

Abstract
The regulatory subunits of cAMP-dependent protein kinase (PKA) are the major receptors of cAMP in most eukaryotic cells. As the cyclic nucleotide binding (CNB) domains release cAMP and bind to the catalytic subunit of PKA, they undergo a major conformational change. The change is mediated by the B/C helix in CNB-A, which extends into one long helix that now separates the two CNB domains and docks onto the surface of the catalytic subunit. We explore here the role of three key residues on the B/C helix that dock onto the catalytic subunit, Arg226, Leu233, and Met 234. By replacing each residue with Ala, we show that each contributes significantly to creating the R:C interface. By also deleting the second CNB domain (CNB-B), we show furthermore that CNB-B is a critical part of the cAMP-induced conformational switch that dislodges the B/C helix from the surface of the catalytic subunit. Without CNB-B the Ka for activation by cAMP increases from 80 to 1000 nM. Replacing any of the key interface residues with Ala reduces the Ka to 25–40 nM. Leu233 and M234 contribute to a hydrophobic latch that binds the B/C helix onto the large lobe of the C-subunit, while Arg226 is part of an electrostatic switch that couples the B/C helix to the phosphate binding cassette where the cAMP docks.
Keywords: protein kinase A, cAMP signaling, RIα
Introduction
cAMP has been conserved throughout biology as a second messenger that allows both prokaryotes and eukaryotes to respond to extracellular signals. The cyclic nucleotide binding (CNB) domain has coevolved with cAMP as the primary receptor for cAMP.1–3 It is an ancient signaling domain that is coupled to many biological responses such as the regulation of transcription, channel opening and closing, guanidine nucleotide exchange, and kinase inhibition. cAMP-dependent protein kinase (PKA), where activation of kinase activity is coupled to cAMP binding, demonstrates how two major signaling mechanisms, cAMP second messenger signaling and protein phosphorylation, have converged. PKA is expressed ubiquitously in every mammalian cell and was the first cAMP signaling system to be discovered in mammalian cells.4,5 PKA is conserved in every cell as a mechanism that regulates basic functions such as growth, development, metabolism, differentiation, memory, and death in response typically to an external signal.
While cAMP is generated in the cytoplasm through a GPCR coupled mechanism, it is binding to a CNB domain that translates the second messenger signal into a biological response. The CNB domain is a compact and highly dynamic signaling module that undergoes major conformational changes on binding to cAMP. It contains a rigid β subdomain, specifically an eight-stranded β-sandwich, that forms a binding pocket for the cyclic phosphate of cAMP shielding it from solvent and phosphodiesterases. Each CNB domain also has a malleable helical subdomain, which consists of two noncontiguous helical elements. There are three essential motifs embedded within each CNB domain. The phosphate binding cassette (PBC), which is the signature motif for binding cAMP, is nested within the β subdomain between β strands 6 and 7.6,7 The two dynamic helical subdomains flank the β subdomain. Fused to the C-terminus of the β subdomain is the B/C helix, which serves as a dynamic switch that couples cAMP binding to recognition of other proteins or domains.8,9 N-terminal to the β sandwich is the αN-310-loop-αA (N3A) motif,10 which coordinates the conformational changes that take place when cAMP binds to the PBC and then causes the release of the catalytic subunit. The β-sandwich with its PBC and the two flanking motifs, the N3A motif and the B/C helix thus constitute the basic signaling domain, and unlike most other signaling domains such as SH2 domains, SH3 domains, and PH domains, this domain is highly dynamic. It is the dynamic interplay of these three motifs that is the essence of cyclic nucleotide signaling.
In the case of PKA, binding of cAMP is coupled to kinase activation. The cAMP binding domains are part of the PKA regulatory (R) subunits, where each R subunit contains a dimerization/docking domain at the N-terminus, a flexible linker that contains an inhibitor site, and two contiguous CNB domains at their C-terminus. In the absence of cAMP, the R subunit dimers are bound tightly to two catalytic (C) subunit, and this maintains PKA in an inactive state. Allosteric binding of cAMP to the PBC breaks the interface between the R and C-subunits and unleashes the catalytic activity. Figure 1 illustrates how cAMP binding is coupled to kinase inhibition through the dynamic conformational changes of the B/C helix and the coordinated movements of the N3A motif in domain A of the RIα subunit. In the holoenzyme the B/C helix is extended into a single long helix that is uncoupled, extended away from the PBC and docked to the surface of the PKA catalytic subunit where it helps to lock an inhibitory peptide into the active site cleft of the kinase.11,12 When cAMP binds, the B/C helix undergoes a major conformational change as it is recruited away from the C-subunit to the PBC. When the B/C helix moves “out” and away from the PBC, the N3A motif moves “in” toward the PBC whereas when the B/C helix moves “in,” the N3A motif moves “out.”
Figure 1.
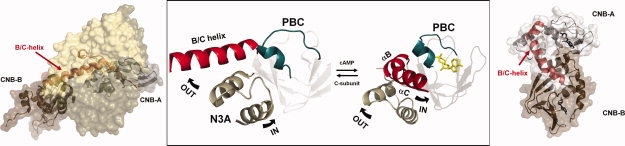
Conformational changes of the B/C helix in the CNB A domain of RIα. The major changes in conformation in RIα when the cAMP-bound conformation (right) is compared with the holoenzyme (left) are mediated by the unusual dynamics of the B/C helix show in red. In the center are highlighted the three conserved motifs in the CNB domain. In blue is the PBC, in tan is the N3A motif, and in red is the B/C helix. While the PBC, which is embedded in the middle of the stable β sandwich moves slightly, the helical subdomain undergoes major changes. Most significant is the extension of the B/C helix. Structures are from PDB: 1RGS and 2QCS.
The CNB A domain of the RIα subunit of PKA, coupled to the inhibitor motif, represents the smallest module that retains the ability to inhibit the activity of the catalytic subunit and to be activated in response to cAMP.13 In the RIα subunit, the CNB A domain plus ATP and two magnesium ions are sufficient to form a high affinity complex while the B domain serves as a “gatekeeper” that modulates access of cAMP to the A domain. It was the structure of this module, RIα(91–244), bound to the catalytic subunit that provided the first glimpse of how cAMP binding was actually coupled directly to kinase inhibition,12 and it was this structure that first revealed the dynamic properties of the B/C helix as it shuttles between its “in” and “out” conformations. The NMR characterization of the CNB domain minus the inhibitor segment, RIα(119–244), provides a comprehensive understanding of how the cAMP binding signal is propagated throughout the CNB domain,14 whereas the addition of the inhibitor segment which is completely disordered in the cAMP bound state, allows us to appreciate how cAMP binding is actually coupled to kinase inhibition. Activation of PKA is, however, highly cooperative, and to understand this complex allostery we need to appreciate the role of the B domain. In RIα it is the first CNB domain, domain A, that is coupled to kinase inhibition while the second domain is allosterically coupled to the A domain and is important for regulating access to the A domain.12,15,16 The ordered pathway for the activation of the RIα holoenzyme involves binding first to the CNB-B domain, and then to the CNB-A domain, which is then followed by release of the catalytic subunit.
To understand how cAMP binding is coupled to kinase activation, and in particular, how the conformation of the dynamic B/C helix is regulated, we focused initially on the B/C helix of CNB-A and used two strategies. We looked first for residues that were exposed to solvent in the cAMP bound state and part of the R:C interface in the holoenzyme state. We then identified two residues that fulfilled these criteria, Leu233 and Met234, and demonstrated their importance for docking to the C-subunit. In addition, we explored the importance of Arg226 which is part of an electrostatic switch that links the B/C helix to R209 in the PBC through D170. D170 is part of β2–β3 loop and has been identified by mutagenesis17 and NMR18 as a critical allosteric residue that toggles between R209 in the cAMP bound state and R226 in the holoenzyme state. By expressing these mutations initially in RIα(91–244), which contains only one CNB domain, we show the importance of all three residues for this conformational electrostatic switch. By expressing the same mutation in RIα(91–379), which contains both CNB domains, we show that the B-domain is a critical part of this allosteric mechanism. Without the B domain it is difficult to dislodge the B/C helix from the catalytic subunit as evidenced by a Ka(cAMP) for activation of 1 μM when only the A domain is present versus 80 nM when both domains are present. Mutagenesis of any one of these three B/C helix residues restores the Ka to 25–50 nM, which is in the range of the Kd for cAMP binding to free RIα(91–244).19,20
Results
The B/C helix in the RIα subunit is the switch that mediates the global changes in conformation of the R-subunit as toggles between an “in” and “out” conformation in the cAMP-bound state and in the holoenzyme, respectively. In its “in” conformation, it is kinked and anchored to the PBC that is bound to cAMP. In its “out” conformation, it is extended and docked to the large lobe of the catalytic subunit. To understand what controls the conformation of the B/C helix as the protein toggles between its ligand bound state and its protein bound state we used several strategies. Crystallography initially defined the cAMP bound state7 and the C-subunit bound conformation.12 Unfolding studies21 and more recently an NMR analysis of a construct containing both CNB domains (McNicholl et al., submitted for publication) and molecular dynamic simulations22 suggest that the unbound state is unstable suggesting that simply removing cAMP does not generate a stable “out” conformation by simply removing cAMP. This was confirmed by crystallization of an apo form of RIα(91–244) (unpublished results).
We initially focused on the B helix of domain A and searched for residues that were exposed to solvent in the cAMP-bound state but part of the hydrophobic interface with the catalytic subunit in the holoenzyme. Two residues were identified, and we used mutagenesis to establish their importance. In addition, we explored the role of Arg226 which is highly conserved in all RIα A domains.2 D170 is part of a three way relay switch involving D170 in the β2–β3 loop and R209, which is the docking site for the phosphate of cAMP in the PBC. Initially, we characterized these sites in only the A domain which is the smallest functional CNB domain that can bind to both the catalytic subunit and the regulatory subunit. This domain in RIα is the major site for interaction with the catalytic subunit whereas the B domain serves as a gatekeeper that mediates access of cAMP to the A domain. By expressing the same mutation in RIα(91–379) that contains both CNB domains, we could evaluate how access of cAMP to the A domain is regulated.
Mutagenesis of B/C helix residues
The extension of the B/C helix creates a large hydrophobic surface that is anchored to the C-subunit by several key residues, some which play no apparent role in the cAMP-bound conformation (Fig. 2). Two conserved hydrophobic residues in the B/C helix, Leu233 and Met234, appear to serve as a clamp that locks the B/C helix onto the large lobe of the kinase where it is buttressed up against the activation loop, and we replaced both these residues with Ala. Another key residue at the beginning of the B helix is Arg226, which is conserved in all PKA R-subunit A domains. The side chain of Arg226, in addition to making hydrophobic interactions, also interacts with the backbone carbonyl of Ala202 in the PBC. Asp170 is predicted to be critical for the allosteric coupling of the PBC to the B/C helix.17,18 In the presence of cAMP it binds to Arg209 in the PBC while in the holoenzyme it binds to Arg226. We also mutated R226 to Ala.
Figure 2.
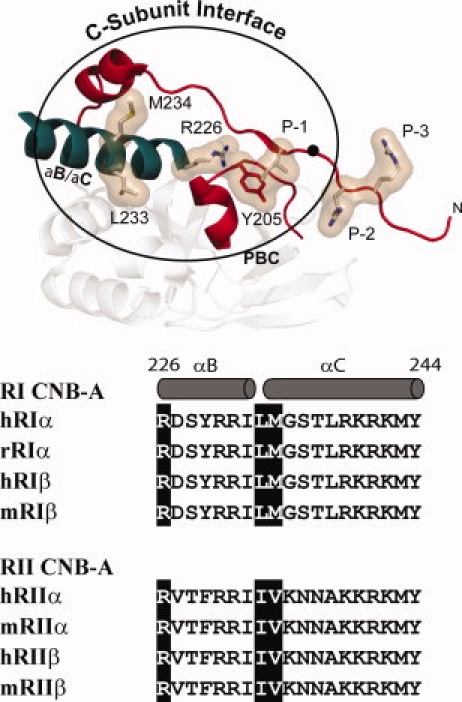
C-Subunit interface in the CNB-A domain of RIα and sequence alignment of B/C helix. Top: Structure of the hydrophobic interface that is created by the conformational changes in CNB-A is shown. The hydrophobic residues that dock onto the C-subunit are highlighted. B/C helix is shown in teal while the PBC and the ordered linker and inhibitory site (residues 92–113 are shown in red). Bottom: Sequence comparison of the B/C helices in the four R-subunit isoforms. Residues mutated in this study are highlighted.
The three mutations were introduced into RIα(91–244), which contains only one CNB domain, and into RIα(91–379), which contains two CNB domains. All six mutant proteins expressed reasonably well in E. coli and were purified by affinity chromatography using cAMP-Sepharose as described previously followed by gel filtration. Holoenzyme was formed by adding a slight excess of regulatory subunit, and the complex was then purified by gel filtration.7
Consequences of mutating Leu233 and Met234
To evaluate the effects of each mutation we measured the Ka(cAMP) for activation of the holoenzyme by cAMP, the Kd using Surface Plasmon Resonance, the IC50 for inhibition, and binding of cAMP using a fluorescent analog of cAMP.
Activation of RIα holoenzymes by cAMP
Activation of wild type and mutant holoenzyme complexes was measured by titrating in cAMP and monitoring activity. This assay is sensitive both to the affinity of the R and C subunits for each other and the ability of the regulatory subunits to bind cAMP (Table I). Leu233A and M234A showed similar effects and required less cAMP to activate than wild-type RIα. In RIα(91–379) 2- to 3-fold less cAMP was also required to activate the holoenzyme (26 and 38 nM, respectively, for Leu233A and M234A), compared with the wild-type RIα(91–379) where the Ka was 80 nM. However, in RIα(91–244), which only has CNB-A, the effects were greatly amplified (Fig. 3, Table I). WT RIα(91–244) has a Ka of 1 μM, while L233A had a Ka of 20 nM, a decrease of 50-fold. M234A in the same construct had a Ka of 28 nM, a decrease of 36-fold. These values for the mutant proteins are comparable with the affinity of CNB-A for cAMP. These mutations suggest that addition of the B-domain does not, in fact, increase the affinity of the R-subunit for the C-subunit, but instead is there to facilitate release of the B/C helix. When Leu233 or Met234 in RIα(91–244) is mutated to Ala, the hydrophobic interface is weakened. Essentially, the construct replaces the allosteric coupling of CNB-B with regards to cAMP activation.
Table I.
Activation of RIα holoenzymes by cAMP and inhibition of PKA by mutant and wild type of R-subunits
| Ka (nM) | IC50 (nM) | |
|---|---|---|
| RIα (91–379) | ||
| WT | 80 ± 10 | 6.4 ± 0.8 |
| R226A | 84 ± 12 | 6.6 ± 0.7 |
| L233A | 26 ± 8 | 8.8 ± 0.6 |
| M234A | 38 ± 10 | 10.8 ± 0.7 |
| RIα (91–244) | ||
| WT | 1000 ± 150 | 10 ± 1.5 |
| R226A | 54 ± 10 | 12 ± 3.0 |
| L233A | 20 ± 7 | 12 ± 2.0 |
| M234A | 28 ± 8 | 9 ± 1.5 |
Figure 3.

Effect of mutating Leu233 and Met234 in RIα(91–244) on activation by cAMP. Holoenzyme (25 nM) was incubated with increasing concentrations of cAMP and catalytic activity was measured. Catalytic activity was measured using a coupled Kemptide assay.
Surface plasmon resonance
To quantitate the binding of the L233A and M234A mutant proteins to the catalytic subunit, we used Surface Plasmon Resonance (SPR) using established methods. SPR provides not only the binding affinities (KD) but also the association (kassoc) and dissociation (kdis) rates. All mutants showed lower affinity for the chip-bound catalytic subunit compared with the wild-type RIα protein. For RIα(91–379), L233A showed a 3-fold increase in KD, entirely due to an increase in off-rate. This is consistent with the decrease in the Ka for activation. M234A showed a 2.5-fold increase in KD, with both a small increase in on-rate and slightly larger increase in off-rate (Table II).
Table II.
Interaction of mutant R-subunits and WT C-subunit by surface plasmon resonance
| kassoc (M−1 s−1) | kdiss (s−1) | KD (nM) | |
|---|---|---|---|
| RIα 91–379 | |||
| WT RIα | 0.8 ± 0.1 × 106 | 0.3 ± 0.02 × 10−3 | 0.41 |
| L233A | 0.8 ± 0.1 × 106 | 1.0 ± 0.1 × 10−3 | 1.28 |
| M234A | 1.2 ± 0.2 × 106 | 1.3 ± 0.1 × 10−3 | 1.08 |
| R226A | 1.8 ± 0.2 × 106 | 1.3 ± 0.1 × 10−3 | 0.72 |
| RIα 91–244 | |||
| WT RIα | 15.0 ± 0.6 × 106 | 2.7 ± 0.1 × 10−3 | 0.18 |
| L233A | 8.7 ± 0.3 × 106 | 6.1 ± 0.2 × 10−3 | 0.70 |
| M234A | 7.5 ± 0.3 × 106 | 8.2 ± 0.2 × 10−3 | 1.09 |
| R226A | 17.0 ± 0.5 × 106 | 9.1 ± 0.1 × 10−3 | 0.54 |
In RIα(91–244), the differences for L233A and M234A were similar to RIα(91–379). L233A showed a 3.5-fold increase in KD, due to a combination of decreased on rate and increased off-rate. M234A had the same on and off rate effects, but to a somewhat greater extent, with an end result of a 5-fold higher KD. The changes seen in M234A are comparable with mutating its interaction partner, W196 in the catalytic subunit, which when mutated to Arg showed a 6-fold increase in KD for RIα WT.23 In both W196RC and M234A, the differences came primarily from an increased off rate.
These results show that there is a modest disruption in the hydrophobic surface of the R:C complex in these mutants, similar to that seen in other hydrophobic mutations (Sjoberg, manuscript in preparation).22 However, this increase in KD is not sufficient to explain the changes in cAMP activation for RIα(91–244). Furthermore, the KD for the shorter construct is, if anything, less than for RIα(91–379). Domain B does not appear to convey enhanced affinity for the C subunit.
Inhibition of catalytic activity by B/C helix mutants
The capacity of mutant L233A and M234A proteins to inhibit catalytic activity was also monitored. In both RIα(91–379) and RIα(91–244), the L233A and M234A mutants the IC50 values showed only minor decreases in their capacity to inhibit catalytic activity as summarized in Table I. All of the differences seen in this assay were <2-fold. These data suggest that while the complex is easier to activate by cAMP, in the absence of cAMP the mutants are still able to fully inhibit activity in a stoichiometric fashion.
cAMP binding
To determine if the mutant proteins were deficient in cAMP binding, we used 8-Fl cAMP and measured the KD using fluorescence polarization. Both constructs of L233A and M234A, along with R226A RIα(91–244) and RIα(91–379) were indistinguishable from the wild type with regards to cAMP binding affinity (data not shown). These data show that a decrease in cAMP binding is not the cause of differences in activation.
Consequences of mutating Arg226
In addition to hydrophobic interactions, anchoring of the B/C helix to the C-subunit also involves Arg226. There are many basic residues in the B/C helix, but most of them, in contrast to Arg226, face outward. Arg241 in the C helix, for example, is coupled to the B domain through Asp267 but then moves over to bridge to the Glu200 in the PBC in the cAMP-bound conformation.
Activation of the R226A RIα holoenzyme by cAMP
To examine the activation by cAMP of R226A, R:C holoenzyme complexes of R226A RIα(91–244) and RIα(91–379) were formed and activated by titration as with L233A and M234A. R226A RIα(91–379) showed Ka values comparable with WT of the same construct (Table I, Fig. 4). However, as with the previous mutants, R226A RIα(91–244) showed a significant decrease in cAMP needed for activation, from 1 to 54 nM, a change of almost 20-fold. The major difference when the long and short constructs are compared is that it is much more difficult to activate the shorter construct when the B domain is missing, but this difference vanishes when Arg226 is mutated to Ala. The Ka for activation is 54 nM which is very similar to the other two mutants. It is also similar to the intrinsic KD for binding cAMP.
Figure 4.
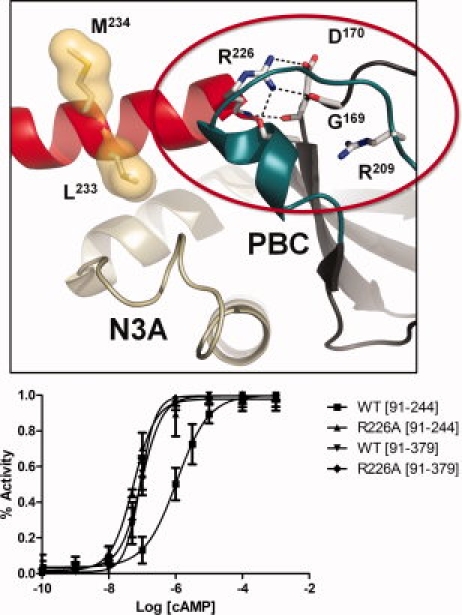
Activation of the Arg226Ala mutant by cAMP. The top panel shows the interactions of Arg226 in the holoenzyme structure of RIα(91–379):C:Mn2ATP (2QCS). The bottom panel shows activation of R226A holoenzyme by cAMP. Holoenzyme (25 nM) was formed with wild type and mutant forms of RIα(91–244) and RIα(91–379) and then incubated with increasing concentrations of cAMP (lower panel).
Interaction of Arg226Ala with catalytic subunit
The effects of the R226A mutation in RIα(91–244) and R226A RIα(91–379) were analyzed by SPR to determine on and off rates as we did with the Leu233Ala and Met234Ala mutants. The KD increased by <2-fold in the longer construct, and almost 3-fold for RIα(91–244) relative to the respective WT constructs (Table II). However, while R226A RIα(91–244) saw changes mostly in off-rate, in R226A RIα(91–379) there were increases in both on and off rate. These results are similar to those seen in D170A, R226's binding partner in the R:C holoenzyme.17
Like the L233A and M234A mutants, the binding affinity of R226A for the catalytic subunit for both RIα(91–244) and RIα(91–379) was measured. Like the other mutants, both R226A constructs showed no change relative to WT in IC50 values by 32P inhibition assay for either construct (Table I).
Discussion
CNB domains are highly conserved in nature, and we now have representative structures of several CNB domain families such as EPAC,24 the catabolite activator protein,25 the HCN channel,26 cyclic nucleotide regulated potassium channel27 in addition to the various PKA R-subunit isoforms.6,7,11,28,29 Each CNB domain has a contiguous β subdomain and a noncontiguous α-helical subdomain. The β subdomain, which harbors the PBC, is remarkably stable. With the exception of the tip of the PBC, the entire eight-stranded β sandwich does not appear to change its conformation in response to the bound partner. In contrast, the helical subdomain undergoes significant conformational changes, and these changes are unique for each CNB domain. In the A domain of the PKA R-subunits this conformational change is profound. There are two motifs embedded in the helical subdomain that change in different ways and their movement is coordinated. The N3A motif that precedes β strand 110 moves more as a rigid body whereas the B/C helix that follows β strand 8 undergoes a significant change in its conformation. In the A domain of RIα and RIIα, the B/C helix is kinked when cAMP is bound and goes to a fully extended helix when the catalytic subunit is bound (Fig. 1).
Such an extreme conformational change is a unique feature of the A domains of the R-subunits of PKA. The B/C helix in the B domain also undergoes a conformational change where the B, C, and C′ helices rearrange but they do not extend into one long helix. In Epac the helix does extend and is fused to a β-strand, in CAP the B/C-helix is a modified, cAMP sensitive, leucine zipper and undergoes moderate changes.30 In HCN channel, the N3A motif is fused to the channel. The A domain of the PKA R-subunits is unusual for several reasons. It is the only example so far when two CNB domains are contiguously fused. Specifically, the B/C helix of domain A is fused to the N3A motif of domain B. Thus, when the B/C helix extends, it includes the N-terminal helix of domain B. In addition the capping residue for cAMP bound to the A domain is located in domain B. In all other CNB domains, including domain B of the R-subunits, the capping residue is self-contained within its own CNB domain. Our goal here was to explore the properties of this unusual B/C helix in domain A of RIα. Is it simply a docking motif between R and C subunits, or is it also a critical part of an allosteric signaling network between the A and B domains that is coupled to the kinase inhibition?
To understand the switch mechanism for the B/C helix in domain A, we mutated a set of residues in the B/C helix that appeared to uniquely stabilize the C-bound conformation. There are two hydrophobic residues, Leu233 and Met234, which apparently serve as hydrophobic clamp as they dock strategically onto the regulatory surface of the kinase large lobe that includes the activation loop and the P + 1 loop. To understand both the role of these two residues and the importance of the B domain, we expressed mutants of these residues in RIα(91–244) that contains a single CNB domain and RIα(91–379) that contains two CNB domains. Mutating either Leu233 or Met234 to Ala in RIα(91–379) increases the KD from 0.4 nM to 1.3 and 1.1 nM, and the Ka for activation is accordingly reduced from 80 nM to about 20 nM. The intrinsic KD for cAMP binding to the A domain is 20 nM. What was surprising, however, was to discover how important the B-domain was for dislodging the B/C helix.
Earlier studies by Huang et al.13 showed that the smallest functional unit capable of binding both cAMP and C-subunit was RIa(91–244), and we subsequently confirmed that residues 245–255 correspond to the N helix and are functionally a part of the B domain. When the entire B domain is removed, including the N3A motif, activation of the kinase with cAMP becomes very difficult. The Ka for activation by cAMP goes from 80 nM to 1 μM. However, simply changing one of the hydrophobic clamp residues to Ala reduces the Ka to 30–40 nM. This demonstrates not only the importance of these two key residues in forming the R:C interface but also the importance of the B domain for dislodging the native B/C helix from the C-subunit. Without the B domain, it is very difficult to activate this complex.
Another residue that contributes to the interface is Arg226 at the beginning of the B/C helix. As seen in Figure 4, Arg226 several important bonds in RIα in the holoenzyme form. It interacts with Ala202 from the PBC and with Gly169 and Asp170 from β2 to β3 loop. Asp170 is a key allosteric residue in the A domain of RIα. In the cAMP bound conformation Asp170 interacts with Arg209, which is also bound to the exocyclic oxygen of cAMP. Therefore, Asp170 plays a critical role in allowing the rest of the CNB domain to sense binding of cAMP.18 Competition for Asp170 is thus one of the important switches when the two conformations are compared. When Asp170 is replaced with Ala, holoenzyme can form even when cAMP is bound to the C-subunit whereas ordinarily we need to remove cAMP either with urea or by replacing it with cGMP to rapidly form holoenzyme.17 Ala202 and Gly169 are also very important residues as they are located in the PBC and the β2–β3 loop, elements that control cAMP binding.31 Replacing Arg226 with Ala confirms its critical role as part of the allosteric switch that is associated with holoenzyme formation and activation. The Ka is reduced to 20 nM in both RIα(91–244) and RIα(91–379). This 50-fold reduction in Ka for the shorter construct means that the KD for binding cAMP and the Ka are nearly identical.
These mutations in the B/C helix highlight clearly the important role of the B domain. We showed previously that the cooperative and ordered pathway for activation of the RIα holoenzyme required binding of cAMP first to the B domain and then to the A domain. This was predicted earlier32 and then confirmed by mutagenesis once we saw the holoenzyme structure.11 We mutated two key residues in the B-domain. One set of mutations broke the ion pair between Glu261 and Arg366, which made activation easier. The other mutated the capping residues for cAMP bound to the A domain (Trp260) and the B domain (Tyr371). Both of these residues are located in the B domain and form a hydrophobic cap for the adenine ring of cAMP. CNB domain A in the PKA regulatory subunits is actually quite unique compared with other CNB domains where the capping residue is typically located in the same CNB domain. CNB domain B is similar to the other CNB domains where the capping residue is located in its own C helix. We learned from the earlier studies that recruiting the capping residue in domain B to the binding site for cAMP of the A domain is a critical step in activation. What we show here is that the B domain has another function. It is essential for dislodging the B/C helix from the large lobe of the C-subunit. Without the B domain separation of the R and C subunits becomes much more difficult. If we now consider the allosteric effects of cAMP on the holoenzyme (Fig. 5), we see that both ends of the B/C helix are in fact sensitive to cAMP. Previously, we demonstrated that cAMP binding to the A domain unlocks the inhibitor peptide. Here, for the first time, we show that cAMP binding to the B domain is essential for unlocking the C-terminal end of the B/C helix in the A domain. Thus the B/C helix serves a polyvalent sensor for cAMP and appears to be an important part of the allosteric communication line between the two CNB domains of regulatory subunits in PKA.
Figure 5.
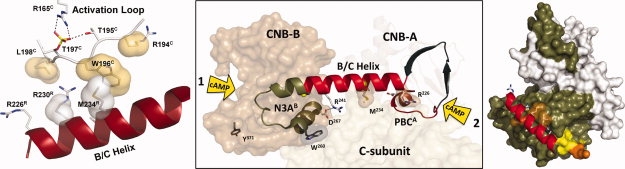
Both ends of the B/C helix are regulated by cAMP. The first cAMP binds to the B domain and helps to unleash the B/C helix from the large lobe of the C-subunit. This allows a second molecule of cAMP to bind to domain A, and this second cAMP is what disrupts the inhibitor peptide. This highly ordered and bivalent interaction of two molecules of cAMP with the B/C helix (center panel) provides a concerted allosteric mechanism for activation and release of kinase inhibition. The panel on the right shows how the B/C helix is anchored onto the large lobe of the C-subunit while the panel on the left shows how this B/C helix is buttressed up against Trp196 and Leu198 in the activation loop. Leu198 is the major determinant for the P + 1 docking site, and these two residues, Leu198 and Trp196, flank the essential phospho-Thr197 which is anchored to the active site through its interactions with Arg165 in the HRD motif in the catalytic loop.
Materials and Methods
Site-directed mutagenesis
Site-directed mutants were prepared using QuikChange kits (Stratagene) using standard protocols for 16 cycles of thermal cycling. All mutants were made in pRSET with ampicillin resistance. Three mutations were engineered L233A, Met234A, and R226A, and each was introduced into two different bovine RIα constructs (91–379 and 91–244). The products were transformed into XL-1 blue Escherichia coli supercompetent cells, and plasmid DNA was purified using the QiaPrep Spin Miniprep kit (Qiagen). Mutant sequences were confirmed by DNA sequencing (Eton Biosciences).
Protein expression and purification
For the regulatory subunit proteins, BL-21 DE3 E. coli cells (Stratagene) were transformed with mutant and wild-type plasmid DNA and purified by established methods. For each construct, cells were grown, centrifuged and lysed by French press into lysis buffer containing a mix of protease inhibitors. Lysate was centrifuged and supernatant was precipitated with 45% ammonium sulfate. The precipitated solution was centrifuged, and the precipitate was re-suspended in lysis buffer with protease inhibitors and applied to 5 mL cAMP Sepharose resin (Sigma), which was previously equilibrated with lysis buffer. This mixture was batch bound on a rotator overnight at 4°C. The resin was washed and eluted with lysis buffer containing high concentrations of cGMP. Eluted protein was concentrated and then purified on a S75 16/60 size exclusion column (BioRad).
Catalytic subunit was expressed in E. coli BL-21 DE3 cells and purified by established protocols. Three peaks of phosphotransferase activity were detected following purification on a Mono S 5/5 column; the second and largest peak was used for these studies. This corresponds to isoform II, which is phosphorylated at S10, T197, and S338. Holoenzyme heterodimers were formed by mixing purified R-subunit and C-subunits in a 1.2:1 ratio and dialyzing overnight against a buffer containing 50 mM MOPS, 150 mM NaCl, 2 mM MgCl2, and 0.2 mM ATP (pH 7.0) and purified by gel filtration (Superdex 75) to separate holoenzyme from free R-subunit.
Holoenzyme activation by cAMP
Protein kinase activity was measured using a coupled spectrophotometric kinase assay. The oxidation of NADH, monitored spectrophotometrically as an absorbance decrease at 340 nm, is coupled to the production of ADP by lactate dehydrogenase and pyruvate kinase. The holoenzyme complexes at concentrations of 25 nM were incubated for 5 min in the assay mix (500 μL of holoenzyme in above buffer, with 1 mM phosphoenolpyruvate, 0.3 mM NADH, 12 units of lactate dehydrogenase, and four units of pyruvate kinase, with varying concentrations of cAMP (Sigma) ranging from 1 nM to 100 μM. The reaction was initiated by adding 100 μM Kemptide (LRRASLG), a synthetic peptide substrate, and the activity of the free C-subunit was followed using the spectrophotometric assay. Nonlinear regression using the Graphpad Prism 4 software was used to determine the activation constant (Ka) for wild type and mutant holoenzymes.
Inhibition of catalytic activity
Inhibition of the catalytic subunit by the wild-type or mutant regulatory subunits was carried out. The catalytic subunit (1 nM) was incubated with different concentrations of regulatory subunit for 30 min at 30°C. A mixture of substrate (Kemptide), γ-32P-ATP was then added to start the phosphorylation reaction. After 30 min, the reaction was quenched by 30% acetic acid. To separate unreacted γ-32P-ATP from the protein-bound radioactivity, 20 μL of the quenched reaction mix was spotted onto a phosphocellulose paper disk (Whatman P81). The disk was then washed with 5% phosphoric acid to remove the unreacted γ-32P-ATP. The spot area was cut out, dried, and put into scintillation vials for radioactivity detection by Cherenkov counting on Beckman LS 6000SC liquid scintillation system. Specific activity of γ-32P-ATP was 500–1000 cpm/pmol. Curve fitting and IC50 calculations were obtained by GraphPad Prism 4.
Surface plasmon resonance
SPR was used to measure the interaction kinetics of the C-subunit and the various RIα subunits using a Biacore 3000 instrument (GE Healthcare Life Sciences). The C-subunit was immobilized to a CM Dextran surface sensor chip (Biosensor amine coupling kit). All binding interactions were performed in 20 mM Mops (pH 7.0), 150 mM KCl, 1 mM TCEP buffer containing 0.2 mM ATP and 1 mM MgCl2. Following injection of the R-subunit, the C-subunit surface was regenerated by injection of 2 min (50 μl) of running buffer with 30 μM cAMP and 1 mM EDTA added. Kinetic constants were calculated using the Biacore pseudo-first-order rate equation and affinity constants (KD) were calculated from the equation KD = kdiss/kassoc.
Fluorescent cAMP affinity
The ability of the wild type and mutant regulatory subunits to bind cAMP was monitored using a fluorescent cAMP analog as follows. 8-Fluo-cAMP (BioLog) was diluted in a 200 μL quartz cuvette to a final concentration of 1 nM. As a control a second cuvette was prepared with a 100× excess of unlabeled cAMP (100 nM final concentration). Both were dissolved in holoenzyme buffer (10 mM MES, 50 mM NaCl, 0.5mM ATP, 5 mM MgCl2, 5 mM DTT, pH = 6.5). Using a FluroroMax-2 (Instruments S.A.) the cuvette was excited with a wavelength of 467 nm and the emission at 516 nm was monitored. The various regulatory subunits were titrated in triplicate to final concentrations in the cuvette from 0.01 to 100 nM. The changes in fluorescence were zeroed using the changes in the cuvette with excess unlabeled cAMP as a baseline and the modified change in fluorescence as a final output. GraphPad Prism 4 was used to calculate apparent binding constants.
Acknowledgments
The authors thank Juniper Pennypacker and Mira Sastri for their help with mutagenesis, Mike Deal for providing purified catalytic subunit, and C.J. Allison for his technical assistance. Structural figures were created using PyMol. Graphs were created with GraphPad Prism.
Glossary
Abbreviations:
- cAMP
cyclic adenosine monophosphate
- CAP
catabolite activator protein
- CNB
cyclic nucleotide binding domain
- PBC
phosphate binding cassette
- PKA
cAMP-dependent protein kinase
- SPR
surface plasmon resonance
References
- 1.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domaan ancient signaling module. Proc Natl Acad Sci USA. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canaves JM, Taylor SS. Classification and phylogenetic analysis of the cAMP-dependent protein kinase regulatory subunit family. J Mol Evol. 2002;54:17–29. doi: 10.1007/s00239-001-0013-1. [DOI] [PubMed] [Google Scholar]
- 3.Kannan N, Wu J, Anand GS, Yooseph S, Neuwald AF, Venter JC, Taylor SS. Evolution of allostery in the cyclic nucleotide binding module. Genome Biol. 2007;8:R264. doi: 10.1186/gb-2007-8-12-r264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gill GN, Garren LD. Role of the receptor in the mechanism of action of adenosine 3′:5′-cyclic monophosphate. Proc Natl Acad Sci USA. 1971;68:786–790. doi: 10.1073/pnas.68.4.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho RJ, Sutherland EW. Action of feedback regulator on adenylate cyclase. Proc Natl Acad Sci USA. 1975;72:1773–1777. doi: 10.1073/pnas.72.5.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- 7.Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 8.Vigil D, Lin JH, Sotriffer CA, Pennypacker JK, McCammon JA, Taylor SS. A simple electrostatic switch important in the activation of type I protein kinase A by cyclic AMP. Protein Sci. 2006;15:113–121. doi: 10.1110/ps.051723606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herberg FW, Zimmermann B, McGlone M, Taylor SS. Importance of the A-helix of the catalytic subunit of cAMP-dependent protein kinase for stability and for orienting subdomains at the cleft interface. Protein Sci. 1997;6:569–579. doi: 10.1002/pro.5560060306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kornev AP, Taylor SS, Ten Eyck LF. A generalized allosteric mechanism for cis-regulated cyclic nucleotide binding domains. PLoS Comput Biol. 2008;4:e1000056. doi: 10.1371/journal.pcbi.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 13.Huang LJ, Taylor SS. Dissecting cAMP binding domain A in the RIalpha subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J Biol Chem. 1998;273:26739–26746. doi: 10.1074/jbc.273.41.26739. [DOI] [PubMed] [Google Scholar]
- 14.Das R, Abu-Abed M, Melacini G. Mapping allostery through equilibrium perturbation NMR spectroscopy. J Am Chem Soc. 2006;128:8406–8407. doi: 10.1021/ja060046d. [DOI] [PubMed] [Google Scholar]
- 15.Herberg FW, Taylor SS, Dostmann WR. Active site mutations define the pathway for the cooperative activation of cAMP-dependent protein kinase. Biochemistry. 1996;35:2934–2942. doi: 10.1021/bi951647c. [DOI] [PubMed] [Google Scholar]
- 16.Ogreid D, Doskeland SO, Gorman KB, Steinberg RA. Mutations that prevent cyclic nucleotide binding to binding sites A or B of type I cyclic AMP-dependent protein kinase. J Biol Chem. 1988;263:17397–17404. [PubMed] [Google Scholar]
- 17.Gibson RM, Ji-Buechler Y, Taylor SS. Interaction of the regulatory and catalytic subunits of cAMP-dependent protein kinase. Electrostatic sites on the type Ialpha regulatory subunit. J Biol Chem. 1997;272:16343–16350. doi: 10.1074/jbc.272.26.16343. [DOI] [PubMed] [Google Scholar]
- 18.Abu-Abed M, Das R, Wang L, Melacini G. Definition of an electrostatic relay switch critical for the cAMP-dependent activation of protein kinase A as revealed by the D170A mutant of RIalpha. Proteins. 2007;69:112–124. doi: 10.1002/prot.21446. [DOI] [PubMed] [Google Scholar]
- 19.Leon DA, Canaves JM, Taylor SS. Probing the multidomain structure of the type I regulatory subunit of cAMP-dependent protein kinase using mutational analysis: role and environment of endogenous tryptophans. Biochemistry. 2000;39:5662–5671. doi: 10.1021/bi992819z. [DOI] [PubMed] [Google Scholar]
- 20.Zorn M, Fladmark KE, Ogreid D, Jastorff B, Doskeland SO, Dostmann WR. Ala335 is essential for high-affinity cAMP-binding of both sites A and B of cAMP-dependent protein kinase type I. FEBS Lett. 1995;362:291–294. doi: 10.1016/0014-5793(95)00261-7. [DOI] [PubMed] [Google Scholar]
- 21.Canaves JM, Leon DA, Taylor SS. Consequences of cAMP-binding site mutations on the structural stability of the type I regulatory subunit of cAMP-dependent protein kinase. Biochemistry. 2000;39:15022–15031. doi: 10.1021/bi001563q. [DOI] [PubMed] [Google Scholar]
- 22.Vigil D, Lin JH, Sotriffer CA, Pennypacker JK, McCammon JA, Taylor SS. A simple electrostatic switch important in the activation of type I protein kinase A by cyclic AMP. Prot Sci. 2006;15:113–121. doi: 10.1110/ps.051723606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson RM, Taylor SS. Dissecting the cooperative reassociation of the regulatory and catalytic subunits of cAMP-dependent protein kinase. Role of Trp-196 in the catalytic subunit. J Biol Chem. 1997;272:31998–32005. doi: 10.1074/jbc.272.51.31998. [DOI] [PubMed] [Google Scholar]
- 24.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- 25.Passner JM, Steitz TA. The structure of a CAP-DNA complex having two cAMP molecules bound to each monomer. Proc Natl Acad Sci USA. 1997;94:2843–2847. doi: 10.1073/pnas.94.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 27.Clayton GM, Silverman WR, Heginbotham L, Morais-Cabral JH. Structural basis of ligand activation in a cyclic nucleotide regulated potassium channel. Cell. 2004;119:615–627. doi: 10.1016/j.cell.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 28.Brown SH, Wu J, Kim C, Alberto K, Taylor SS. Novel isoform-specific interfaces revealed by PKA RIIbeta holoenzyme structures. J Mol Biol. 2009;393:1070–1082. doi: 10.1016/j.jmb.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Brown SH, von Daake S, Taylor SS. PKA type IIalpha holoenzyme reveals a combinatorial strategy for isoform diversity. Science. 2007;318:274–279. doi: 10.1126/science.1146447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popovych N, Tzeng SR, Tonelli M, Ebright RH, Kalodimos CG. Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc Natl Acad Sci USA. 2009;106:6927–6932. doi: 10.1073/pnas.0900595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das R, Esposito V, Abu-Abed M, Anand GS, Taylor SS, Melacini G. cAMP activation of PKA defines an ancient signaling mechanism. Proc Natl Acad Sci USA. 2007;104:93–98. doi: 10.1073/pnas.0609033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herberg FW, Dostmann WR, Zorn M, Davis SJ, Taylor SS. Crosstalk between domains in the regulatory subunit of cAMP-dependent protein kinase: influence of amino terminus on cAMP binding and holoenzyme formation. Biochemistry. 1994;33:7485–7494. doi: 10.1021/bi00189a057. [DOI] [PubMed] [Google Scholar]