

Abstract
AIMS
To develop a population pharmacokinetic model for mycophenolic acid (MPA) in children with idiopathic nephrotic syndrome (INS) treated with mycophenolate mofetil (MMF), identify covariates that explain variability and determine the Bayesian estimator of the area under the concentration–time curve over 12 h (AUC0–12).
METHODS
The pharmacokinetic model of MMF was described from 23 patients aged 7.4 ± 3.9 years (range 2.9–14.9) using nonlinear mixed-effects modelling (NONMEM) software. A two-compartment model with lag–time and first-order absorption and elimination was developed. The final model was validated using visual predictive check. Bayesian estimator was validated using circular permutation method.
RESULTS
The population pharmacokinetic parameters were apparent oral clearance 9.7 l h−1, apparent central volume of distribution 22.3 l, apparent peripheral volume of distribution 250 l, inter-compartment clearance 18.8 l h−1, absorption rate constant 5.16 h−1, lag time 0.215 h. The covariate analysis identified body weight and serum albumin as individual factors influencing the apparent oral clearance. Accurate Bayesian estimation of AUC0–12 was obtained using the combination of three MPA concentrations measured just before (T0), 1 and 4 h (T1 and T4) after drug intake with a small error of 0.298 µg h−1 ml−1 between estimated and reference AUC0–12.
CONCLUSIONS
The population pharmacokinetic model of MPA was developed in children with INS. A three-point (T0, T1 and T4h) Bayesian estimator of AUC0–12 was developed and might be used to investigate the relation between MPA pharmacokinetic and pharmacodynamics in children with INS and determine if there is any indication to monitor MPA exposure in order to improve patient outcome based on individual AUC-controlled MMF dosing.
Keywords: Bayesian estimation, MMF, MPA, nephrotic syndrome, paediatrics, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
MMF has been proposed as a treatment of steroid-dependent nephrotic syndrome and in the recent years, several studies have suggested its positive effect in preventing relapses.
WHAT THIS PAPER ADDS
The population pharmacokinetics of MPA was first evaluated in children with idiopathic nephrotic syndrome and data fitted well with a two-compartment model with first-order absorption and lag time.
Body weight and serum albumin had a significant impact on oral clearance.
A three-point (T0, T1 and T4h) Bayesian estimator of AUC0–12 was developed.
Introduction
Idiopathic nephrotic syndrome (INS) is the most frequent glomerular nephropathy in children. Steroids are administered for the treatment of the first episode of INS. Approximately 90% of children respond to steroids, presenting with steroid-sensitive nephrotic syndrome (SSNS). In 10% of patients, the absence of remission occurs, the INS being steroid resistant (SRNS). Among steroid-sensitive children, 60% will experience frequent relapses or steroid-dependent nephrotic syndrome (SDNS) [1].
In the recent years, ciclosporin has been administered in patients with SDNS. It inhibits cytokine production from T-helper cells (Th1 and Th2) and also has an inhibitory effect on antigen-presenting cells, which are the main agents of T-cell stimulation. A further effect of interleukin (IL)-2 inhibition is a reduction in B-cell activation and subsequent antibody production. IL-2 levels are known to increase during proteinuria and to normalize during remission in patient with INS [2]. The use of ciclosporin allows reduction or suppression of relapses and reduction of steroid requirements [3, 4] However, in a limited number of patients, repeated relapses occur despite daily ciclosporin administration. In addition, as ciclosporin is associated with major side-effects, primarily nephrotoxicity, alternatives to ciclosporin are required in INS [5].
Mycophenolate mofetil (MMF) is an immunosuppressive drug used for the prevention of acute and chronic rejection after solid organ transplantation, as well as haematopoietic stem cell transplantation. MMF is rapidly and completely absorbed after oral intake, hydrolysed and transformed into the active metabolite mycophenolic acid (MPA), MPA inhibits inosine monophosphate dehydrogenase (IMPDH), an enzyme required for the de novo pathway of purine synthesis, which, in turn, is essential for the proliferation of B and T lymphocytes [6]. This perturbation of lymphocyte populations and functions might be crucial in the pathogenesis of INS [7]. Based on this theory, MMF has been proposed as a treatment of SDNS and, in recent years, several studies have suggested its positive effect in preventing relapses [8–14].
Among pathological disorders, INS induces hypoalbuminaemia and lipid disturbances with overproduction of lipoproteins, with important changes in plasma composition [15]. This may result in important pharmacokinetic changes, as shown with prednisolone [16, 17] and ciclosporin [18]. INS may also have an impact on MPA pharmacokinetics, as the drug is highly bound to serum albumin [19].
To date, information on the pharmacokinetics of MPA in paediatric patients with INS is limited [20]. The aims of the present study were: (i) to develop a population pharmacokinetic model of MPA in children with INS; (ii) to identify the patients' characteristics that influence pharmacokinetic parameters; and (iii) to define the Bayesian estimator of AUC0–12.
Patients and methods
Study population
Paediatric patients with INS were enrolled in this prospective multicentre clinical trial to evaluate the efficacy, pharmacokinetics and safety of MPA in children treated with MMF. The patients were recruited from six French centres. This clinical trial was designed in accordance with legal requirements and the Declaration of Helsinki and approved by the Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale (Saint-Louis Hospital, France). The parents of our patients signed an informed consent to the participation of their children.
Study protocol
Children were eligible for entry into this study when they had evidence of idiopathic SDNS and had relapses, despite cyclophosphamide therapy. Patients' recruitment was from 2004 to 2006. MMF (Cellcept®; Roche Pharma A.G, Grenzach-Wyhlen, Germany) was administered at doses of 1200 mg m−2 day−1 in two divided doses. After the start of MMF therapy, the dose of prednisone was reduced according to the following protocol: initial dose of 60 mg m−2 day−1 until 7 days after remission, then given at the same dose every other day for 2 weeks and decreased to 50% and 25% of the threshold prednisone dose (defined as the dose received at last relapse of NS) at the end of month 3 and month 6, respectively. Then prednisone dosage remained unchanged until month 12.
Pharmacokinetic study – assay for mycophenolic acid
Pharmacokinetic study was performed at month 1 (M1) and month 6 (M6) after inclusion. The blood samples were obtained before, 0.5, 1, 2, 4, 8 and 12 h after administration of MMF. Samples were immediately centrifuged and plasma was kept frozen at −20°C until analysis. The following covariates were also recorded prospectively on the pharmacokinetic day: age, weight, sex, height, body surface area, creatinine clearance (calculated according to the Schwartz formula), urine protein, serum albumin, haemoglobin, cholesterol, alkaline phosphatase, aspartate amino transferase (ASAT), alanine amino transferase (ALAT), prednisone dose and time after start of therapy.
MPA concentrations were measured by high-performance liquid chromatography with ultra-violet detection (HPLC-UV). The HPLC-UV system consisted of a quaternary P1000 XR pump (ThermoQuest TQ, San Jose, CA, USA), a TQ auto sampler, and a TQ UV 6000 detector (detection wavelength 254 nm) linked to a TQ Spectranet for recording and storing throughout analysis. The system used a Hypersil BDS C18, 5 µm (250 × 4.6 mm) analytic column (CIL Cluzeau, Sainte Foix la Grande, Gironde, France) with a mobile phase of acetonitrile and 0.05% aqueous phosphoric acid (38:62 v/v, increased to 43/57 v/v at 10 min and decreased at 28/72 v/v at 18 min). The assay showed linearity from 0.1 to 40 µg ml−1. The lower limit of quantification for MPA was 0.1 µg ml−1. The coefficients of variation (CV) of intraday and interday precision were <6%.
Model development
Pharmacokinetic analysis was carried out using the nonlinear mixed-effects modelling program NONMEM (V6.1.1; GloboMax LLC, Ellicott City, MD, USA) with the interface Wings for NONMEM (http://wfn_sourceforge.net/). The first-order conditional estimation (FOCE) method with INTERACTION option was used to estimate pharmacokinetic parameters and variabilities.
The model was parameterized in terms of absorption rate constant (Ka), lag time between intake and absorption start (lag time), apparent oral clearance (CL/F), apparent central volume of distribution (V1/F), apparent peripheral volume of distribution (V2/F) and inter-compartment clearance (Q/F).
Interindividual variability of the pharmacokinetic parameters was estimated by use of an exponential error model and could be expressed as follows:
where θA is the typical population value of pharmacokinetic parameter, ηjθA represents the difference between the jth individual's θA values and the predicted values, ηjθA values are independent, identically distributed random variables and normally distributed around 0 with variance ω[2] and j is the variable for the jth individual. A proportional model was used for the residual random effects. This selection was based on objective function (OFV) value and diagnostic plot of predicted concentrations vs. observed concentrations.
Covariate analysis
The individual pharmacokinetic parameter estimates were plotted against demographic and clinical factors (age, weight, sex, height, body surface area, creatinine clearance, urine protein, serum albumin, haemoglobin, cholesterol, alkaline phosphatase, ASAT, ALAT, prednisone dose, time after start of therapy) for visual inspection according to the method described by Maitre et al. [21].
Covariates that showed a correlation with a pharmacokinetic parameter were entered into the model for further tests. The selection of covariates used a forward and backward selection process. During forward selection, a covariate was selected only if a significant (P < 0.05) decrease (reduction >3.84) in OFV from the covariate-free model was obtained. Then all the covariates found to be significant during the forward selection were added simultaneously into a ‘full’ model. The importance of each covariate was re-evaluated by backward selection. Each covariate was independently removed from the full model to confirm its relevance. An increase in the objective function of >6.635 (P < 0.01) was required to confirm that the covariate was significant. The resulting model was called the ‘final’ population pharmacokinetic model that included all the significant covariates.
Model validation
The final model was assessed by visual predictive check. A visual predictive check was performed by simulating 1000 subjects from the final model. The 50th percentile concentration (as an estimator of the population-predicted concentration) and the 5th–95th percentile concentrations were processed by R for NONMEM (v.20070911, http://wfn.sourceforge.net/) and then plotted against original data [22, 23].
Bayesian estimator
For clinical practice, it is admitted that the best Bayesian estimator should be selected within 4 h post dose and the number of samplings should not exceed three. The selection was made based on their respective prediction performance. According to the method described by Sheiner and Beal [24], the predictive performance of Bayesian estimator was evaluated by calculating the prediction bias – percentage of prediction error (PE%) and prediction precision – percentage of the absolute prediction error (APE%). The reference AUC values were calculated by Bayesian estimation (‘MAXEVAL = 0’ and ‘Posthoc’ in the $ESTIMATION step of NONMEM software) using all the available time points.
 |
 |
Because of the limited number of patients in this study, the circular permutation method was used to validate Bayesian estimator. The full dataset was randomly divided into four subsets (each one contains 25% pharmacokinetic profiles) [25]. The population parameters obtained in each combination of three subsets (building group) were used as priors to calculate the individual pharmacokinetic parameters of the remaining subset (validation group), respectively. This procedure was repeated four times and the data were analysed using NONMEM software. Estimated AUCs using Bayesian estimator from circular permutation, repeated four times, were then compared with the reference AUCs.
Results
Patients
Twenty-three children with INS, consisting of 18 boys and five girls, aged 7.4 ± 3.8 years (range 2.9–14.9) and weighing 29.9 ± 18.0 kg (range 14.0–83.2), were enrolled in this prospective multicentre trial. A total of 41 pharmacokinetic profiles from 23 patients were analysed (18 patients had both pharmacokinetic profiles at M1 and M6; three patients had only one pharmacokinetic profile at M1; two patients had only one pharmacokinetic profile at M6). The characteristics of the patients on the day of pharmacokinetic study are presented in Table 1. Prednisone dose record was missing in five patients. As no relapse was reported in these five patients during the study period, the doses of prednisone were set to standard values according to the steroid therapy protocol in the step of covariate analysis.
Table 1.
Characteristics of the 23 children with nephrotic syndrome
| PK1 | PK2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patients' characteristics | Number | Mean | SD | Median | Range | Number | Mean | SD | Median | Range |
| No. of patients | 21/23 | 20/23 | ||||||||
| Male/female | 17/4 | 15/5 | ||||||||
| Age (years)* | 7.5 | 4.1 | 5.2 | 2.9–14.5 | 7.3 | 3.8 | 5.4 | 3.3–14.9 | ||
| Time after start of therapy* | 33 | 7 | 31 | 26–61 | 176 | 14 | 181 | 149–196 | ||
| Body weight (kg)* | 30.3 | 17.1 | 23.6 | 14.0–78.4 | 29.6 | 18.0 | 23.3 | 14.8–83.2 | ||
| Height (cm)* | 120.0 | 21.6 | 114.8 | 90.6–156.0 | 119.4 | 20.2 | 112.3 | 93.0–157.0 | ||
| Body surface area (m2)* | 1.01 | 0.37 | 0.89 | 0.61–1.90 | 0.99 | 0.38 | 0.88 | 0.63–1.96 | ||
| Creatinine clearance (ml min−1)*† | 125.6 | 26.2 | 121.6 | 95.1–210.2 | 130.4 | 37.1 | 120.4 | 87.6–249.5 | ||
| Urine protein (g l−1)* | 0.12 | 0.13 | 0.07 | <0.05–0.59 | 0.08 | 0.03 | 0.08 | <0.05–0.15 | ||
| Albumin (g l−1)* | 36.0 | 5.0 | 37.3 | 26.5–45.5 | 39.6 | 4.2 | 40.5 | 25.6–44.0 | ||
| Haemoglobin (mg dl−1) | 13.2 | 1.2 | 13.7 | 10.3–15.3 | 12.6 | 1.1 | 12.5 | 10.7–14.8 | ||
| Cholesterol (mmol l−1)* | 5.3 | 1.2 | 5.0 | 3.7–9.0 | 4.1 | 0.7 | 4.0 | 2.9–5.2 | ||
| Alkaline phosphatase (UI l−1)* | 152 | 59 | 139 | 75–314 | 193 | 73 | 168 | 122–397 | ||
| Aspartate amino transferase (IU l−1)* | 22 | 7 | 22 | 8–39 | 25 | 13 | 21 | 8–68 | ||
| Alanine amino transferase (IU l−1)* | 15 | 5 | 14 | 9–26 | 15 | 4 | 15 | 9–26 | ||
| Mycophenolate mofetil dose (mg)* | 563 | 212 | 500 | 300–1000 | 540 | 209 | 470 | 300–1000 | ||
| Prednisone (mg per 2 days)* | 47 | 14 | 40 | 25–65 | 13 | 8 | 8 | 5–35 | ||
Pharmacokinetic day.
Schwartz formula.
Population modelling
A total of 285 plasma concentrations were available for population modelling. MPA concentrations ranged from 0.35 µg ml−1 to 65.17 µg ml−1. All the measured concentrations were above the lower limit of quantification. The data fitted a two-compartment model with first-order absorption and lag time. In the diagnostic plot of weighted residuals (WRES) vs. time, no trends were observed. Interindividual variability was best described by an exponential model and was then estimated for V1/F, Q/F, CL/F and lag time. Intraindividual variability was not included in the model because of imprecision in the estimates of variance. Residual variability was best described by a proportional model. After testing different values for V2/F, between 100 and 600.V2/F was fixed to 250 l, as this gave the best estimation for both pharmacokinetic parameters and residual variability and was in reasonable agreement the reported values in transplant patients ranging from 137 l to 496 l [26–28].
During the covariate analysis, age, weight, sex, height, body surface area, creatinine clearance, urine protein, serum albumin, haemoglobin, cholesterol, alkaline phosphatase, ASAT, ALAT, prednisone dose at M1 and M6, time after start of therapy were tested. Plots of the individual pharmacokinetic parameters (Ka, V1/F, Q/F, CL/F and lag time) vs. potential covariates indicated that CL/F was related to weight, age, body surface area, serum albumin, haemoglobin and alkaline phosphatase, and that Q/F was related to serum albumin. In the forward selection processes, the inclusion of bodyweight (kg), body surface and serum albumin on systemic clearance produced a significant decrease in OFV. In the backward selection process, only body weight and serum albumin were retained. Body weight and serum albumin caused a decrease in OFV of 23.6 and 15.8 units, respectively. The forward and backward covariates selection processes are summarized in Table 2. The interindividual variability of CL/F decreased from 40.7% to 22%. No covariates significantly influenced V1/F, Q/F or lag time. Finally, the relationship between CL/F and covariates was expressed with the equation:
Table 2.
Covariates forward and backward selection processes
| Pharmacokinetic parameter | OFV | ||
|---|---|---|---|
| Forward | Backward | ||
| Two-compartment with first-order absorption and lag-time no covariate | 834.6 | ||
| Weight | CL/F | 811.0 (P < 0.01) | +14.5 (P < 0.01) |
| Serum albumin | CL/F | 818.8 (P < 0.01) | +6.7 (P < 0.01) |
| Body surface area | CL/F | 824.5 (P < 0.01) | +1.4 (P > 0.05) |
 |
The median body weight and albumin in the population were 23.5 kg and 38.6 g l−1, respectively.
Model validation
The parameters of the final model are presented in Table 3. Routine diagnostic weighted residuals vs. model-predicted values (Figure 1a) were symmetrically distributed and were mostly within about 3 units (expressed in standard deviation) of the null ordinate, indicating a good fit of the model to the data. Plots of weighted residuals vs. time (Figure 1b) were distributed symmetrically with no obvious trend and were mostly within approximately 3 units of the null ordinate, indicating that no time-related factor affected the data and that no subject's data contributed to any marked deviation from the model.
Table 3.
Population pharmacokinetic parameters of MMF
| Model final | ||
|---|---|---|
| Parameters | Final estimate | SE (%) |
| Lag time (h) | 0.215 | 37.9 |
| Absorption rate constant (h−1) Ka | 5.16 | 43.4 |
| Apparent central volume of distribution (l) V1/F | 22.3 | 22.6 |
| Apparent peripheral volume of distribution (l) V2/F | 250 Fixed | |
| Intercompartment clearance (l h−1) Q/F | 18.8 | 14.8 |
| Apparent oral clearance (l h−1) CL/F | ||
| CL/F =θ1 × (Weight/23.5) ×θ2 ×[1–θ3 × (Albumin/38.6)] | ||
| θ1 | 22.5 | 16.9 |
| θ2 | 0.753 | 11.2 |
| θ3 | 0.570 | 12.9 |
| Residual proportional | 44.6 | 9.4 |
| Interindividual variability | ||
| Lag time | 54.0 | 100.3 |
| V1/F | 79.9 | 67.6 |
| Q/F | 57.6 | 35.2 |
| CL/F | 22.0 | 42.6 |
Figure 1.
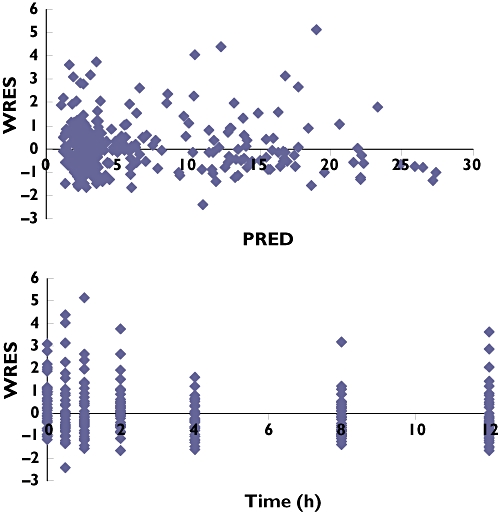
Goodness-of-fit plots for the final population pharmacokinetic model of mycophenolic acid (MPA). (a) Weighted residuals (WRES) vs. model-predicted concentration (PRED). (b) Weighted residuals (WRES) vs. time
For the visual predictive check, the 5th and 95th percentiles as well as the median 50th percentile of these simulations and the patients' data are shown in Figure 2. This graph, representing a visual internal validation of the model, shows that approximately 90% of the data of MPA fitted well within the 5th–95th percentiles [exact binomial test, 6.52% out of limits, 95% confidence interval (CI) 3.91, 10.1].
Figure 2.
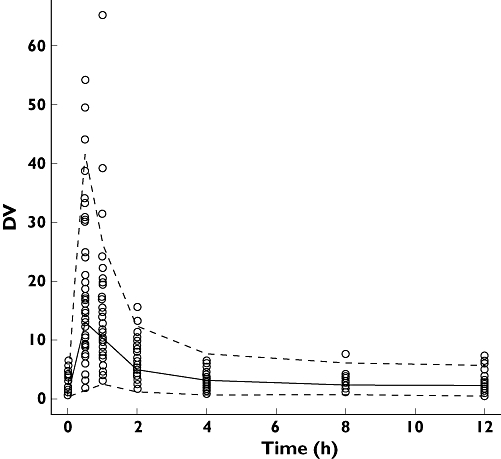
Visual predictive check of the model. The 50th percentile concentration is shown by a solid line and the 5th–95th percentiles simulated from the final model are encompassed by the broken lines
Bayesian estimator of AUC0–12
The reference AUC0–12 values were ranged from 31.925 to 73.669 µg h−1 ml−1 with a median value of 48.372 µg h−1 ml−1. As a first step, the Bayesian estimator was tested with our original dataset using estimated pharmacokinetic parameters of the final population model. Of the combinations of MPA concentrations measured at T0, T1, T4 and T0.5, T1 and T2 gave the better Bayesian estimations of AUC0–12 (small prediction error with mean AP% <1.7% and good precision with mean APE% <9.5). Further validation was conducted using the circular permutation method described above. As presented in Table 4, the performance of the combination of T0, T1 and T4 is better than that of T0.5, T1 and T2. Bland and Altman analysis show that only two of 41 values (4.9%) did not fall within the 95% CI of the mean AUC value (mean ± 2 SD). The average error between estimated AUC and referenced AUC was 0.298 µg h−1 ml−1 (range −20.911 to 9.294 µg h−1 ml−1, Figure 3).
Table 4.
Determination of Bayesian estimator of MMF
| Original data | Circular permutation validation | |||
|---|---|---|---|---|
| PE% | APE% | PE% | APE% | |
| Limited sampling strategies | mean ± SD | mean ± SD | mean ± SD | mean ± SD |
| 0 h, 1 h, 4 h | 1.7 ± 12.0 | 9.5 ± 7.4 | 0.3 ± 15.6 | 12.0 ± 9.8 |
| 0 h, 0.5 h, 4 h | 2.5 ± 14.4 | 11.5 ± 8.8 | ||
| 0 h, 0.5 h, 2 h | 5.6 ± 9.6 | 9.2 ± 6.1 | ||
| 0.5 h, 1 h, 4 h | −7.3 ± 12.8 | 12.3 ± 7.9 | ||
| 0.5 h, 1 h, 2 h | 1.5 ± 11.3 | 7.8 ± 8.2 | 2.6 ± 22.2 | 17.0 ± 14.3 |
Figure 3.
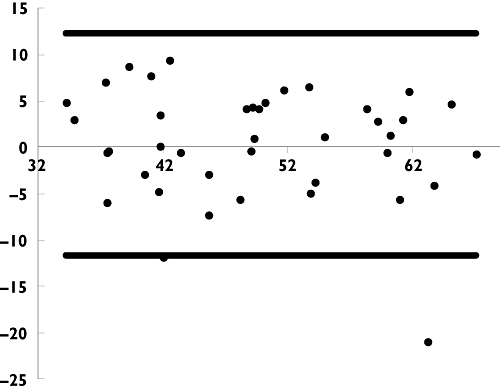
Bland and Altman plot of difference against mean for mycophenolic acid (MPA) AUC0–12 determined by Bayesian estimation (‘MAXEVAL = 0’ and ‘Posthoc’ in the $ESTIMATION step) using all the available time points and Bayesian estimator (T0, T1 and T4 after drug intake). Difference (•); Mean Diff. ± 2SD (—)
Discussion
This is the first population pharmacokinetic study of MPA in children with INS treated with MMF. Our results showed that a two-compartment model with first-order absorption and lag time was adequate for data modelling and that body weight and serum albumin had a significant impact on CL/F.
MMF is rapidly and completely absorbed after oral intake, hydrolysed and transformed into MPA [19]. MPA is primarily metabolized by glucuronidation to form the 7-O glucuronide conjugate (7-O-MPAG). Among other metabolites, the acylglucuronide metabolite of MPA (AcMPAG) was found to inhibit rh-IMPDH II in vitro with a similar efficacy to that of MPA [29]. The pharmacokinetic properties of MMF have been largely studied in organ transplant patients [30–32]. In the present study, the dose-normalized AUC0–12 (mean AUC0–12 49.3 µg h−1 ml−1 resulted from the dose of 1200 mg m−2 day−1) was slightly lower than that recently reported in renal transplant children by Filler et al. (median AUC0–12 of 49.8 µg h−1 ml−1 with a mean dose of 549 mg m−2 day−1 in 27 MMF-tacrolimus patients [33]) or by David-Neto et al. [median AUC0–12 of 63.2 µg h−1 ml−1 with a mean dose of 785 mg m−2 day−1 in 20 children (16 being treated with MMF-ciclosporin) [34]. The corresponding mean weight-normalized CL/F 0.43 l h−1 kg−1 is higher than that previously reported in renal transplanted adults (0.246 l h−1 kg−1) by Le Guellec et al. [26]. This observation will have to be confirmed. However, it might be explained by the physiopathological changes associated with INS. The basic functional defect in INS is a change of selectivity of the glomerular filtration barrier, leading to binding protein lost in the urine but also to reduced binding ability [35, 36]. As MPA is bound extensively to serum albumin [19], the loss of binding protein might lead more MPA to be eliminated by the kidney and then high CL/F is expected. Similarly, Gugler et al. have shown that the clearance of phenytoin (bound primarily to albumin) was higher in patients with INS than in healthy volunteers and decreased with the increase of albumin concentrations [36]. However, the impact of weight also has to be considered. Our patients were younger and had a lower weight (mean 29.9 kg) than that of patients studied by Le Guellec et al. (median 64 kg). According to the exponential relationship between CL/F and weight in our model, the linear weight-normalized CL/F is expected to be higher in children.
The fact that is an exponential function of body weight should be discussed further. MMF was given twice daily at doses of 1200 mg m−2 day−1. However, after the forward and backward covariate selection process, body weight was found to be better correlated with CL/F than body surface area. This finding was in agreement with our recent results in paediatric kidney transplant children [37], indicating that dose regimen should be based on weight. In addition, although the number of children is limited and the estimated exponential coefficient was very close to allometric scaling for CL/F, the fixed allometric scaling model (0.75 for clearance and 1 for volume of distribution) failed to describe the MPA pharmacokinetic data as the population pharmacokinetic parameters (V1/F, Q/F and V2/F) could not be precisely estimated. We also tested different maturation models, including exponential, asymptotic, and sigmoid maximum effect (Emax), but no significant effect of age was found.
In our patients, the duration of MMF treatment had no impact on MPA CL/F as the AUCs were not statistically different at M1 and M6 (P= 0.2, Wilcoxon test). In contrast, MPA exposures increase with increasing postoperative delay in renal transplanted patients [38] and might reflect the impact of renal function on MPA pharmacokinetics, as suggested by Van Hest et al. [32]. In patients with impaired renal function, acidosis and uraemia decrease MPA binding to albumin [39] and the accumulation of the glucuronidated metabolite MPAG displaces MPA from its albumin binding sites [40], resulting in high concentrations of free MPA and a high MPA clearance in the initial post-transplant period. As renal function improves, MPA clearance progressively increases over the first months after transplantation [32]. Van Hest et al. also demonstrated that the impact of renal function was significant only in adult kidney-transplanted patients having a creatinine clearance <25 ml min−1[31]. In our study, all the patients had a creatinine clearance >25 ml min−1, which might explain why neither time after start of therapy nor renal function significantly influenced MMF CL/F.
The MPA enterohepatic recirculation (EHC) was widely reported in adult kidney transplant patients [41]. As we had to limit the number of samples drawn during this paediatric study, only T8 and T12 were included in the pharmacokinetic profiles. The concentrations at T8 and T12 did not show evidence of EHC (2.3 ± 1.2 µg ml−1vs. 2.2 ± 1.7 µg ml−1) and no obvious trend was observed in the time period 6–12 h in the plot of WRES vs. time [19]. EHC is not expected to have played a large part in this study. However, the co-medication influence on transporters of MPA should be evaluated in INS children, as nonsteroidal anti-inflammatory drugs [42] have been shown to inhibit the ABCC2, which might potentially reduce the EHC.
In a previous study, Cattaneo et al. [43] reported that the withdrawn of steroids reduced MMF CL/F in adult kidney transplant patients. As all our patients received prednisone in addition to MMF at M1 and M6, withdrawn could not be tested. However, reduction of the daily prednisone dose between M1 and M6 had no influence on the pharmacokinetic parameters of MPA.
MMF is free of major toxicities and when it was introduced, therapeutic drug monitoring (TDM) was considered unnecessary and was not recommended by the manufacturer. However, pharmacokinetic studies revealed substantial inter- and intraindividual variability in pharmacokinetic parameters and significant drug interactions [26, 30]. TDM of MPA is now an important tool in transplantation to optimize therapy in organ transplantation [27, 44, 45]. Recently, Dorresteijn et al. reported a randomized controlled trial of MMF vs. ciclosporin for remission maintenance in children with INS. In the MMF group, seven of 12 patients' AUC0–12 were calculated using the limited sampling strategy (T0, T75 min and T360 min), which was developed from paediatric kidney transplant recipients [46]. They reported that relapses were associated with lower AUC, an argument for TDM in children with INS [20]. Therefore, using the population pharmacokinetic parameters of final model and NONMEM software, it is now possible to estimate the AUC0–12 based on MPA concentrations at T0, T1 and T4, and covariates of weight and albumin to optimize MPA exposure in INS children.
This clinical trail was designed in 2002, at a time when the efficacy of MMF in children with INS was not evaluated. In our study, the first pharmacokinetic profile was taken 1 month after initiation of treatment, at a time when remission was already obtained, in order to be under steady-state conditions for both MPA and its glucuronidated metabolite MPAG. However, additional studies will have to investigate the impact of biological disturbances of INS on the pharmacokinetics of MMF at the initiation of treatment with MMF.
In summary, the population pharmacokinetic model developed for MPA in children with INS showed that MPA weight-normalized clearance was higher than that in kidney transplant recipients. Body weight and serum albumin were significant covariates influencing MPA CL/F. The Bayesian estimator of AUC0–12 was developed from the final model in order to be used in a prospective pharmacokinetic/pharmacodynamic study of MPA and TDM in children with INS.
Competing interests
None to declare.
We would like to acknowledge the contribution of Ying Wang in the conduct of the study. Technical support from Michel Popon was very much appreciated.
REFERENCES
- 1.Churg J, Habib R, White RHR. Pathology of nephrotic syndrome in children: a report for the International Study of Kidney Disease in Children. Lancet. 1970;760:1299–302. doi: 10.1016/s0140-6736(70)91905-7. [DOI] [PubMed] [Google Scholar]
- 2.Cattran DC, Alexopoulos E, Heering P, Hoyer PF, Johnston A, Meyrier A, Ponticelli C, Saito T, Choukroun G, Nachman P, Praga M, Yoshikawa N. Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome: workshop recommendations. Kidney Int. 2007;72:1429–47. doi: 10.1038/sj.ki.5002553. [DOI] [PubMed] [Google Scholar]
- 3.Hino S, Takemura T, Okada M, Murakami K, Yagi K, Fukushima K, Yoshioka K. Follow-up of children with nephrotic syndrome treated with a long-term moderate dose of cyclosporine. Am J Kidney Dis. 1998;31:932–9. doi: 10.1053/ajkd.1998.v31.pm9631836. [DOI] [PubMed] [Google Scholar]
- 4.Okada M, Hino S, Takemura T, Fukushima K, Yoshioka K. Cyclosporine therapy in children with steroid-resistant nephrotic syndrome. Clin Exp Nephrol. 1999;3:S34–9. [Google Scholar]
- 5.Moudgil A, Bagga A, Jordan SC. Mycophenolate mofetil therapy in frequently relapsing steroid-dependent and steroid-resistant nephrotic syndrome of childhood: current status and future directions. Pediatr Nephrol. 2005;20:1376–81. doi: 10.1007/s00467-005-1964-z. [DOI] [PubMed] [Google Scholar]
- 6.Sollinger HW. Mycophenolates in transplantation. Clin Transplant. 2004;18:485–92. doi: 10.1111/j.1399-0012.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 7.Afzal K, Bagga A, Menon S, Hari P, Jordan SC. Treatment with mycophenolate mofetil and prednisolone for steroid-dependent nephrotic syndrome. Pediatr Nephrol. 2007;22:2059–65. doi: 10.1007/s00467-007-0617-9. [DOI] [PubMed] [Google Scholar]
- 8.Mendizabal S, Zamora I, Berbel O, Sanahuja MJ, Fuentes J, Simon J. Mycophenolate mofetil in steroid/cyclosporine-dependent/resistant nephrotic syndrome. Pediatr Nephrol. 2005;20:914–9. doi: 10.1007/s00467-005-1877-x. [DOI] [PubMed] [Google Scholar]
- 9.Ulinski T, Dubourg L, Said MH, Parchoux B, Ranchin B, Cochat P. Switch from cyclosporine A to mycophenolate mofetil in nephrotic children. Pediatr Nephrol. 2005;20:482–5. doi: 10.1007/s00467-004-1778-4. [DOI] [PubMed] [Google Scholar]
- 10.Gellermann J, Querfeld U. Frequently relapsing nephrotic syndrome: treatment with mycophenolate mofetil. Pediatr Nephrol. 2004;19:101–4. doi: 10.1007/s00467-003-1300-4. [DOI] [PubMed] [Google Scholar]
- 11.Barletta GM, Smoyer WE, Bunchman TE, Flynn JT, Kershaw DB. Use of mycophenolate mofetil in steroid-dependent and -resistant nephrotic syndrome. Pediatr Nephrol. 2003;18:833–7. doi: 10.1007/s00467-003-1175-4. [DOI] [PubMed] [Google Scholar]
- 12.Novak I, Frank R, Vento S, Gauthier B, Trachtman H. Efficacy of mycophenolate mofetil in pediatric patients with steroid-dependent nephrotic syndrome. Pediatr Nephrol. 2005;20:1265–8. doi: 10.1007/s00467-005-1957-y. [DOI] [PubMed] [Google Scholar]
- 13.Fujinaga S, Ohtomo Y, Umino D, Takemoto M, Shimizu T, Yamashiro Y, Kaneko K. A prospective study on the use of mycophenolate mofetil in children with cyclosporine-dependent nephrotic syndrome. Pediatr Nephrol. 2007;22:71–6. doi: 10.1007/s00467-006-0294-0. [DOI] [PubMed] [Google Scholar]
- 14.Bagga A, Hari P, Moudgil A, Jordan SC. Mycophenolate mofetil and prednisolone therapy in children with steroid dependent nephrotic syndrome. Am J Kidney Dis. 2003;42:1114–20. doi: 10.1053/j.ajkd.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Kaysen GA. Plasma composition in the nephrotic syndrome. Am J Nephrol. 1993;13:347–59. doi: 10.1159/000168649. [DOI] [PubMed] [Google Scholar]
- 16.Bergrem H. Pharmacokinetics and protein binding of prednisolone in patients with nephrotic syndrome and patients undergoing hemodialysis. Kidney Int. 1983;23:876–81. doi: 10.1038/ki.1983.110. [DOI] [PubMed] [Google Scholar]
- 17.Miller PF, Bowmer CJ, Wheeldon J, Brocklebank JT. Pharmacokinetics of prednisolone in children with nephrosis. Arch Dis Child. 1990;65:196–200. doi: 10.1136/adc.65.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacqz-Aigrain E, Montes C, Brun P, Loirat C. Cyclosporine pharmacokinetics in nephrotic and kidney-transplanted children. Eur J Clin Pharmacol. 1994;47:61–5. doi: 10.1007/BF00193480. [DOI] [PubMed] [Google Scholar]
- 19.Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet. 1998;34:429–55. doi: 10.2165/00003088-199834060-00002. [DOI] [PubMed] [Google Scholar]
- 20.Dorresteijn EM, Kist-van Holthe JE, Levtchenko EN, Nauta J, Hop WC, van der Heijden AJ. Mycophenolate mofetil versus cyclosporine for remission maintenance in nephrotic syndrome. Pediatr Nephrol. 2008;23:2013–20. doi: 10.1007/s00467-008-0899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maitre PO, Bührer M, Thomson D, Stanski DR. A three-step approach combining Bayesian regression and NONMEM population analysis: application to midazolam. J Pharmacokinet Biopharm. 1991;19:377–84. doi: 10.1007/BF01061662. [DOI] [PubMed] [Google Scholar]
- 22.Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001;28:171–92. doi: 10.1023/a:1011555016423. [DOI] [PubMed] [Google Scholar]
- 23.Zhao W, Baudouin V, Zhang D, Deschênes G, Guellec CL, Jacqz-Aigrain E. Population pharmacokinetics of ganciclovir following administration of valganciclovir in paediatric renal transplant patients. Clin Pharmacokinet. 2009;48:321–8. doi: 10.2165/00003088-200948050-00004. [DOI] [PubMed] [Google Scholar]
- 24.Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9:503–12. doi: 10.1007/BF01060893. [DOI] [PubMed] [Google Scholar]
- 25.Djebli N, Rousseau A, Hoizey G, Rerolle JP, Toupance O, Le Meur Y, Marquet P. Sirolimus population pharmacokinetic/pharmacogenetic analysis and Bayesian modelling in kidney transplant recipients. Clin Pharmacokinet. 2006;45:1135–48. doi: 10.2165/00003088-200645110-00007. [DOI] [PubMed] [Google Scholar]
- 26.Le Guellec C, Bourgoin H, Büchler M, Le Meur Y, Lebranchu Y, Marquet P, Paintaud G. Population pharmacokinetics and Bayesian estimation of mycophenolic acid concentrations in stable renal transplant patients. Clin Pharmacokinet. 2004;43:253–66. doi: 10.2165/00003088-200443040-00004. [DOI] [PubMed] [Google Scholar]
- 27.Staatz CE, Duffull SB, Kiberd B, Fraser AD, Tett SE. Population pharmacokinetics of mycophenolic acid during the first week after renal transplantation. Eur J Clin Pharmacol. 2005;61:507–16. doi: 10.1007/s00228-005-0927-4. [DOI] [PubMed] [Google Scholar]
- 28.Shum B, Duffull SB, Taylor PJ, Tett SE. Population pharmacokinetic analysis of mycophenolic acid in renal transplant recipients following oral administration of mycophenolate mofetil. Br J Clin Pharmacol. 2003;56:188–97. doi: 10.1046/j.1365-2125.2003.01863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schutz E, Shipkova M, Armstrong VW, Wieland E, Oellerich M. Identification of a pharmacologically active metabolite of mycophenolic acid in plasma of transplant recipients treated with mycophenolate mofetil. Clin Chem. 1999;45:419–22. [PubMed] [Google Scholar]
- 30.Payen S, Zhang D, Maisin A, Popon M, Bensman A, Bouissou F, Loirat C, Gomeni R, Bressolle F, Jacqz-Aigrain E. Population pharmacokinetics of mycophenolic acid in kidney transplant pediatric and adolescent patients. Ther Drug Monit. 2005;27:378–88. doi: 10.1097/01.ftd.0000159784.25872.f6. [DOI] [PubMed] [Google Scholar]
- 31.van Hest RM, van Gelder T, Vulto AG, Mathot RA. Population pharmacokinetics of mycophenolic acid in renal transplant recipients. Clin Pharmacokinet. 2005;44:1083–96. doi: 10.2165/00003088-200544100-00006. [DOI] [PubMed] [Google Scholar]
- 32.Van Hest RM, Van Gelder T, Bouw R, Goggin T, Gordon R, Mamelok RD, Mathot RA. Time-dependent clearance of mycophenolic acid in renal transplant recipients. Br J Clin Pharmacol. 2007;63:741–52. doi: 10.1111/j.1365-2125.2006.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filler G, Foster J, Berard R, Mai I, Lepage N. Age-dependency of mycophenolate mofetil dosing in combination with tacrolimus after pediatric renal transplantation. Transplant Proc. 2004;36:1327–31. doi: 10.1016/j.transproceed.2004.05.043. [DOI] [PubMed] [Google Scholar]
- 34.David-Neto E, Pereira Araujo LM, Sumita NM, Mendes ME, Ribeiro Castro MC, Alves CF, Kakehashi E, Romano P, Yagyu EM, Queiroga M, Nahas WC, Ianhez LE. Mycophenolic acid pharmacokinetics in stable pediatric renal transplantation. Pediatr Nephrol. 2003;18:266–72. doi: 10.1007/s00467-002-1057-1. [DOI] [PubMed] [Google Scholar]
- 35.Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. 1998;338:1202–11. doi: 10.1056/NEJM199804233381707. [DOI] [PubMed] [Google Scholar]
- 36.Gugler R, Shoeman DW, Huffman DH, Cohlmia JB, Azarnoff DL. Pharmacokinetics of drugs in patients with the nephrotic syndrome. J Clin Invest. 1975;55:1182–9. doi: 10.1172/JCI108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W, Fakhoury M, Deschênes G, Roussey G, Brochard K, Niaudet P, Tsimaratos M, André JL, Cloarec S, Cochat P, Bensman A, Azougagh S, Jacqz-Aigrain E. Population pharmacokinetics and pharmacogenetics of mycophenolic acid following administration of mycophenolate mofetil in de novo pediatric renal transplant patients. J Clin Pharmacol. 2010 doi: 10.1177/0091270009357429. DOI: 10.1177/0091270009357429. [DOI] [PubMed] [Google Scholar]
- 38.Hale MD, Nicholls AJ, Bullingham RE, Hené R, Hoitsma A, Squifflet JP, Weimar W, Vanrenterghem Y, Van de Woude FJ, Verpooten GA. The pharmacokinetic–pharmacodynamic relationship for mycophenolate mofetil in renal transplantation. Clin Pharmacol Ther. 1998;64:672–83. doi: 10.1016/S0009-9236(98)90058-3. [DOI] [PubMed] [Google Scholar]
- 39.Shaw LM, Korecka M, Aradhye S, Grossman R, Bayer L, Innes C, Cucciara A, Barker C, Naji A, Nicholls A, Brayman K. Mycophenolic acid area under the curve values in African American and Caucasian renal transplant patients are comparable. J Clin Pharmacol. 2000;40:624–33. doi: 10.1002/j.1552-4604.2000.tb05988.x. [DOI] [PubMed] [Google Scholar]
- 40.Nowak I, Shaw LM. Mycophenolic acid binding to human serum albumin: characterization and relation to pharmacodynamics. Clin Chem. 1995;41:1011–7. [PubMed] [Google Scholar]
- 41.van Gelder T, Klupp J, Barten MJ, Christians U, Morris RE. Comparison of the effects of tacrolimus and cyclosporine on the pharmacokinetics of mycophenolic acid. Ther Drug Monit. 2001;23:119–28. doi: 10.1097/00007691-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 42.El-Sheikh AA, van den Heuvel JJ, Koenderink JB, Russel FG. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther. 2007;320:229–35. doi: 10.1124/jpet.106.110379. [DOI] [PubMed] [Google Scholar]
- 43.Cattaneo D, Perico N, Gaspari F, Gotti E, Remuzzi G. Glucocorticoids interfere with mycophenolate mofetil bioavailability in kidney transplantation. Kidney Int. 2002;62:1060–7. doi: 10.1046/j.1523-1755.2002.00531.x. [DOI] [PubMed] [Google Scholar]
- 44.van Gelder T, Le Meur Y, Shaw LM, Oellerich M, DeNofrio D, Holt C, Holt DW, Kaplan B, Kuypers D, Meiser B, Toenshoff B, Mamelok RD. Therapeutic drug monitoring of mycophenolate mofetil in transplantation. Ther Drug Monit. 2006;28:145–54. doi: 10.1097/01.ftd.0000199358.80013.bd. [DOI] [PubMed] [Google Scholar]
- 45.Le Meur Y, Büchler M, Thierry A, Caillard S, Villemain F, Lavaud S, Etienne I, Westeel PF, Hurault de Ligny B, Rostaing L, Thervet E, Szelag JC, Rérolle JP, Rousseau A, Touchard G, Marquet P. Individualized mycophenolate mofetil dosing based on drug exposure significantly improves patient outcomes after renal transplantation. Am J Transplant. 2007;7:2496–503. doi: 10.1111/j.1600-6143.2007.01983.x. [DOI] [PubMed] [Google Scholar]
- 46.Schütz E, Armstrong VW, Shipkova M, Weber L, Niedmann PD, Lammersdorf T, Wiesel M, Mandelbaum A, Zimmerhackl LB, Mehls O, Tönshoff B, Oellerich M. Limited sampling strategy for the determination of mycophenolic acid area under the curve in pediatric kidney recipients. German Study Group on MMF Therapy in Pediatric Renal Transplant Recipients. Transplant Proc. 1998;30:1182–4. doi: 10.1016/s0041-1345(98)00200-0. [DOI] [PubMed] [Google Scholar]