

Abstract
Osteogenesis imperfecta is a clinically and genetically heterogeneous brittle bone disorder that results from defects in the synthesis, structure, or posttranslational modification of type I procollagen. Dominant forms of OI result from mutations in COL1A1 or COL1A2, which encode the chains of the type I procollagen heterotrimer. The mildest form of OI typically results from diminished synthesis of structurally normal type I procollagen, whereas moderately severe to lethal forms of OI usually result from structural defects in one of the type I procollagen chains. Recessively inherited OI, usually phenotypically severe, has recently been shown to result from defects in the prolyl-3-hydroxylase complex that lead to the absence of a single 3-hydroxyproline at residue 986 of the α1(I) triple helical domain. We studied a cohort of five consanguineous Turkish families, originating from the Black Sea region of Turkey, with moderately severe recessively inherited OI and identified a novel locus for OI on chromosome 17. In these families, and in a Mexican-American family, homozygosity for mutations in FKBP10, which encodes FKBP65, a chaperone that participates in type I procollagen folding, was identified. Further, we determined that FKBP10 mutations affect type I procollagen secretion. These findings identify a previously unrecognized mechanism in the pathogenesis of OI.
Introduction
Osteogenesis imperfecta (OI [MIM 166200, 166210, 259420, 166220, 610967, 61098, 610682, 610915, 259440]) is a heritable brittle bone disease that varies in severity from mild to neonatal lethal. In addition to clinical heterogeneity, there is genetic heterogeneity with autosomal-dominant and -recessive forms of the disease. The vast majority of individuals with OI have dominantly inherited mutations in COL1A1 or COL1A2 (MIM 120150, 120160) that encode type I procollagen and result in defects in the synthesis or structure of type I collagen.1 OI has been historically classified into subtypes primarily based on clinical and, recently, molecular findings. Individuals with OI type I, the mildest phenotype, have normal or near-normal height and the phenotype usually results from mutations that lead to decreased synthesis of structurally normal type I collagen. The more severe OI types (II–IV) typically result from mutations that lead to structural defects in type I collagen.2 Less common forms include OI type V,3 an autosomal-dominant form of unknown etiology with hypertrophic callus formation, dense metaphyses, and a mesh-like histological appearance of the bone; OI type VI,4 a moderately severe, recessively inherited type of unknown cause that includes increased serum alkaline phosphatase (ALP) levels and bone histology described as exhibiting a fish scale-like lamellar pattern; and OI type VII, moderately severe, recessively inherited forms resulting from decreased expression of the cartilage associated protein (CRTAP) (MIM 605497) gene.5,6
Type I procollagen, a heterotrimer composed of two α1(I) and one α2(I) chains, is assembled in the lumen of the endoplasmic reticulum (ER). The three chains associate noncovalently at their carboxyl termini, with chain selection determined by the C-terminal propeptides. Disulfide bonds form between the chains, mediated by protein-disulfide isomerase (PDI). The unfolded chains undergo several posttranslational modifications including 4-prolyl hydroxylation, which is a critical modification for increasing the stability of the folded protein. Additional modifications include 3-hydroxylation of proline residue 986 of the α1(I) triple helical domain, which is accomplished by the prolyl-3-hydroxylase complex,5 and hydroxylation of some lysine residues by lysyl hydroxylases, followed by glycosylation by collagen galactosyl- and glucosyl-transferases.7 Chaperones, including PDI, BiP, and HSP47, assist with proper chain registration and folding of the molecules into the collagen triple helix.8 Once the chains fold, they are inaccessible to further posttranslational modification.9 The triple helical procollagen molecules leave the ER, traffic to the Golgi, and are then packaged into secretory vesicles and released into the extracellular matrix. They are then processed by removal of the amino- and carboxy-terminal propeptides and form the substrate for bone mineralization.10
Recently, the importance of the processes involved in type I collagen posttranslational modification in OI has been uncovered. Morello et al.5 showed in a knockout mouse model that cartilage associated protein (CRTAP) is essential for prolyl-3-hydroxylation and that reduced or absent activity leads to OI. CRTAP is part of the collagen prolyl-3-hydroxylase complex along with prolyl-3-hydroxylase-1 (P3H1 [MIM 610339]) and cyclophilin B or peptidyl-prolyl isomerase B (CYPB or PPIB [MIM 123841]) in a 1:1:1 ratio.11 Mutations in LEPRE1, which encodes P3H1, have also been shown to be associated with recessively inherited severe OI.5,6,12 In a subsequent study, Baldridge et al.13 screened a cohort of 78 individuals with presumed recessive forms of OI and found mutations in either CRTAP or LEPRE1 in 19 individuals. The absence of mutations in most cases suggests that there are additional genes responsible for recessive OI that have yet to be discovered.
We ascertained and studied five families that originated from Northern Turkey with a severe, progressive deforming type of OI. In these families, autosomal-recessive epidermolysis bullosa simplex (EB [MIM 131900]), resulting from a defect in keratin 14, cosegregated with the progressive, deforming OI.14 Homozygosity by descent mapping demonstrated that all affected individuals shared a 0.83 Mb region on chromosome 17q21 containing KRT14, the gene that encodes keratin 14. Sequence analysis of genes in the interval revealed that the OI phenotype resulted from homozygosity for an in-frame deletion in FKBP10, the gene that encodes the immunophilin FKBP65 (MIM 607063). In a second OI family, homozygosity for an FKBP10 null mutation was identified. FKBP65 has been shown to interact with type I collagen and tropoelastin, and we demonstrate that FKBP65 mutations affect the secretion of type I collagen. Thus, in addition to defects leading to alterations in the structure, synthesis, and posttranslational modification of type I procollagen, loss of FKBP65 chaperone activity can also produce a progressive deforming OI phenotype.
Material and Methods
Clinical Information, DNA Isolation, and Genotyping
Patients and their family members were assessed under approved human subjects protocols and all participants provided informed consent. Clinical information, radiographs, serum alkaline phosphatase levels, and skin and bone histology were reviewed to determine the findings in the affected individuals. DNA was isolated from either blood or cultured fibroblasts via a standard manufacturer's protocol (QIAGEN). Genotypes were determined with the GeneChip Mapping 250K array set and standard protocols (Affymetrix), and SNPs surrounding KRT14 on chromosome 17q21 were analyzed with a standard bioinformatics package (Vigenos, Hemosoft).
Bone Immunohistochemistry
Bone biopsies from the iliac crest of patient R06-116A were obtained during surgery to correct severe scoliosis. Biopsy specimens were fixed in 10% buffered formalin and processed for paraffin embedding after decalcification. Decalcified 6-μm-thick sections were cut and stained with Masson trichrome and toluidine blue (pH 3.7). Bone tissue obtained from the autopsy of an age-matched patient was used as a control. Sections were visualized under polarized light at 200× magnification.
Mutation Detection for KRT14 and FKBP10 and RT-PCR
With 50 ng of genomic DNA per reaction, exon 3 of KRT14 (Ensembl; ENSG0000018684) and the ten coding exons of FKBP10 (Ensembl: ENSG00000321562) were amplified by PCR (oligonucleotide sequences and conditions are available upon request). Nucleotide sequences of the PCR products were determined by bidirectional sequence analysis (MC Labs) and compared with the reference sequence for KRT14 and FKBP10 via Sequencher (Gene Codes). Nucleotides were numbered starting from the A of the ATG initiation codon. Fibroblasts were propagated in DMEM with 10% fetal bovine serum (FBS). RNA was isolated with Trizol (Invitrogen) and RT-PCR for FKBP10 was performed via SuperScript III (Invitrogen) with oligonucleotides that spanned introns 8 and 9.
Cell Culture and Analysis of Collagenous Proteins
Dermal fibroblast cultures were established from explanted skin biopsies from three OI patients (International Skeletal Dysplasia Registry reference numbers R06-113A from family 2, R06-016 from family 3, and R93-188A and R93-188B from family 6). Fibroblasts from affected individuals and controls were plated at confluence (250,000 cells) in 35 mm tissue culture dishes in Dulbecco-Vogt Modified Eagle Medium supplemented with 10% FBS allowed to attach overnight, and then incubated in the presence of 50 μM ascorbic acid for 18 hr prior to labeling. For pulse-chase studies, confluent cells were starved of proline in serum-free medium supplemented with 50 μM ascorbic acid for 1 hr, then labeled in serum-free medium with 100 μCi/ml [2,3,4,5-3H] proline. After 60 min in label, cells were rinsed twice with complete medium with 50 μM ascorbic acid, and fresh chase medium was added that contained 12 mM proline as previously described.15 At 0, 20, 40, 60, and 120 min, the medium was decanted and the cell layer scraped into a separate tube in PBS. Protease inhibitors were added to both, and the collagenous proteins were precipitated with 50% v:v ethanol and carrier collagen in the presence of 15 mM iodoacetamide added to prevent new disulfide bond formation. Precipitated proteins were resuspended in SDS-sample buffer and separated on 5% SDS-PAGE under nonreducing conditions.15 All gels were fixed, treated with Enhance, and dried. Radiolabeled bands were visualized by autoradiography and densities were quantified with Image J.
3-hydroxylation of proline at position 986 of the triple helix in the α1(I) chains of type I collagen was determined on collagen prepared from cell cultures of skin fibroblasts. Either CNBR-derived peptides or pepsinized collagens were isolated by SDS-PAGE and tryptic digests prepared for MS/MS. Electrospray MS was performed on an LCQ Deca XP ion-trap mass spectrometer with in-line liquid chromatography (LC) (ThermoFinnigan) with a C8 capillary column (300 mm × 150 mm; Grace Vydac 208MS5.315) eluted at 4.5 ml/min. The mobile gradient was made from buffer A (0.1% formic acid plus 2% buffer B in MilliQ water) and buffer B (0.1% formic acid in 3:1 acetonitrile:n-propanol v/v). An electrospray ionization source (ESI) under atmospheric pressure was used to introduce the sample stream into the mass spectrometer. The spray voltage was 3 kV and the inlet capillary temperature 160°C.
Transmission Electron Microscopy
Cells were fixed in 2% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer and washed. After postfixation in 1% OsO4 in buffer for 1 hr, the cells were dehydrated in a graded series of ethanol, treated with propylene oxide, and embedded in Eponate 12 (Ted Pella). Thin sections (60 nm) were cut, placed on formvar-coated grids, and counterstained with 7% methanolic uranyl acetate and lead citrate. Sections were viewed with a Tecnai 12 transmission electron microscope at 120 kV and images were digitally captured.
Immunohistochemistry
Cultured fibroblasts were seeded on 4-well chamber slides (Lab-Tek) and grown in 10% FBS to near confluence. Slides were washed with ice-cold PBS and fixed in 4% paraformaldehyde for 15 min at room temperature. Cells were permeabilized with PBS plus 0.1% Tween-20 (PBST), blocked with 10% goat serum (Zymed) for 1 hr, and incubated with the following antibodies: msFKBP65 (BD), type I procollagen, LF41 (a kind gift from Dr. L.W. Fisher, National Institutes of Health), diluted 1/50, 1/25, and 1/50, respectively, in 1% goat serum/PBST overnight at 4°C. Primary antibodies were detected with either Alexa Fluor 488 or 568 and goat anti-mouse antibody (Molecular Probes, Invitrogen) diluted 1/400 in 1% goat serum/PBST at room temperature for 1 hr. Slides were coverslipped with Vectashield with DAPI. Confocal images were taken sequentially on a Leica TCS-SP2 AOBS inverted Confocal Microscope (Mannheim) equipped with a 405 nm blue diode laser, argon laser (488 nm blue excitation: JDS Uniphase), and three helium neon lasers (543 nm, green; 594 nm, near red; 633 nm, far red excitations).
Results
OI Clinical Findings and Bone Histology
Affected individuals from five separately ascertained Turkish families had clinically normal consanguineous parents (Figure 1). These families originated from neighboring cities about 120 km apart in the Black Sea region of Turkey. A geneological link was established between two small nuclear families (Families 1 and 2, Figure 1), whereas the remaining three families declared no relationship. All affected individuals had normal birth length and weight but were born with skin blisters on their hands and feet, which later developed into generalized bullous lesions (Figure 2). Epidermolysis bullosa (EB) simplex was diagnosed and histologically confirmed by absence of keratin 14 (data not shown).
Figure 1.

Pedigrees of OI Families
Turkish OI families originating from the Black Sea region of Turkey (Families 1–5) and the Mexican-American OI family (Family 6).
Figure 2.

Clinical Phenotype
(A) EB at the ankle (arrow) in case R06-113A.
(B) Upper extremity joint laxity and long fingers in R06-113A.
(C and D) Radiographs showing scoliosis (arrows) in R06-113A and R93-188A, respectively.
(E and F) Wedge vertebrae (arrows) in R06-113A and R93-188A, respectively.
(G and H) Fractures, bent bones, and osteopenia (arrows) in R06-113A.
All affected individuals had a history of recurrent long bone fractures, beginning in infancy, which led to severe deformities in the extremities, and they were wheelchair bound by early childhood. Progressive kyphoscoliosis developed during childhood and some of the patients underwent corrective spinal surgery. Radiographic evaluation demonstrated severe osteopenia, fractures leading to deformities in the long bones, and kyphoscoliosis with flattening and wedging of the vertebrae (Figure 2). None of the affected individuals had dentinogenesis imperfecta, their sclerae were grayish white, not blue, and hearing was normal. Ligamentous laxity with long fingers and toes was noted in two patients. Elevated alkaline phosphatase levels were detected in two of the ascertained individuals at ages 6 and 16, respectively (levels were 300 IU and 400 IU, normal < 100 IU). All of the affected individuals ascertained had both OI and EB and, in extended pedigree analysis (Figure 1), there were no historically reported individuals who had either OI or EB alone.
Histologic examination of an iliac crest sample from R06-113A showed loss of normal orientation of the lamellae (Figure 3). The lamellae were thinner than normal and a pattern resembling fish scales was observed in some areas under polarized light.
Figure 3.

Bone Histomorphology
Bone histology from the iliac crest from control and an affected individual (R06-113A) demonstrating normal (A) and abnormal (B) lamellar bone patterns (arrows).
A second family (R93-188), originating from Mexico (Family 6, Figure 1), with severe progressive deforming OI in three affected siblings, was also identified. They were included in the analysis because no mutations had been identified in COL1A1, COL1A2, CRTAP, or LEPRE1. They did not have any manifestations of EB. At the time of ascertainment, the affected individuals were 12, 16, and 18 years of age. Their respective heights were 109, 124, and 132 cm (<5th percentile). The two older siblings were wheelchair bound, whereas the younger child walked with assistance. All three siblings had short trunks resulting from severe kyphoscoliosis, and additionally had coxa vara and extensive hyperextensibility of their joints, especially the elbows and fingers. They had neither dentinogenesis imperfecta nor hearing loss. Radiographic evaluation demonstrated severe osteopenia, wormian bones, scoliosis, and wedge vertebrae (Figure 2).
Homozygosity Mapping Identifies a New OI Locus
EB simplex was confirmed histologically by absence of keratin 14, and a previous report in one case14 (Family 3, Figure 1) demonstrated that the affected individual was homozygous for a null mutation in the keratin 14 gene (KRT14 [MIM 148066]). Analysis of DNA from all of the Turkish affected individuals demonstrated that all were homozygous for the previously identified c.612T>A KRT14 mutation predicted to lead to the amino acid substitution Y204X (data not shown). Because all of the affected individuals from the Turkish cohort segregated both EB and OI, under the hypothesis that there were two recessive disorders cosegregating in these families, the KRT14 region on chromosome 17q21 became a candidate region for the OI disease gene. Analysis of markers at loci near KRT14 identified a 0.83 Mb region of homozygosity between 39.63 and 40.46 Mb containing 38 genes (Genome Reference Consortium Human Build 37), one of which was presumed to contain the OI mutation.
Mutations in FKBP10 Cause Recessive OI
Mutation analysis was carried out for two genes in the interval, NKIRAS2 and FKBP10. NKIRAS2 screening was negative but mutations were found in FKBP10. The FKBP10 gene (MIM 607063), which encodes FKBP65, a 65 kD rough ER FK506-binding protein that is part of a large family of FKBPs that have been implicated in a variety of cellular processes that include peptidyl-prolyl cis-trans isomerization (PPIase) and chaperone function, was among the genes in the interval.16–18 KRT14 and FKBP10 are separated by about 230,000 bp. Mutation analysis in one of the Turkish cases (R06-113A) demonstrated homozygosity for a 33 base pair deletion (c.321_353del) in FKBP10, which is predicted to result in deletion of 11 amino acids, p. Gly107_Leu117del, in the first PPIase domain of the molecule (Figuress 4A, 4C, and 4G). All affected Turkish individuals were shown to be homozygous for the same mutation, consistent with a founder mutation in the population. RT-PCR products derived from fibroblasts of case R06-113A showed that this mutation leads to synthesis of FKBP65 mRNA containing the deletion (Figure 4E).
Figure 4.

FKBP10 Mutation Detection
(A and B) Sequence analysis of exon 2 in control and a representative affected individual (R06-113A) with the mutation in the Turkish families.
(C and D) Exon 5 sequence analysis in control and a representative affected individual (R93-188A) with the mutation in the Mexican-American family (R93-188A).
(E) RT-PCR of FKBP10 cDNA from control and R06-113A fibroblasts showing that a FKBP10 cDNA is synthesized.
(F) RT-PCR of FKBP10 cDNA from control and R93-188A fibroblasts showing that a FKBP10 cDNA is not synthesized; lower band demonstrates control cDNA synthesis.
(G) Cartoon of the FKBP65 molecule with predicted protein consequences for each mutation. PPIase, peptidyl-prolyl cis-trans isomerase; EF/Hand domain; HEEL, putative ER-retention sequence.
In the Mexican-American family, all affected individuals were homozygous for a c.831_832insC sequence change producing a translational frameshift (p.Gly278ArgfsX295) predicted to result in a stop codon 17 amino acids downstream in the third PPIase domain of the protein (Figures 4B, 4D, and 4G). RT-PCR performed on mRNA obtained from skin fibroblasts from an affected individual showed absence of FKBP65 cDNA, demonstrating that the mutation leads to a functional null allele (Figure 4F). Neither of the two mutations was found in panels of 210 alleles from ethnically matched unaffected individuals, indicating that they are not common polymorphisms in their respective populations.
FKBP10 Mutations Affect Type I Collagen Synthesis
Because FKBP65 is known to localize to the ER,8 tandem mass spectrometry was used to examine the possibility that it could be functionally involved in the P3H1/CRTAP/cyclophilin B complex and prolyl-3-hydroxylation.5 These studies showed normal 3-hydroxylation of proline residue 986 (data not shown), indicating that the mechanism of disease in this form of OI is distinct from that affecting prolyl-3-hydroxylation.
To examine the effect of the mutations on type I procollagen synthesis, cultured fibroblasts from one of the Turkish cases (R06-016A) and two of the Mexican-American siblings (R93-188A and R93-188B) were studied. In all three cases, type I procollagens and collagens showed normal electrophoretic migration on denaturing SDS-PAGE gels, with no evidence of increased posttranslational modification (Figure 5). These data suggested that the type I procollagen molecules synthesized by cultured mutant cells in vitro folded in a similar fashion as compared to molecules synthesized by normal cells. However, transmission electron microscopy demonstrated dilation of the ER in cultured fibroblasts from the affected individuals relative to control cells (Figure 6A), suggesting a defect in protein secretion. To determine whether this represented a defect in the intracellular processing of mature type I procollagen, pulse chase studies were carried out and showed a delay in the secretion of trimeric procollagen in the cells from affected individuals in both families compared to the control cells (Figures 6B and 6C).
Figure 5.
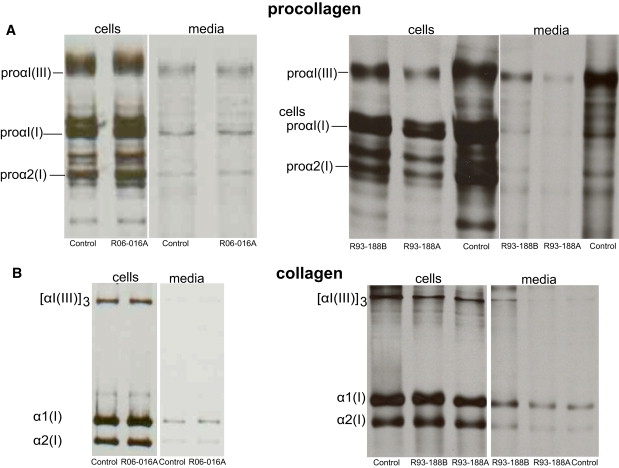
Type I Procollagen/Collagen Biochemical Analysis
(A) Cells synthesized normal procollagenous proteins. Normal amounts and mobilities were seen for proα1(I) and proα2(I) chains in control and affected fibroblasts in both the cell layer and the media (R06-016A, Family 3; R93-188A and B, Family 6).
(B) Cells also synthesized normal collagenous proteins, with normal amounts and mobilities seen for α1(I) and α2(I) chains in control and affected fibroblasts in both the cell layer and the media (R06-016A, Family 3; R93-188A and B, Family 6).
Figure 6.

Transmission Electron Microscopy, Pulse-Chase of Collagenous Proteins
(A) TEM of cultured fibroblasts from WT and an affected individual (R06-113A) showing dilated ER in the affected cells (labeled).
(B) Pulse-chase. The bands in each set represent the collagen trimers that migrated into the gel under nonreducing conditions. In all cell lines, label was seen in the monomeric proα chains. For controls, by 40 min the amount of trimer in the medium exceeded that remaining in the cells. In R06-113 and R93-188 cells, the amount in the medium exceeded that in the cells by 40 min of chase, but not to the extent seen in control cells. In all experiments, the amount of labeling in the cells with FKBP10 mutations was slightly reduced compared to that in the control cells.
(C) Graph of band intensities from each pulse-chase experiment (control, R06-113A, and R93-188A) showing a delay in secretion of the type I collagen trimer.
FKBP65 and Type I Procollagen Colocalize in the ER
FKBP65 is a known chaperone for type I procollagen.19,20 Confocal analysis of the intracellular distribution of FKBP65 by immunofluorescence showed that FKBP65 and type I procollagen colocalized in the ER in control fibroblasts (Figures 7A and 7B). Analysis of type I procollagen in cultured fibroblasts from individuals with the predicted in-frame deletion (R06-113A) and the null mutation (R93-188A) of FKBP10 showed that type I procollagen appeared to accumulate in aggregates within the cells, especially in the FKBP65 null cells (Figures 7C and 7D).
Figure 7.

Intracellular Localization of FKBP65 and Type I Procollagen
(A–C) Confocal microscopy demonstrating ER localization of both FKBP65 (green) and type I procollagen (red) and colocalization of the two proteins in control fibroblasts and R06-113 (merge).
(C and D) Confocal microscopy of fibroblasts from cases R06-113A and R93-188A, respectively, demonstrating a speckled pattern of type I procollagen suggestive of intracellular aggregates containing this protein.
Discussion
Biosynthesis and maturation of type I procollagen involves a large number of posttranslational steps involving many RER-resident proteins. Together these molecules mediate disulfide bond formation, peptidyl-prolyl cis-trans isomerization, prolyl and lysyl hydroxylation, and hydroxylysyl glycosylation. Molecular chaperones guide and provide quality control for these processes as they interact with the folding chains to prevent premature aggregation and/or nonspecific interactions.8,10 Some chaperones, including HSP47 and prolyl-4-hydroxylase, are collagen specific,21,22 whereas others including calnexin, BiP, GRP94, and PDI are general ER chaperones.21 ER chaperones including HSP47 travel with type I procollagen from the ER to the cis-Golgi, which facilitates the exit of mature proteins from the ER for transport through the Golgi apparatus, with HSP47 and procollagen dissociating between the post-ER and cis-Golgi networks.23
The RER-localized FK506-binding protein FKBP65 is part of a family of FKBPs that have been implicated in cellular processes that include peptidyl-prolyl cis-trans isomerization and chaperone functions. FBKP65 has some PPIase activity that may assist in the folding of the proline-rich tropoelastin monomer.24 Although the PPIase activity of FKBP65 alone on collagen triple helix formation is marginal,16 in vitro studies have demonstrated the ability of FKBP65 to interact with the collagen triple helix and function as a chaperone.19 Dilation of the ER in FKBP65-related OI, along with delayed type I procollagen secretion, is consistent with a defect in this process. The well-established chaperone HSP47 is known to preferentially bind to the triple helix of type I procollagen, rather than to the unfolded chains. Evidence from HSP47 null cells demonstrates that in the absence of a chaperone, type I procollagen aggregates in the ER and there is deficient extracellular fibrillogenesis.25 Similar to the HSP47 null cells, the data presented here show aggregated intracellular type I procollagen in FKBP65 mutant cells, suggesting a functional defect in the chaperone activity of the FKBP65.
The OI phenotype resulting from FKBP65 deficiency was consistent among the patients studied to date and characterized by severe osteopenia, with fractures that began in infancy and led to long bone deformities during childhood that prevented ambulation. Progressive kyphoscoliosis with platyspondyly and wedging of especially the thoracic vertebrae was a consistent and distinctive finding in this form of OI. Dentinogenesis imperfecta was absent, hearing was normal, and blue sclerae, which is characteristic of some types of OI because of mutations in the type I procollagen genes, was not present. Because of the moderately severe and progressive nature of the phenotype, FKBP65-related OI most closely resembles OI type III. However, the phenotype does not fit neatly into this category and the histological features, with a distorted lamellar structure and a fish scale-like pattern, along with elevated serum alkaline phosphatase, was also similar to OI type VI.4,26
Among the Turkish cases, all of the families studied were derived from cities in the eastern Black Sea region and all affected individuals had a second recessive disorder, EB simplex. This strongly suggested a founder effect, an inference supported by the conserved disease haplotype among the families and the identification of homozygosity for both the KRT14 EB simplex mutation and the FKBP65 OI mutation among all of the affected individuals. A case of EB simplex resulting from the same Y204X mutation in a girl from the Black Sea region of Turkey in whom OI was not present suggests that the FKBP10 mutation occurred on an allele carrying the KRT14 EB mutation.27
At least three distinct mechanisms are now known to produce OI. Primary defects in the type I procollagen genes that lead to reduced synthesis and/or abnormal structure of type I collagen produce the vast majority of OI cases, most of which are dominantly inherited. Defects in posttranslational modification of type I procollagen resulting from loss of the prolyl-3-hydroxylation complex have been found in a proportion of recessively inherited severe to lethal OI cases.5,12 In the present study, abnormalities in type I procollagen chaperone activity, resulting from defects in the FKBP65, have been identified in a moderately severe form of OI. Despite the different OI inheritance patterns and diverse molecules involved in these disorders, the unifying principle among all of the forms of OI appears to be abnormalities that affect the synthesis, assembly, and secretion of mature type I collagen to form the template necessary for normal bone mineralization.
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Accession Numbers
Acession numbers for data presented in this paper are as follows: Keratin 14: NCBI_3861, Ensembl_ENSG0000018684; FKBP65: NCBI_9606, Ensembl_ENSG00000321562
Acknowledgments
Y.A. was supported by TUBITAK (2219 Program). D.K., D.H.C., D.R.E., and B.L. were supported by NIHCD HD22657. D.K. and D.H.C. were also supported by NIH R01DE019567. B.Z. and A.S.F. are supported by SKELNET and EuroGrow. E.C.D. is a Canada Research Chair. We also acknowledge the support of the Facility for Electron Microscopy Research (FEMR) at McGill University.
References
- 1.Byers, P. Disorders of Collagen Biosynthesis and Structure. In The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID), D. Valle, ed. (New York: McGraw-Hill, Inc). Chapter 205, http://www.ommbid.com/.
- 2.Wenstrup R.J., Willing M.C., Starman B.J., Byers P.H. Distinct biochemical phenotypes predict clinical severity in nonlethal variants of osteogenesis imperfecta. Am. J. Hum. Genet. 1990;46:975–982. [PMC free article] [PubMed] [Google Scholar]
- 3.Glorieux F.H., Rauch F., Plotkin H., Ward L., Travers R., Roughley P., Lalic L., Glorieux D.F., Fassier F., Bishop N.J. Type V osteogenesis imperfecta: A new form of brittle bone disease. J. Bone Miner. Res. 2000;15:1650–1658. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- 4.Glorieux F.H., Ward L.M., Rauch F., Lalic L., Roughley P.J., Travers R. Osteogenesis imperfecta type VI: A form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002;17:30–38. doi: 10.1359/jbmr.2002.17.1.30. [DOI] [PubMed] [Google Scholar]
- 5.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Barnes A.M., Chang W., Morello R., Cabral W.A., Weis M., Eyre D.R., Leikin S., Makareeva E., Kuznetsova N., Uveges T.E. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kivirikko K.I., Myllylä R. Collagen glycosyltransferases. Int. Rev. Connect. Tissue Res. 1979;8:23–72. doi: 10.1016/b978-0-12-363708-6.50008-4. [DOI] [PubMed] [Google Scholar]
- 8.Makareeva E., Leikin S. Procollagen triple helix assembly: An unconventional chaperone-assisted folding paradigm. PLoS ONE. 2007;2:e1029. doi: 10.1371/journal.pone.0001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myllyharju J., Kivirikko K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Canty E.G., Kadler K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 11.Vranka J.A., Sakai L.Y., Bächinger H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004;279:23615–23621. doi: 10.1074/jbc.M312807200. [DOI] [PubMed] [Google Scholar]
- 12.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T.K., Pace J.M., Pepin M.G., Weis M., Eyre D.R., Walsh J. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008;29:1435–1442. doi: 10.1002/humu.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan Y., Anton-Lamprecht I., Yu Q.C., Jäckel A., Zabel B., Ernst J.P., Fuchs E. A human keratin 14 “knockout”: The absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev. 1994;8:2574–2587. doi: 10.1101/gad.8.21.2574. [DOI] [PubMed] [Google Scholar]
- 15.Pace J.M., Kuslich C.D., Willing M.C., Byers P.H. Disruption of one intra-chain disulphide bond in the carboxyl-terminal propeptide of the proalpha1(I) chain of type I procollagen permits slow assembly and secretion of overmodified, but stable procollagen trimers and results in mild osteogenesis imperfecta. J. Med. Genet. 2001;38:443–449. doi: 10.1136/jmg.38.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng B., MacDonald J.R., Bann J.G., Beck K., Gambee J.E., Boswell B.A., Bächinger H.P. Chicken FK506-binding protein, FKBP65, a member of the FKBP family of peptidylprolyl cis-trans isomerases, is only partially inhibited by FK506. Biochem. J. 1998;330:109–114. doi: 10.1042/bj3300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patterson C.E., Schaub T., Coleman E.J., Davis E.C. Developmental regulation of FKBP65. An ER-localized extracellular matrix binding-protein. Mol. Biol. Cell. 2000;11:3925–3935. doi: 10.1091/mbc.11.11.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patterson C.E., Gao J., Rooney A.P., Davis E.C. Genomic organization of mouse and human 65 kDa FK506-binding protein genes and evolution of the FKBP multigene family. Genomics. 2002;79:881–889. doi: 10.1006/geno.2002.6777. [DOI] [PubMed] [Google Scholar]
- 19.Ishikawa Y., Vranka J., Wirz J., Nagata K., Bächinger H.P. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- 20.Patterson C.E., Abrams W.R., Wolter N.E., Rosenbloom J., Davis E.C. Developmental regulation and coordinate reexpression of FKBP65 with extracellular matrix proteins after lung injury suggest a specialized function for this endoplasmic reticulum immunophilin. Cell Stress Chaperones. 2005;10:285–295. doi: 10.1379/CSC-118R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamandé S.R., Bateman J.F. Procollagen folding and assembly: the role of endoplasmic reticulum enzymes and molecular chaperones. Semin. Cell Dev. Biol. 1999;10:455–464. doi: 10.1006/scdb.1999.0317. [DOI] [PubMed] [Google Scholar]
- 22.Nagata K. HSP47 as a collagen-specific molecular chaperone: Function and expression in normal mouse development. Semin. Cell Dev. Biol. 2003;14:275–282. doi: 10.1016/j.semcdb.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 23.Satoh M., Hirayoshi K., Yokota S., Hosokawa N., Nagata K. Intracellular interaction of collagen-specific stress protein HSP47 with newly synthesized procollagen. J. Cell Biol. 1996;133:469–483. doi: 10.1083/jcb.133.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis E.C., Broekelmann T.J., Ozawa Y., Mecham R.P. Identification of tropoelastin as a ligand for the 65-kD FK506-binding protein, FKBP65, in the secretory pathway. J. Cell Biol. 1998;140:295–303. doi: 10.1083/jcb.140.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishida Y., Kubota H., Yamamoto A., Kitamura A., Bächinger H.P., Nagata K. Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol. Biol. Cell. 2006;17:2346–2355. doi: 10.1091/mbc.E05-11-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Land C., Rauch F., Travers R., Glorieux F.H. Osteogenesis imperfecta type VI in childhood and adolescence: Effects of cyclical intravenous pamidronate treatment. Bone. 2007;40:638–644. doi: 10.1016/j.bone.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 27.Yiasemides E., Trisnowati N., Su J., Dang N., Klingberg S., Marr P., Melbourne W., Tran K., Chow C.W., Orchard D. Clinical heterogeneity in recessive epidermolysis bullosa due to mutations in the keratin 14 gene, KRT14. Clin. Exp. Dermatol. 2008;33:689–697. doi: 10.1111/j.1365-2230.2008.02858.x. [DOI] [PubMed] [Google Scholar]