

Abstract
Objective
The dose-response effects of dysferlin transgenesis were analyzed to determine if the dysferlin-deficient myopathies are good candidates for gene replacement therapy.
Methods
We have generated three lines of transgenic mice, expressing low, mid and high levels of full-length human dysferlin from a muscle-specific promoter. Transgenic skeletal muscle was analyzed and scored for morphological and functional deficits.
Results
Overexpression of dysferlin in mice resulted in a striking phenotype of kyphosis, irregular gait and reduced muscle mass and strength. Moreover, protein dosage correlated with phenotype severity. In contrast to dysferlin-null skeletal muscle, no evidence of sarcolemmal impairment was revealed. Rather, increased levels of Ca2+-regulated, dysferlin-binding proteins and ER stress chaperone proteins were observed in muscle lysates from transgenic mice as compared to controls.
Interpretation
Expression levels of dysferlin are important for appropriate function without deleterious or cytotoxic effects. As a corollary, we propose that future endeavors in gene replacement for correction of dysferlinopathy should be tailored to take account of this.
Introduction
The muscular dystrophies (MD) are a heterogeneous group of inherited muscle disorders, defined by progressive loss of muscle strength and integrity. Autosomal recessive forms of MD include the clinically divergent limb-girdle muscular dystrophy type 2B and distal Miyoshi myopathy. While distinct in terms of weakness onset pattern, both disorders arise from defects in the gene encoding dysferlin (1, 2). Gene mutations result in partial to complete loss of dysferlin in affected individuals, though protein abundance does not stringently correlate with disease severity (3).
Dysferlin is a member of the muscle-specific repair complex that permits rapid resealing of membranes disrupted by mechanical stress (4, 5). Membrane repair is a widely conserved pro-survival cellular function, mechanistically analogous to Ca2+-dependent exocytosis (6). Re-sealing occurs within seconds of wounding and extracellular Ca2+ influx, and requires an internal membrane source in the form of aggregated exocytotic vesicles (7). In mature myofibers, dysferlin is expressed predominantly at the surface membrane, while also localized to cytoplasmic vesicles (4). Enrichment of dysferlin at injury sites is thought to reflect docking and fusion of an endomembrane patch comprising, in part, dysferlin-containing organelles. Dysferlin binding proteins (5, 8-10) facilitate this process through cytoskeletal rearrangement and patch trafficking. In dysferlinopathic muscle, membrane thickening and subsarcolemmal vesicle accumulation is apparent (8-10) supporting a role for dysferlin in membrane fusion. Furthermore, mouse models of dysferlin deficiency develop a progressive muscular dystrophy, characterized by attenuation of membrane repair in response to microinjury (4, 5). These findings implicate dysferlin as a vital component for continuous muscle cell repair, absence of which leads to progressive muscle degeneration.
Gene replacement strategies for MD have recently evolved to achieve efficient systemic delivery of therapeutic genes, critical to effectively targeting most affected muscle (11, 12). Promising findings have emerged from studies using adeno-associated virus (AAV) packaged genes in dystrophic mice and dogs (13-15). While the limited size of most AAV serotypes preclude their use for dysferlin, optimized design of trans-splicing AAV vectors has recently permitted whole-body transduction of reporter genes, raising hope for use of such a system in dysferlinopathy (16). However, to date, dose-response effects dysferlin transgenesis have not been examined. Toward this end, we generated transgenic mice that express different levels of dysferlin driven by a muscle-specific promoter. We report here that high level overexpression of dysferlin induces a dystrophic process that is pathogenically distinct from impaired sarcolemmal repair associated with dysferlinopathy.
Materials and Methods
Generation of hDYSF Tg mice
The human dysferlin ORF (NM_003494) was subcloned into pBSX-HSAvpa. This expression vector incorporates the human skeletal α-actin promoter (17), which has been extensively characterized (18). The 8.8kb transgene was isolated by PvuI/KpnI digest for microinjection. Mice were generated in the BL6/C3H background by the MGH Transgenic Mouse Core using standard protocols. All procedures involving animals were performed according to NIH guidelines and approved by the MGH animal care committee. hDysf-transgenic mice were identified by PCR amplification of genomic tail DNA extracted with the DNeasy kit (Qiagen, Valencia, CA). Primers used for genotyping and transcript analysis are summarized in supplementary Table S1.
Antibodies
Monoclonal antibodies with specificity for human dysferlin alone (clone Ham3/17B2) or for both mouse and human dysferlin (clone Ham1/7B6) were purchased from Vector Laboratories (Burlingame, CA). Antibodies against dystrophin (NCL-DYS), β-dystroglycan (NCL-β-DG) and calpain (NCL-12A2) were purchased from Novocastra Laboratories (Newcastle-Upon-Tyne, UK), and anti-caveolin-3 was from Transduction Labs. Antibodies against annexins A1 and A2, and GADD153 were purchased from Santa Cruz Biotechnology Inc. Rabbit anti- GRP78 and anti-calreticulin were from Affinity Bioreagents (Golden, CO).
Protein lysate preparation and western blotting
For whole cell lysate preparation, freshly harvested hind limb muscle was homogenized in cold sample lysis buffer (19). Crude muscle membrane isolation and subcellular fractionation,were carried out as previously described (20). Protein concentration was determined using Coomassie protein assay reagent (Bio-Rad, Hercules, CA), and immunoblotting performed by standard procedures (21). For densitometric analysis, band size and pixel intensity were quantified using the NIH ImageJ program.
Electron Microscopy
8-week old mice were perfused with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer (pH 7.4). Quadriceps and gastrocnemius muscles were bilaterally dissected and post-fixed overnight. After secondary fixation in 1% osmium tetroxide and dehydration in ethanol, samples were embedded in epon resin for ultrathin sectioning, and visualized on a JEOL 1200 EX transmission electron microscope.
Histology and immunofluorescence analysis
Histological and immunofluorescence analyses of heart and skeletal muscles were carried out on unfixed tissue snap-frozen in liquid nitrogen-cooled isopentane using established methods. Images were captured using an Eclipse E800 M microscope (Nikon, Mellville, NY) and Spot RT software (Sterling Heights, MI).
In vivo membrane integrity assays
Evans Blue dye (EBD) was used to evaluate sarcolemmal integrity as previously described (22). Myofibers positive for EBD uptake were observed under a fluorescent microscope with rhodamine filters. For plasma creatine kinase measurements, blood samples were collected by submandibular (cheek pouch) bleed using heparinized capillary tubes (Sarstedt Inc, Newton, NC). Plasma samples were analyzed using a Heska Spotchem Analyzer.
Isolation and culture of primary myoblasts
Primary myoblasts were cultured from 2-5 day old pups as outlined previously (23), and maintained in primary cell growth medium. For assay of myoblast fusion, cells were counted after pre-plating and plated in primary culture medium at 1200 cells per well on ECL-coated 8-well chamber slides (LabTek, Nalge Nunc, Rochester, NY). After 12 hours cells were induced to differentiate by switching to serum-deprived medium. Six days post-differentiation cells were fixed and analyzed for desminpositivity and nuclei number (24). Twenty fields at 10× magnification were examined for quantification, using five fields in duplicate wells from each of two separate cultures of wild-type and hDysf-mid transgenic neonates.
Grip-strength and fiber force measurement
Hindlimb strength of wild-type and age matched hDysf transgenic mice was quantified using a grip strength meter (Columbus Instruments, Columbus, OH), and expressed as Newton of tension per kilogram body weight. Chemically skinned single muscle fiber preparations were used to assess contractile properties as previously described in detail (25, 26). Maximum unloaded shortening velocity (VO) was measured with the slack test (27). Force velocity curves were generated by performing a series of isotonic contractions of the muscle fiber after completion of the slack test. The MyHC composition of single fibers was determined by SDS-PAGE.
Results
Expression of hDysf transgene in skeletal and cardiac muscle
To generate mice over-expressing wild-type dysferlin, the human coding sequence for dysferlin (hDysf) was subcloned into plasmid pBSX-HSAvpa (Figure 1a). We elected to use human cDNA to allow us to distinguish between endogenous murine and transgenic dysferlin, given the availability of a human-specific dysferlin antibody. Mouse (NP_067444) and human dysferlin peptide sequences (NP_003485) share 93% homology over the entire protein, with >75% sequence homology for each predicted functional C2 domain. Three lines were selected in which the transgene was overexpressed at a range of levels, designated hDysf-low, -mid and –high (Figure 1). Transgene copy number was determined by TaqMan quantitative PCR (hDysf-low, 5 copies; hDysf-mid, 2 copies; hDysf–high, 4 copies), and transcript levels were analyzed by RT-PCR (Figure 1b). To confirm expression of transgenic dysferlin protein, hindlimb muscle from eight week-old mice was subjected to Western blot analysis. Dysferlin antibodies directed against human (H), or against both human and mouse (M; H) detected a band of approximately 230kDa in both wild-type and transgenic homogenates. Varying supraphysiological levels of dysferlin protein were detected in transgenic lines (Figure 1c) ranging from 2-fold (low) to approximately 36-fold (mid) and 176-fold (high) endogenous levels, as quantified by densitometry (Figure 1d). To further scrutinize subcellular expression of transgenic dysferlin, crude muscle microsomal homogenates were subjected to a density gradient to enrich for sarcoplasmic reticular and sarcolemmal membranes (Figure 1e).
Figure 1. Generation of transgenic mice overexpressing dysferlin.

a, Schematic diagram of hDysf transgene used to generate dysferlin transgenic mice. The human skeletal actin promoter (2.2kb) drives skeletal muscle-specific expression. The hybrid HSA/vp1 intron is situated between the promoter and cDNA, containing splice donor and acceptor sequences. The SV40 poly(A) signal follows the dysferlin cassette. b, RT-PCR analysis of hDysf transgene (HSA-hDysf), total dysferlin including mouse endogenous (mDysf/hDysf) and ribosomal gene RPS7 expression levels in skeletal muscle from wild-type (WT) mice and three transgenic lines overexpressing hDysf at different levels: hDysf-low, hDysf-mid and hDysf-high. c, Upper panel, Western blot analysis of human dysferlin protein expression in hindlimb skeletal muscle lysates from wild-type mice, and three hDysf transgenic lines. Immunoblotting was performed with monoclonal antibodies that recognize either human-specific dysferlin alone (Dysf (H)) or human and mouse dysferlin (Dysf (M; H)). Actin was monitored as a loading control. Lower panel, Protein lysates were prepared from hDysf-high transgenic mice tissues and immunoprobed with human-specific dysferlin antibody. Exogenous, transgenic Dysf expression is observed only in cardiac and skeletal muscle lysates. d, Immunoblot analysis of dysferlin expression (endogenous and transgenic) in muscle lysates from wild-type and hDysf mice. e, Subcellular fractionation of microsomal homogenates from wild-type and hDysf-low mice reveal that transgenic dysferlin is enriched in sarcoplasmic reticular fractions as well as in the sarcolemma. SL, sarcolemma; LSR, light sarcoplasmic reticulum; TRI, triads; HSR, heavy sarcoplasmic reticulum; P, pellet.
Increased expression of dysferlin induces a dystrophic phenotype
While hDysf-transgenic mice were born viable and at the expected Mendelian ratio, high levels of dysferlin overexpression were found to correlate with a reduction in body weight compared to wild-type littermates, evident by 7 weeks and persisting with age (Figure 2a). This discrepancy was not attributable to reduced caloric intake (Figure 2b). Gross examination of adult (8 week-old) transgenic animals revealed atrophy of selective muscles, obvious both from visualization of hindlimb musculature (Figure 2c) from reduced muscle mass (Figure 2d). hDysf-high transgenic mice failed to thrive had to be euthanized between 5-7 months of age due to severe kyphosis, hind-limb atrophy and dehydration.
Figure 2. Increased transgenic dysferlin overexpression correlates with muscle atrophy and reduced body mass.

a, Body weight gain of wild-type (WT) and hDysf transgenic mice (n=5 per group) was monitored biweekly from 4-16 weeks of age. Values are means ± SD. Transgenic animals displayed consistently reduced body weight compared to wild-type controls (group hDysf low (P < 0.05) and groups hDysf-mid and -high (P < 0.01 by ANOVA)). b, Ratio of food consumption to body mass in WT and hDysf transgenic mice (n=3 mice per group). Values are means ± SD. c, Photographs of wild-type control and hDysf over-expressing transgenic mice at 8 weeks of age. hDysf-mid and –high level transgenic mice are kyphotic with an irregular gait and display atrophy of the limb musculature. d, Relative muscle weights. The weights of five muscle groups (n=4 mice per group) from hDysf transgenic mice were measured and normalized to weights of corresponding muscles in age-matched littermate wild-type controls. Values are means ± standard error (*P < 0.05; **P <0.001, t-test).
Histological analysis of skeletal muscle from hDysf-overexpressing mice revealed hallmarks of a non-necrotic, congenital myopathy. hDysf-mid and –high transgenic muscle cryosections showed marked reduction in fiber diameter, together with increased connective tissue and centralized nuclei (Supplementary Figure 1a). Cardiac muscle cryosections from hDysf-high transgenic animals revealed areas of endomysial fibrosis and dystrophic calcification (Supplementary Figure 1b). Quantification of quadriceps fiber cross sectional area revealed a 50% and 70% reduction in hDysf-mid and hDysf-high myofibers compared to wild-type , respectively, with fiber size distribution showing a progressive shift to smaller myofiber size with increasing dysferlin expression (Supplementary Figure 1c).
Ultrastructural analysis of hDysf-overexpressing muscle reveals altered fiber composition and sarcolemmal vesicle accumulations
Given the over-representation of smaller fibers in hDysf transgenic muscle, we investigated whether fiber-type composition was also affected. NADH-tetrazolium reductase staining of transgenic quadriceps cryosections revealed the selective loss of more fast-twitch fiber types (Supplementary Figure 2a). In mice, fast type II fibers consist of type IIa, IIx hybrid IIb/IIx, and IIb, listed from slowest to fastest, with type IIx representing an intermediate between IIa and IIb fibers in terms of oxidative enzyme activity, fatigue resistance and maximum shortening velocity. We further investigated the fiber composition of isolated hDysf-mid gastrocnemius muscle. Transgenic fibers (n=16) comprised 62% type IIb, 19% IIb/IIx, 6% IIx, and 12% IIx/IIa fibers, while all wild-type fibers analyzed (n=18) were type IIb.
In contrast to dysferlin-deficient membranes (13), electron micrographs of hDysf-high muscle did not reveal sarcolemmal disruption or thickening as compared to wild-type (Supplementary Figure 2b). However, higher magnification of sarcolemmal membranes revealed a large number of associated vesicular structures of unknown origin (Supplementary Figure 2b), although their size and flask-shaped appearance may indicate caveolar organelles.
Decreased hindlimb strength and reduced myofiber force and shortening velocity in hDysf transgenic mice
To investigate the functional consequences of dysferlin overexpression, hindlimb strength was measured. Grip-strength was found to be consistently and significantly reduced in hDysf-high animals over the course of the trial (Figure 3a). To further verify this functional deficit, chemically skinned myofibers from age-matched hDysf-mid transgenic mice and controls were subjected to the slack test. Fibers isolated from transgenic muscle were much smaller, generated less force than wild-type fibers and had a reduced Vo (Figure 3b).
Figure 3. Hindlimb strength and single muscle fiber contractile properties in hDysf-transgenic mice.

a, Hindlimb grip strength of WT and hDysf-transgenic mice (n=3 per group) was measured from 10 to 25 weeks of age and grip strength in newtons (N) was normalized to body weight (kg). Each bar represents the mean ± standard error of values measured. hDysf-mid and hDysf-high transgenic mice displayed attenuated hindlimb strength compared to wild-type controls (P < 0.05 and P < 0.01, respectively, by ANOVA) b, The slack test was performed on isolated single myofibers from hDysf-mid transgenic and wild-type mice from quadriceps (WT, n=14 fibers; hDysf Tg, n=10 fibers) and gastrocnemius muscle (WT, n=18 fibers; hDysf Tg, n=10 fibers). Data are presented for Type IIb fibers only because other fiber types were not present in both the wild type and transgenic mice. Maximal force (Po) and maximum unloaded shortening velocity (Vo) was measured. Values are means ± SD (*P < 0.05).
Sarcolemmal and dysferlin-binding protein localization and abundance in hDysf-overexpression
To examine the influence of dysferlin overexpression on global protein levels, we employed a panel of antibodies against sarcolemmal and dysferlin-binding proteins in immunohistochemical and immunoblot analyses. Immunostaining of quadriceps cryosections revealed localization of dysferlin to the sarcolemma and cytoplasmic vesicles (Figure 4a). Dysferlin staining was also detected in intracellular aggregate structures, and at the periphery of individual centralized nuclei, in transgenic muscle. Expression and localization of caveolin-3 and architectural DGC proteins was unaltered. Of the proteins analyzed, annexin A2 levels were found to be most significantly increased in hDysf-transgenic muscle by Western blot (5.6-fold ± 0.64) (Figure 4b, c).
Figure 4. Immunofluorescence and western blot analyses of sarcolemmal and dysferlin-binding proteins in hDysf transgenic muscle.

a, Immunofluorescence staining of quadriceps muscle cross-sections from wild-type (WT) and hDysf-mid transgenic mice with human-specific (Dysf (h)) and mouse/human specific antibodies to dysferlin (Dysf (m/h)). Dysferlin immunostaining is both sarcolemmal and intracellular, visible as punctate vesicular, perinuclear (arrowhead) and cytoplasmic aggregate staining (arrows). b, Normal immunofluorescnce staining for caveolin-3 (Cav-3) and DGC components β-dystroglycan (β-DYS) and dystrophin (DYS). Scale bar, 100μm. c, Upper panel Immunoblot analysis of quadriceps microsomal lysates from from wild-type (WT) and hDysf-mid transgenic mice (n=3 per group), using antibodies to annexin A1 (AnxA1), annexin A2 (AnxA2), caveolin-3 (Cav-3), calpain-3, β-dystroglycan (β-DYS); β-tubulin staining was monitored as a loading control. Lower panel, Densitometric analysis of band intensity normalized to β-tubulin, expressed as fold change over wild-type (WT) levels. Values are means ± standard error (*P < 0.05; **P < 0.01; ***P <0.001, unpaired t-test).
Sarcolemmal integrity is minimally compromised by overexpression of hDysf
Given the central role of dysferlin in the sarcolemmal repair complex, we reasoned that altered stoichiometry of this complex resulting from forced dysferlin overexpression may compromise the integrity of the membrane microenviroment. Membrane integrity was evaluated by Evans blue dye uptake and plasma creatine kinase levels. Rare individual EBD-positive fibers were observed microscopically in both wild-type and transgenic proximal and distal muscles, consistent with an intact plasma membrane (Supplementary Figure 3a). Furthermore, plasma creatine kinase levels were largely comparable between wild-type and hDysf-overexpressing mice (Supplementary Figure 3b). In contrast, plasma creatine kinase in dysferlin-null A/J mice was significantly higher compared to wild-type, consistent with previous reports (8).
Dysferlin deficiency has also been linked to altered myogenic factor expression and delayed fusion of human myoblasts in culture (28). Furthermore, dysferlin expression in activated satellite cells suggests a role in regenerative satellite cell recruitment and fusion (29). Therefore, we assayed the fusion ability of isolated primary myoblasts from hDysf-transgenic neonates in vitro. We found no significant difference in the fusogenicity of hDysf-mid myoblasts compared to wild-type (Supplementary Figure 3c). These findings strongly indicate that that the inherent pathogenicity associated with dysferlin overexpression is mediated through a mechanism distinct from that of dysferlin deficiency.
Dysferlin overexpression causes increase in ER stress markers
The finding that transgenic hDysf protein co-fractionates with SR and tubular structures, and is enriched in cytoplasmic aggregates led us to question the involvement of endoplasmic reticulum stress response pathways in the pathophysiology of these mice. Metabolic stresses such as impaired SR Ca2+ homeostasis, or ER protein accumulation have been shown to induce upregulation of ER chaperone proteins (30). Differential expression of these chaperones is governed by the specific stressing stimulus; increased levels of heat shock proteins GRP78 and GRP94, for example, are observed in myopathies presenting tubular aggregate (TA) formation. Furthermore, tubular aggregates have been found to strongly express dysferlin (31). Fibers with TA-like structures and intra-sarcoplasmic vacuoles were observed in hDysf-mid transgenic muscle (Figure 5a). Immunoblotting with ER stress marker antibodies revealed increased expression of GRP-78 and calreticulin in transgenic muscle lysates (Figure 5b). Expression of the pro-apoptotic ER stress protein GADD153/CHOP was not increased as compared to wild-type.
Figure 5. Expression of ER stress proteins in hDysf transgenic muscle.
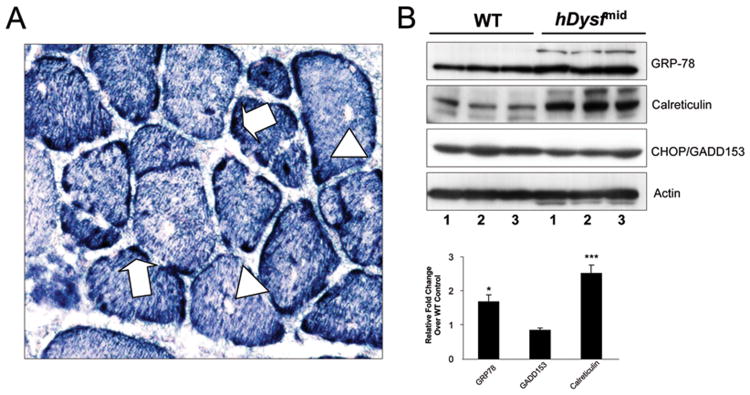
a, Representative NADH-tetrazolium reductase histochemical staining of hDysf-mid transgenic quadriceps muscle reveals tubular aggregate-like structures (arrows) and intra-sarcoplasmic vacuoles (arrowheads). b, Western blot analysis of whole cell protein lysates from wild-type (WT) and hDysf-mid transgenic mice (n=3 per group), using antibodies to the endoplasmic reticulum stress proteins glucose-related protein 78 (GRP78), GADD150/CHOP and calreticulin; α-actin used as a loading control. Lower panel, Densitometric analysis of band intensity normalized to α-actin, expressed as fold change over wild-type (WT) levels. Values are means ± standard error (*P < 0.05; ***P <0.001, unpaired t-test).
Discussion
Dysferlin deficiency defines a class of muscular dystrophies resulting from defective muscle membrane repair and maintenance. Prospective therapeutic paradigms for dysferlinopathies include virally-mediated gene replacement to restore normal protein to affected muscles. In an effort to define a therapeutic window for gene delivery, we have generated transgenic lines to express various levels of dysferlin. In accordance with a recently published study (32), we observed that low levels of dysferlin overexpression (∼2-fold endogenous) are well tolerated by recipient skeletal muscle, with no evidence of cytotoxicity. However, excessive dysferlin levels (>30-fold normal) result in a non-necrotic, progressive dystrophy, characterized by marked atrophy and weakness. Functional and ultrastructural analyses suggest that while sarcolemmal integrity is largely conserved, there is increased abundance of Ca2+-regulated, dysferlin-binding proteins and ER stress chaperone proteins.
Differential toxicity of transgenic or virally-mediated overexpression of MD-associated proteins has been well documented. Studies focused on components of the DGC have proven highly informative in terms of the stoichiometric capacity of recipient muscle membrane complexes. Interestingly, clinical severity resulting from deficit of the implicated gene is not predictive of response in these forced ‘stoichiometry-myopathies’. Cytotoxicity associated with sarcoglycan overexpression correlates with the hierarchial assembly of the sarcoglycan complex; 5-fold endogenous levels of γ-sarcoglycan were better tolerated than low level overexpression (2-fold) of the α- subunit (33, 34). Conversely, 50-fold overexpression levels of dystrophin corrected dystrophic symptoms in the mdx mouse without deleterious side effects (35). It may be proposed that the myopathy observed in our studies reflects an adverse consequence of forcing high levels of expression of human dysferlin in mice, rather than fundamental biological consequence of altered dysferlin-stoichiometry. We feel this is unlikely, given the high level (>93%, as noted above) of amino acid sequence homology between the murine and human dysferlin proteins.
We reasoned that dysferlin overexpression may alter homeostatic interactions or stoichiometric assembly of membrane repair complex proteins. Dysferlin interacts with cytosolic calpain-3 in normal muscle (36); reduced levels of calpain-3 and dysferlin protein have been reported in cases of LGMD2B/MM and LGMD2A, respectively (37, 38). Although a moderate increase in calpain-3 expression was found in transgenic muscle, given the observation that severe calpain-3 overexpression is tolerated without toxicity (39) we deem this unlikely to account for the dystrophic pathology in our model. In contrast, a dramatic increase in annexin A2 expression was observed in transgenic muscle, with negligible change in annexin A1 levels. This finding is consistent with a discrete, non-redundant role for each annexin in dysferlin binding and patch repair processes. Although both interactions are Ca2+-dependent, sarcolemmal injury in normal muscle has been found to selectively disrupt annexin A1-dysferlin association (5). While supra-physiological dysferlin and annexin expression could be anticipated to disturb sarcolemmal fluidics and normal lipid microdomain assembly, sarcolemmal integrity and repair capacity appear relatively unaffected, indicating that enhanced protein production induces muscle wasting by other mechanisms.
Immunohistochemical analysis of transgenic muscle revealed normal sarcolemmal and vesicular dysferlin localization. However, we also observed perinuclear dysferlin staining and accumulation in cytoplasmic aggregate structures in isolated myofibers. Dysferlin association with TAs and intra-sarcoplasmic vacuoles has been described for several myopathies (31). Formation of TAs, SR-derived organelles with Ca2+-loading capacity, is thought to reflect an underlying disturbance in intra-sarcoplasmic Ca2+ homeostasis. Furthermore, upregulation and co-expression of GRP78 and GRP94 with dysferlin in TAs is evidence of an ER-stress response in SR remodeling (31). In this study, structures reminiscent of TAs were observed in transgenic muscle cryosection, with GRP78 mildly upregulated compared to control. However, significant upregulation of the Ca2+-binding ER chaperone calreticulin was observed in transgenic muscle. Calreticulin has been shown to act in vitro as a molecular chaperone for both glycosylated and non-glycosylated unfolded proteins (40) and is known to modulate intracellular Ca2+ homeostasis (41). Accumulation of unfolded or misfolded protein in the ER triggers the unfolded protein response (UPR), a signal transduction pathway critical to alleviate ER stress. Myocyte differentiation is thought to be highly dependent on an intact UPR, given the high level of protein synthesis occurring in these cells (42). Although it remains to be determined whether ER stress may be a causative factor in dysferlin overexpression-induced myopathy, or reflects a more general downstream effect of dysferlin overexpression, it is compelling that in the presence of a proteosomal inhibitor, wild-type dysferlin accumulates in the ER/Golgi (43). Furthermore, cytoplasmic polyQ aggregates stimulated ER accumulation of dysferlin, suggesting that dysferlin is a substrate for the ubiquitin/proteasome ER-associated degradation (ERAD) system (43). It is feasible that overwhelming of the ERAD and UPR machinery under conditions of supraphysiological dysferlin levels may lead to dysregulated intrasarcoplasmic Ca2+ homeostasis and induction of apoptotic pathways, culminating in progressive skeletal muscle damage and dysfunction, such as that observed in other muscle disorders exhibiting elements of the ER stress response (31, 44).
Taken together, these findings suggest that dysferlin delivery strategies should strive to stringently calibrate the dose response of treatment, to avoid deleterious effects associated with high transduction levels.
Supplementary Material
a, Representative hematoxylin-eosin stained transverse cryosections of proximal and distal muscles from hDysf-high transgenic mice. Scale bar, 100μm. b, Representative hematoxylin-eosin stained section of heart muscle from hDysf-high transgenic mice, revealing areas of calcification. Scale bar, 50μm. Bottom panel is magnification of upper inset panel. c, Representative cross-sections of gastrocnemius muscle from hDysf-high transgenic and wild-type. Scale bar, 100μm. Multiple images of transverse cryosections were examined to quantify fiber cross-sectional area of wild-type and transgenic gastrocnemius muscle (3 animals per genotype, 10 images per animal). The distribution of gastrocnemius fiber size was analyzed between transgenic and littermate wild-type mice, revealing a predominance of smaller fibers in hDysf-mid and –high overexpressing muscle.
a, Representative NADH-tetrazolium reductase histochemical staining identifies slow-twitch (dark) and fast-twitch (light) fibers. Fast fibers are selectively affected by hDysf overexpression. b, Electron micrographs of quadriceps muscle from 8-week old wild-type and hDysf-high transgenic mice. Subsarcolemmal vesicles accumulation in hDysf-overexpressing muscle (arrowheads). Left, Scale bar 150nm. Right, Scale bar, 500nm.
a, Evans blue dye uptake in quadriceps and tibialis anterior muscle from wild-type (WT) and hDysf-mid transgenic mice. Scale bar, 200μm. b, Plasma serum levels of creatine kinase in WT, hDysf-transgenic and A/J dysferlin-deficient mice (n=5 per group). Values are means ± SD (*P < 0.0001, t-test). c, Fusogenic capacity of wild-type (WT) and hDysf-mid transgenic primary myoblast cultures. Efficiency of fusion was analyzed by quantification of singly-nucleated desmin-positive myoblast cells, cells containing two to three nuclei and myotubes containing four or more nuclei. Five 10× fields from two separate cultures of each genotype were analyzed, comprising 573 hDysf-mid and 512 wild-type nuclei.
Acknowledgments
The authors would like to thank Dr Jeffrey S. Chamberlain (University of Washington School of Medicine, Seattle, WA, USA) for kindly donating the pBSX-HSAvpa expression vector and Dr Sean Colgan for critical reading of the manuscript and helpful comments. These studies were supported by the CB Day Company, the NINDS at the NIH, and the Muscular Dystrophy Association.
References
- 1.Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I, Marconi G, Passos-Bueno MR, Moreira Ede S, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20(1):37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20(1):31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 3.Saito A, Higuchi I, Nakagawa M, Saito M, Hirata K, Suehara M, Yoshida Y, Takahashi T, Aoki M, Osame M. Miyoshi myopathy patients with novel 5′ splicing donor site mutations showed different dysferlin immunostaining at the sarcolemma. Acta Neuropathol (Berl) 2002;104(6):615–620. doi: 10.1007/s00401-002-0593-x. [DOI] [PubMed] [Google Scholar]
- 4.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423(6936):168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 5.Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278(50):50466–50473. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- 6.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106(2):157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 7.McNeil PL, Terasaki M. Coping with the inevitable: how cells repair a torn surface membrane. Nat Cell Biol. 2001;3(5):E124–129. doi: 10.1038/35074652. [DOI] [PubMed] [Google Scholar]
- 8.Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, Watkins SC, Cox GA, Brown RH., Jr Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Human molecular genetics. 2004;13(18):1999–2010. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- 9.Piccolo F, Moore SA, Ford GC, Campbell KP. Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb--girdle muscular dystrophies. Ann Neurol. 2000;48(6):902–912. [PubMed] [Google Scholar]
- 10.Selcen D, Stilling G, Engel AG. The earliest pathologic alterations in dysferlinopathy. Neurology. 2001;56(11):1472–1481. doi: 10.1212/wnl.56.11.1472. [DOI] [PubMed] [Google Scholar]
- 11.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, Russell DW, Chamberlain JS. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10(8):828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23(3):321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 13.Cerletti M, Negri T, Cozzi F, Colpo R, Andreetta F, Croci D, Davies KE, Cornelio F, Pozza O, Karpati G, Gilbert R, Mora M. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirusmediated utrophin gene transfer. Gene Ther. 2003;10(9):750–757. doi: 10.1038/sj.gt.3301941. [DOI] [PubMed] [Google Scholar]
- 14.Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, Finn E, Adams ME, Froehner SC, Murry CE, Chamberlain JS. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12(7):787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodino-Klapac LR, Shontz KM, Malik V, Montgomery CL, Davis N, Canan B, Hauschka SD, Janssen P, Clark KR, Brown RH, Jr, Mendell JR. rAAV5 Mediated Delivery of Dysferlin as a Therapeutic Strategy for LGMD2B and Miyoshi Myopathy; 12th Annual Meeting of the American Society of Gene Therapy; San Diego, California. American Society of Gene and Cell Therapy; 2009. [Google Scholar]
- 16.Ghosh A, Yue Y, Long C, Bostick B, Duan D. Efficient whole-body transduction with trans-splicing adeno-associated viral vectors. Mol Ther. 2007;15(4):750–755. doi: 10.1038/sj.mt.6300081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muscat GE, Kedes L. Multiple 5′-flanking regions of the human alpha-skeletal actin gene synergistically modulate muscle-specific expression. Mol Cell Biol. 1987;7(11):4089–4099. doi: 10.1128/mcb.7.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268(1):719–725. [PubMed] [Google Scholar]
- 19.Anderson LV, Davison K, Moss JA, Young C, Cullen MJ, Walsh J, Johnson MA, Bashir R, Britton S, Keers S, Argov Z, Mahjneh I, Fougerousse F, Beckmann JS, Bushby KM. Dysferlin is a plasma membrane protein and is expressed early in human development. Human molecular genetics. 1999;8(5):855–861. doi: 10.1093/hmg/8.5.855. [DOI] [PubMed] [Google Scholar]
- 20.Sharp AH, Imagawa T, Leung AT, Campbell KP. Identification and characterization of the dihydropyridine-binding subunit of the skeletal muscle dihydropyridine receptor. J Biol Chem. 1987;262(25):12309–12315. [PubMed] [Google Scholar]
- 21.Ho M, Gallardo E, McKenna-Yasek D, De Luna N, Illa I, Brown RH., Jr A novel, blood-based diagnostic assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann Neurol. 2002;51(1):129–133. doi: 10.1002/ana.10080. [DOI] [PubMed] [Google Scholar]
- 22.Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem (Tokyo) 1995;118(5):959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- 23.Bischoff R, Heintz C. Enhancement of skeletal muscle regeneration. Dev Dyn. 1994;201(1):41–54. doi: 10.1002/aja.1002010105. [DOI] [PubMed] [Google Scholar]
- 24.Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM. Normal myoblast fusion requires myoferlin. Development. 2005;132(24):5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsson L, Moss RL. Maximum velocity of shortening in relation to myosin isoform composition in single fibres from human skeletal muscles. J Physiol. 1993;472:595–614. doi: 10.1113/jphysiol.1993.sp019964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krivickas LS, Fielding RA, Murray A, Callahan D, Johansson A, Dorer DJ, Frontera WR. Sex differences in single muscle fiber power in older adults. Medicine and science in sports and exercise. 2006;38(1):57–63. doi: 10.1249/01.mss.0000180357.58329.b1. [DOI] [PubMed] [Google Scholar]
- 27.Edman KA. The velocity of unloaded shortening and its relation to sarcomere length and isometric force in vertebrate muscle fibres. J Physiol. 1979;291:143–159. doi: 10.1113/jphysiol.1979.sp012804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Luna N, Gallardo E, Soriano M, Dominguez-Perles R, de la Torre C, Rojas-Garcia R, Garcia-Verdugo JM, Illa I. Absence of dysferlin alters myogenin expression and delays human muscle differentiation “in vitro”. J Biol Chem. 2006;281(25):17092–17098. doi: 10.1074/jbc.M601885200. [DOI] [PubMed] [Google Scholar]
- 29.De Luna N, Gallardo E, Illa I. In vivo and in vitro dysferlin expression in human muscle satellite cells. J Neuropathol Exp Neurol. 2004;63(10):1104–1113. doi: 10.1093/jnen/63.10.1104. [DOI] [PubMed] [Google Scholar]
- 30.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikezoe K, Furuya H, Ohyagi Y, Osoegawa M, Nishino I, Nonaka I, Kira J. Dysferlin expression in tubular aggregates: their possible relationship to endoplasmic reticulum stress. Acta Neuropathol. 2003;105(6):603–609. doi: 10.1007/s00401-003-0686-1. [DOI] [PubMed] [Google Scholar]
- 32.Millay DP, Maillet M, Roche JA, Sargent MA, McNally EM, Bloch RJ, Molkentin JD. Genetic Manipulation of Dysferlin Expression in Skeletal Muscle. Novel Insights into Muscular Dystrophy. The American journal of pathology. 2009 doi: 10.2353/ajpath.2009.090107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dressman D, Araishi K, Imamura M, Sasaoka T, Liu LA, Engvall E, Hoffman EP. Delivery of alpha- and beta-sarcoglycan by recombinant adeno-associated virus: efficient rescue of muscle, but differential toxicity. Hum Gene Ther. 2002;13(13):1631–1646. doi: 10.1089/10430340260201725. [DOI] [PubMed] [Google Scholar]
- 34.Zhu X, Hadhazy M, Groh ME, Wheeler MT, Wollmann R, McNally EM. Overexpression of gamma-sarcoglycan induces severe muscular dystrophy. Implications for the regulation of Sarcoglycan assembly. J Biol Chem. 2001;276(24):21785–21790. doi: 10.1074/jbc.M101877200. [DOI] [PubMed] [Google Scholar]
- 35.Cox GA, Cole NM, Matsumura K, Phelps SF, Hauschka SD, Campbell KP, Faulkner JA, Chamberlain JS. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993;364(6439):725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Verheesen P, Roussis A, Frankhuizen W, Ginjaar I, Haldane F, Laval S, Anderson LV, Verrips T, Frants RR, de Haard H, Bushby K, den Dunnen J, van der Maarel SM. Protein studies in dysferlinopathy patients using llama-derived antibody fragments selected by phage display. Eur J Hum Genet. 2005;13(6):721–730. doi: 10.1038/sj.ejhg.5201414. [DOI] [PubMed] [Google Scholar]
- 37.Anderson LV, Harrison RM, Pogue R, Vafiadaki E, Pollitt C, Davison K, Moss JA, Keers S, Pyle A, Shaw PJ, Mahjneh I, Argov Z, Greenberg CR, Wrogemann K, Bertorini T, et al. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies) Neuromuscul Disord. 2000;10(8):553–559. doi: 10.1016/s0960-8966(00)00143-7. [DOI] [PubMed] [Google Scholar]
- 38.Chrobakova T, Hermanova M, Kroupova I, Vondracek P, Marikova T, Mazanec R, Zamecnik J, Stanek J, Havlova M, Fajkusova L. Mutations in Czech LGMD2A patients revealed by analysis of calpain3 mRNA and their phenotypic outcome. Neuromuscul Disord. 2004;14(10):659–665. doi: 10.1016/j.nmd.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Spencer MJ, Guyon JR, Sorimachi H, Potts A, Richard I, Herasse M, Chamberlain J, Dalkilic I, Kunkel LM, Beckmann JS. Stable expression of calpain 3 from a muscle transgene in vivo: immature muscle in transgenic mice suggests a role for calpain 3 in muscle maturation. Proc Natl Acad Sci U S A. 2002;99(13):8874–8879. doi: 10.1073/pnas.132269299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito Y, Ihara Y, Leach MR, Cohen-Doyle MF, Williams DB. Calreticulin functions in vitro as a molecular chaperone for both glycosylated and non-glycosylated proteins. Embo J. 1999;18(23):6718–6729. doi: 10.1093/emboj/18.23.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groenendyk J, Lynch J, Michalak M. Calreticulin, Ca2+, and calcineurin - signaling from the endoplasmic reticulum. Molecules and cells. 2004;17(3):383–389. [PubMed] [Google Scholar]
- 42.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nature reviews. 2008;8(9):663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 43.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Human molecular genetics. 2007;16(6):618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 44.Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, Chang J, Dwivedi S, Mitsak M, Chen YW, Plotz P, Rosen A, Hoffman E, Raben N. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52(6):1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a, Representative hematoxylin-eosin stained transverse cryosections of proximal and distal muscles from hDysf-high transgenic mice. Scale bar, 100μm. b, Representative hematoxylin-eosin stained section of heart muscle from hDysf-high transgenic mice, revealing areas of calcification. Scale bar, 50μm. Bottom panel is magnification of upper inset panel. c, Representative cross-sections of gastrocnemius muscle from hDysf-high transgenic and wild-type. Scale bar, 100μm. Multiple images of transverse cryosections were examined to quantify fiber cross-sectional area of wild-type and transgenic gastrocnemius muscle (3 animals per genotype, 10 images per animal). The distribution of gastrocnemius fiber size was analyzed between transgenic and littermate wild-type mice, revealing a predominance of smaller fibers in hDysf-mid and –high overexpressing muscle.
a, Representative NADH-tetrazolium reductase histochemical staining identifies slow-twitch (dark) and fast-twitch (light) fibers. Fast fibers are selectively affected by hDysf overexpression. b, Electron micrographs of quadriceps muscle from 8-week old wild-type and hDysf-high transgenic mice. Subsarcolemmal vesicles accumulation in hDysf-overexpressing muscle (arrowheads). Left, Scale bar 150nm. Right, Scale bar, 500nm.
a, Evans blue dye uptake in quadriceps and tibialis anterior muscle from wild-type (WT) and hDysf-mid transgenic mice. Scale bar, 200μm. b, Plasma serum levels of creatine kinase in WT, hDysf-transgenic and A/J dysferlin-deficient mice (n=5 per group). Values are means ± SD (*P < 0.0001, t-test). c, Fusogenic capacity of wild-type (WT) and hDysf-mid transgenic primary myoblast cultures. Efficiency of fusion was analyzed by quantification of singly-nucleated desmin-positive myoblast cells, cells containing two to three nuclei and myotubes containing four or more nuclei. Five 10× fields from two separate cultures of each genotype were analyzed, comprising 573 hDysf-mid and 512 wild-type nuclei.