arg163 catalytic tetrad [Fig. 1(B) and Supporting Information Fig. 3]. Structural homologs were identified by Dali using monomer A (holo) as the reference structure.11 The most similar structures present within the PDB were of Streptomyces coelicolor actinorhodin polyketide ketoreductase (KR; PDB: 1X7H, 1X7G, and 1W4Z), with Z-scores of 34.2–34.4, RMSDs of 2.1–2.3 Å over 253–254 Cα atoms, and all contained typical SCOR catalytic residues. Z-scores > 2 are considered nonrandom alignments. Dali searches using apo Q9HYA2 yielded similar results.
The conformation of the atypical catalytic tetrad in the apo and holo Q9HYA2 structures is comparable. Monomer A of the holo Q9HYA2 structure was superimposed on monomer A of the KR-NADP+ structure (1X7H), demonstrating that they contained similar main-chain positions about their catalytic centers. Additionally, Q9HYA2 Ser146 and Arg163 side chains superimposed well upon the KR catalytic residues Ser139 and Lys156, respectively [Fig. 1(D)]. This Ser-Arg combination has been recently observed in a structure of another atypical SCOR.12 The KR Asn114 backbone and side chain Nδ2 overlapped with the corresponding Lys118 backbone and Nζ of Q9HYA2. A characteristic of SCORs is a kinked α-helix 5 at the conserved catalytic asparagine (Asn114 in KR), with its main-chain carbonyl roughly perpendicular to the helical axis rather than involved in the standard α-helical hydrogen-bonding scheme. This kinked helix is critical for organizing a water-mediated proton wire connecting the active site to bulk solvent.10 A similar helical kink is present at Lys118 of Q9HYA2, and there is an ordered water 2.6 Å from its backbone carbonyl. A second water is 3.08 Å away from the first water (Supporting Information Fig. 4). Conformational changes in Q9HYA2's NNAG motif upon partial NADPH binding are reminiscent of those observed in FabG1, where conformational changes within the structure upon cofactor binding are required for completing the proton wire.10 These observations suggest that a complete proton wire to bulk solvent may form in Q9HYA2 upon fully occupied cofactor binding and/or substrate binding.
The most striking difference within the superimposed active sites was Thr159 (Q9HYA2) versus Tyr157 (KR). The main chain atoms of these residues overlapped, and both side chains pointed into the cofactor cavity [Fig. 1(D)]. In the traditional SCOR catalytic tetrad, the tyrosine hydroxyl participates in hydride transfer by interacting with the cofactor, the catalytic lysine, and the substrate.10 The apo Q9HYA2 Thr159 side chain is ill positioned to form any of these interactions, as its Oγ1 atom forms hydrogen bonds to two well-ordered residues (Arg99 N and Glu114 Oɛ2) positioned outside of the usual SCOR catalytic site. However, cofactor binding resulted in Thr159 assuming alternate rotamer conformations, one of which is similar to that observed in the apo structure. The second Thr159 rotamer directs its hydroxyl toward the cofactor cavity, positioning the hydroxyl such that it may interact with bound cofactor and substrate and with other residues likely involved in proton/hydride transfer. The Thr159 hydroxyl is ∼3.2–3.7 Å from the Arg163 guanidinium group, placing the distance between them within what is observed between the analogous residues in a traditional SCOR active site (e.g., 1X7H: Tyr157 Oη to Lys161 Nζ is ∼4.7 Å).
The apo structure had a fully occupied Cl− built into spherical electron density present near (2.9–3.0 Å) the Arg163 Nɛ atom and refined. Other chemical species in the crystallization cocktail were tested in this electron density, but Cl− was judged the best based on refined temperature factors. A similar Cl− ion was included in the holo structure based on the presence of difference density. The Cl− ion is located within ∼1 Å of the position typically occupied by a SCOR's catalytic tyrosine's hydroxyl moiety, poised to interact with a bound substrate and cofactor. The identification of the Cl− ion is tentative, and so water or another small chemical species may occupy this position.
Typical SCOR catalysis occurs through a general acid–base catalysis, where the catalytic lysine lowers the pKa of the catalytic tyrosine's hydroxyl moiety. The pKa of this hydroxyl is 9.7–10.2 in solution and 7.2 in the cofactor-bound local environment of the SCOR active site.13,14 This results in deprotonation of the tyrosine, priming it for proton/hydride transfer between substrate and cofactor.15 Detailed elucidation of the catalytic mechanism for Q9HYA2 awaits the determination of a substrate; however, several scenarios may be suggested. The positively charged Arg163 and/or Lys118 may reduce the normally high pKa of the adjacent Thr159, deprotonating and priming it for potential proton/hydride transfer. Arginine residues in dehydrogenases, lyases, reductases, and oxidases have been observed to act as a general base to facilitate proton abstractions, and so this is another possibility.16 Finally, a threonine residue in l-lactate dehydrogenase interacts with its NADH cofactor to facilitate hydride transfer, and Thr159 may play a similar role in Q9HYA2.17
Catalytic residue conservation and evolution
To understand the evolution of the protein Q9HYA2, the SwissProt/TrEMBL databank was searched for the presence of orthologs containing the atypical catalytic center. A Prosite search vector was used composed of the (1) N-terminal SCOR motif (TGxxxGxG)14, (2) the atypical catalytic tetrad (Kx(25,35) Sx(10,15)TxxxR)2, and (3) a substrate-binding motif. Three loops are implicated in substrate binding: β4α5, β5α6, and β6α7 [Fig. 1(C)]. Positions on these loops have been shown to identify and cluster similar SCORs by substrate preference.9,18 Five positions on the Q9HYA2 substrate-binding loops were used to enhance search selectivity. Two of the five positions are located after the NNAG motif in the β4α5 loop, and the remaining three positions are located just before Q9HYA2's atypical catalytic motif (TxxxR) in the β5α6 loop (Supporting Information Fig. 5), yielding the search pattern TGxxxGxG x(60,100) GxV x(10,30) K x(25,35) S x(5,10) ExHMxxTxxxR. This search pattern identified Q9HYA2, as expected, plus 27 orthologs all from two classes (β and γ) of Proteobacteria (Supporting Information Fig. 6). The majority of bacteria containing a Q9HYA2 ortholog are known human pathogens, or closely related to one. The presence of a Q9HYA2 ortholog in numerous divergent species across two classes suggests that evolutionary pressure exists to select for its atypical catalytic tetrad. Additionally, Q9HYA2 occurs in a predicted operon where it is sandwiched between several genes.19 The genes of note upstream of Q9HYA2 are annotated as encoding a transcriptional regulator (PA3508) and a hydrolase (PA3509). The downstream genes are annotated as l-aspartate dehydrogenase (PA3505) and a conserved hypothetical protein (PA3506). This five-gene cassette is fully conserved in 19 of the species containing a Q9HYA2 ortholog, and in the remaining nine species this cassette contains at least three of the genes.
This study identified a new subfamily of the large and biologically important SCOR family that is primarily present in bacterial pathogens. Sequence alignments identified a P. aeruginosa SCOR, Q9HYA2, that contained an atypical catalytic center of Lys-Ser-Thr-Arg, and crystal structures of this protein with and without the predicted cofactor NADPH were determined. These structures are the first examples of this SCOR subfamily deposited in the PDB. Orthologs have been identified in 27 additional species with all possessing identical substrate fingerprints and catalytic centers. Substrate identification for Q9HYA2 is ongoing in our laboratories.
Materials and Methods
Expression and purification
The open reading frame of the gene PA3507 encoding the protein Q9HYA2 (RefSeq: NP_252197) from P. aeruginosa PAO1 was first cloned into the pGEM-T Easy plasmid (Promega). During cloning, the restriction sites NdeI and XhoI were engineered onto the 5′ and 3′ ends, respectively, of the PCR-amplified fragment. The gene was subcloned into a customized pETDuet vector (Novagen) containing a N-terminal (His)6-SUMO affinity tag using these restriction sites.20 The expression cassette of the pQ9HYA2-SUMO plasmid was confirmed by DNA sequencing. Rossetta (DE3) E. coli (Novagen) were transformed with pQ9HYA2-SUMO. The cells were initially cultured in 150-mL Luria-Bertani (LB) medium supplemented with 100 μg mL−1 ampicillin and 35 μg mL−1 chloramphenicol. The overnight culture was transferred to 6-L LB, further cultured at 37°C to an optical density (600 nm) of ∼0.6, then induced with isopropyl β-d-1-thiogalactopyranoside (0.1 mg mL−1), and incubated overnight at 25°C. Cells were harvested by centrifugation at 6000g for 15 min. Cells were resuspended in lysis buffer (50 mM Tris pH 8.0, 1 mM DTT, 500 mM NaCl, and 10 mM imidazole) and 1 mL per 1 L of protease inhibitor cocktail (Sigma-Aldrich) and lysed via Microfluidizer (Microfluidics). Lysate was centrifuged at 35,000g for 1 h at 4°C. The supernatant was loaded onto a Ni-NTA superflow IMAC column (Qiagen) and eluted with lysis buffer plus 500 mM imidazole. Sample purity was evaluated by SDS-PAGE (BioRad). The affinity tag was cleaved by overnight incubation at 4°C with Ulp1 protease.21 Final purification and buffer exchange (20 mM Hepes pH 7.0, 100 mM NaCl, 5%(v/v) glycerol, and 1 mM EDTA) were accomplished with a Superdex 200 size exclusion column (GE Healthcare). Q9HYA2-containing fractions were pooled and concentrated to 10 mg mL−1 using an Amicon Ultra 30-kDa MWCO concentrator (Millipore). Protein concentration was determined by the Bradford assay, and molecular weight was determined through analytical SEC calibrated using protein standards.
Crystallization, X-ray data collection, structural determination, refinement, and bioinformatics
Crystallization screening was conducted using the high throughput robotics at the Hauptman-Woodward Institute22 and manual hanging drop screens. Diffraction quality apoenzyme crystals were grown by hanging drop vapor diffusion at 20°C, equilibrating a 1:1 droplet containing 10 mg mL−1 Q9HYA2 and reservoir solution, respectively, against a reservoir of 100 mM MES pH 6.0, 100 mM MnCl2, 20% (v/v) MPD, and 5% (w/v) PEG 4000. Crystals were flash cooled in liquid nitrogen. Diffraction data were collected via remote access on Stanford Synchrotron Radiation Laboratory (SSRL) beam line BL9-1 using an ADSC Q315R CCD detector controlled by Blu-Ice23 with a strategy calculated in MOSFLM.24 Intensities were processed with HKL2000.25 Holoenzyme crystal growth required different crystallization conditions. Optimized vapor diffusion crystal growth occurred at 10 mg mL−1 Q9HYA2 with 2 mM NADPH and a reservoir solution of 100 mM MES pH 6.0, 40 mM MnSO4, 20% (v/v) glycerol, and 15% (w/v) PEG 4000. The crystals were flash cooled in liquid nitrogen. Diffraction data were collected via remote access on SSRL BL9-2 on a MAR325 CCD detector.
The apo crystal structure was phased by MR using MolRep,24 using the closest sequence homolog (human peroxisomal trans 2-enoyl CoA reductase; PDB: 1YXM; 32% identity) as the search model. Initial phases for the holo structure were obtained by MR using the refined apo structure as the search model. Structure refinement was preformed in Phenix where the first run included simulated annealing to remove model bias.26 Each chain was defined as an independent TLS group in later refinement stages. Structures were manually fit in Coot.27 All small molecules were included in the models based on FO − FC density. The model was evaluated using MolProbity.28 Images were created with PyMOL.29 Sequence alignments and structural alignments were done with ClustalW and Dali, respectively.11,30 Motif-based searches for Q9HYA2 orthologs were done with the Prosite search tool.31 Cleft analysis was performed using the CASTp server.32 Buried surface areas were calculated by PISA.33 The coordinates of the apo and holo structures were deposited into the Protein Data Bank with the codes 3LF1 and 3LF2, respectively.
Acknowledgments
The Ulp1 protease expression plasmid was a kind gift of Dr. Christopher Lima (Sloan-Kettering Institute). P. aeruginosa PAO1 genomic DNA was a kind gift of Dr. Andrew Gulick (HWI). Initial crystallization hits were obtained with assistance from the Hauptman-Woodward Institute High Throughput Crystallization Laboratory. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
References
- 1.Tanaka N, Nonaka T, Nakamura KT, Hara A. SDR structure, mechanism of action, and substrate recognition. Curr Org Chem. 2001;5:89–111. [Google Scholar]
- 2.Duax WL, Pletnev V, Addlagatta A, Bruenn J, Weeks CM. Rational proteomics. I. Fingerprint identification and cofactor specificity in the short-chain oxidoreductase (SCOR) enzyme family. Proteins. 2003;53:931–943. doi: 10.1002/prot.10512. [DOI] [PubMed] [Google Scholar]
- 3.Duax WL, Thomas J, Pletnev V, Addlagatta A, Huether R, Habegger L, Weeks CM. Determining structure and function of steroid dehydrogenase enzymes by sequence analysis, homology modeling, and rational mutational analysis. Ann NY Acad Sci. 2005;1061:135–148. doi: 10.1196/annals.1336.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossolini GM, Mantengoli E. Treatment and control of severe infections caused by multiresistant Pseudomonas aeruginosa. Clin Microbiol Infect. 2005;11(Suppl 4):17–32. doi: 10.1111/j.1469-0691.2005.01161.x. [DOI] [PubMed] [Google Scholar]
- 5.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 6.Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 7.Shi R, Lin SX. Cofactor hydrogen bonding onto the protein main chain is conserved in the short chain dehydrogenase/reductase family and contributes to nicotinamide orientation. J Biol Chem. 2004;279:16778–16785. doi: 10.1074/jbc.M313156200. [DOI] [PubMed] [Google Scholar]
- 8.Cohen-Gonsaud M, Ducasse S, Hoh F, Zerbib D, Labesse G, Quemard A. Crystal structure of MabA from Mycobacterium tuberculosis, a reductase involved in long-chain fatty acid biosynthesis. J Mol Biol. 2002;320:249–261. doi: 10.1016/S0022-2836(02)00463-1. [DOI] [PubMed] [Google Scholar]
- 9.Duax WL, Huether R, Pletnev V, Umland TC, Weeks CM. Divergent evolution of a Rossmann fold and identification of its oldest surviving ancestor. Int J Bioinform Res Appl. 2009;5:280–294. doi: 10.1504/IJBRA.2009.02642. [DOI] [PubMed] [Google Scholar]
- 10.Price AC, Zhang YM, Rock CO, White SW. Cofactor-induced conformational rearrangements establish a catalytically competent active site and a proton relay conduit in FabG. Structure. 2004;12:417–428. doi: 10.1016/j.str.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamura A, Ichimura T, Mimoto F, Ohtsuka J, Miyazono K, Okai M, Kamo M, Lee WC, Nagata K, Tanokura M. A unique catalytic triad revealed by the crystal structure of APE0912, a short-chain dehydrogenase/reductase family protein from Aeropyrum pernix K1. Proteins. 2008;70:1640–1645. doi: 10.1002/prot.21860. [DOI] [PubMed] [Google Scholar]
- 13.Hwang CC, Chang YH, Hsu CN, Hsu HH, Li CW, Pon HI. Mechanistic roles of Ser-114, Tyr-155, and Lys-159 in 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. J Biol Chem. 2005;280:3522–3528. doi: 10.1074/jbc.M411751200. [DOI] [PubMed] [Google Scholar]
- 14.Kavanagh KL, Jornvall H, Persson B, Oppermann U. Medium- and short-chain dehydrogenase/reductase gene and protein families : the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci. 2008;65:3895–3906. doi: 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gani OA, Adekoya OA, Giurato L, Spyrakis F, Cozzini P, Guccione S, Winberg JO, Sylte I. Theoretical calculations of the catalytic triad in short-chain alcohol dehydrogenases/reductases. Biophys J. 2008;94:1412–1427. doi: 10.1529/biophysj.107.111096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillen Schlippe YV, Hedstrom L. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch Biochem Biophys. 2005;433:266–278. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 17.Sakowicz R, Kallwass HK, Parris W, Kay CM, Jones JB, Gold M. Threonine 246 at the active site of the L-lactate dehydrogenase of Bacillus stearothermophilus is important for catalysis but not for substrate binding. Biochemistry. 1993;32:12730–12735. doi: 10.1021/bi00210a023. [DOI] [PubMed] [Google Scholar]
- 18.Huether R, Liu ZJ, Xu H, Wang BC, Pletnev VZ, Mao Q, Duax WL, Umland TC. Sequence fingerprint and structural analysis of the SCOR enzyme A3DFK9 from Clostridium thermocellum. Proteins. 2010;78:603–613. doi: 10.1002/prot.22584. [DOI] [PubMed] [Google Scholar]
- 19.Pertea M, Ayanbule K, Smedinghoff M, Salzberg SL. OperonDB: a comprehensive database of predicted operons in microbial genomes. Nucleic Acids Res. 2009;37:D479–D482. doi: 10.1093/nar/gkn784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cody V, Mao Q, Queener SF. Recombinant bovine dihydrofolate reductase produced by mutagenesis and nested PCR of murine dihydrofolate reductase cDNA. Protein Expr Purif. 2008;62:104–110. doi: 10.1016/j.pep.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 22.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 23.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 24.Collaborative Computational Project. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLano WL. The PyMOL molecular graphics system. Palo Alto, CA: DeLano Scientific LLC; 2008. [Google Scholar]
- 30.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 31.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, De Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJ. The 20 years of PROSITE. Nucleic Acids Res. 2008;36:D245–D249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]