

Abstract
Lymphocytes that invade nonlymphoid tissues often organize into follicle-like structures known as tertiary lymphoid organs (TLOs). These structures resemble those found in spleen or lymph nodes, but their function is unknown. TLOs are recognized in many autoimmune diseases, including the NOD mouse model of type 1 diabetes. In some cases, TLOs have been associated with the B lymphocyte chemoattractant, CXCL13. Studies presented in this article show that CXCL13 is present in inflamed islets of NOD mice. Ab blockade of this chemokine unraveled B lymphocyte organization in islet TLOs, without reducing their proportion in the islets. These chaotic milieus contained B lymphocytes with the same distinct repertoire of B cell receptors as those found in mice with well-organized structures. Somatic hypermutation, associated with T–B interactions, was not impaired in these disorganized insulitis lesions. Finally, loss of B lymphocyte organization in islets did not provide disease protection. Thus, B lymphocytes infiltrating islets in NOD mice do not require the morphology of secondary lymphoid tissues to support their role in disease.
Tertiary lymphoid organs (TLOs) are found in inflamed tissues of multiple autoimmune diseases, including rheumatoid arthritis, Sjogren’s syndrome, experimental autoimmmune encephalitis, and the NOD mouse model of type 1 diabetes (T1D) (1–8). These structures mimic secondary lymphoid tissues anatomically, and also share functional characteristics, such as germinal center (GC) reactions, critical for B cell isotype class switching and affinity maturation. In the NOD mouse, these ectopic lymphoid structures, consisting of a central T cell zone surrounded by B cells, begin to coalesce even at the early stage of peri-insulitis. This process occurs for each islet independently, and T cells can be seen grouping together at the islet interface of the lymphocyte attack, with B cells tending to flank them. The lymphocytes fully organize into follicle-like structures that include GCs by the time each islet is fully infiltrated (9). Although many of the cellular and molecular mechanisms underlying the formation of TLOs have been delineated, the role of ectopic lymphocyte organization in disease pathogenesis has yet to be determined. The presence of TLOs in NOD mice can be correlated with disease, which sometimes leads to the assumption that they are critical to disease-related processes (10). However, whether autoreactive lymphocytes must be structurally organized to contribute to disease pathogenesis is still an unanswered question.
Previous studies in our laboratory have shown that B cells in islet TLOs of NOD mice have a skewed repertoire of BCRs compared with the pool of recirculating lymphocytes in secondary lymphoid organs, indicating that a selective process for B cells occurs at the inflamed site. This work also showed the presence of GCs, as well as somatic hypermutation (SHM) of BCRs, suggesting that T–B interactions occur within islet TLOs (9). Because B lymphocytes act as essential APCs to support this T cell-mediated autoimmune disease, such interactions raise the possibility that disease-promoting cellular crosstalk may occur within TLOs in the islets.
CXCL13 (B lymphocyte chemoattractant) is a pivotal chemokine responsible for the formation and maintenance of B lymphocyte follicles and GCs in spleen and lymph nodes (11), and has been identified in inflamed autoimmune-associated TLOs (12–14). Transgenic CXCL13 expression in normal mouse islets is sufficient for full formation of ectopic lymphoid aggregates, a process that is lymphotoxin-dependent (15). Lymphotoxin blockade, in turn, stops the development of diabetes in NOD mice (10, 16) and has been shown to reverse insulitis (10). Therefore, the blockade of CXCL13 could reasonably be expected to disrupt this process in a similar fashion. However, in these studies, the data show the CXCL13 blockade largely disrupted B lymphocyte organizational morphology without altering their recruitment to islets. B lymphocytes in these chaotic milieus maintained the same BCR V gene bias found in untreated mice, and SHM of these genes was robust, indicating that T–B interactions were not effectively disrupted. Diabetes progression was unimpaired. Thus, in NOD mice, B lymphocytes in islets under autoimmune attack do not require widespread duplication of the morphology found in secondary lymphoid tissues to promote disease.
Materials and Methods
RT-PCR for CXCL13
Islets were isolated from NOD pancreata as previously described (9). Briefly, pancreata were macerated with scissors and then agitated at 37°C for 12 min in HBSS containing 3 mg/ml collagenase P. Islets were handpicked using a dissecting microscope and placed in overnight culture so those that extruded lymphocytes could be differentiated from those that did not. RNA was extracted from these isolated islets, and first-strand cDNA was synthesized using SuperScript II (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. This cDNA was amplified by PCR using primers for cxcl13 that incorporated most of the gene encoding the expressed molecule (200 bp). Primer pair used: 5′-TCT CTC CAG GCC ACG GTA TTC T-3′ and 5′-ACC ATT TGG CAC GAG GAT TCA C-3′. Annealing temperature was 56°C for 40 cycles. Product was loaded and run on an ethidium bromide 1.2% agarose gel.
Real-time PCR for CXCL13
Islets were isolated from pancreata of the indicated strains of mice as described previously, and RNA was extracted using an RNAqueous Micro Kit (Ambion, Austin, TX). First-strand cDNA was synthesized as previously described and was used as a template in real-time PCR using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) with the cxcl13 primers mentioned previously, and a 56°C annealing temperature, or with primers to amplify hprt as a control: 5′-AGG TTG CAA GCT TGC TGG T-3′, 5′-TGA AGT ACT CAT TAT AGT CAA GGG CA-3′, with a 52°C annealing temperature in a MyiQ single-color real-time PCR detection system (Bio-Rad, Hercules, CA). Data were analyzed with MyiQ software (Bio-Rad). The average of triplicate reactions for each individual sample was used to calculate the relative level of cxcl13 to hprt.
Immunohistochemistry and immunofluorescent staining
Frozen sections were obtained as previously described (9). Briefly, pancreata were fixed in 4% paraformaldehyde in a high-phosphate buffer at 4°C for 2 h immediately after removal from 10- to 12-wk-old female NOD mice. This was followed by overnight soak in 30% sucrose, then immersion in OCT (Sakura Finetek, Torrance, CA), and freezing/storing at −80°C. Sections, 5 μm thick, were obtained using a cryostat microtome (Leica Microsystems, Deerfield, IL). For CXCL13 staining, primary Ab was polyclonal, goat anti-mouse CXCL13 IgG (Santa Cruz Biotechnologies, Santa Cruz, CA), followed by biotin-conjugated rabbit-derived anti–goat-IgG from BD Pharmingen (San Diego, CA) (after biotin-block). Streptavidin-HRP and DAB were then applied for visualization. For immunofluorescent staining, 8 μm sections were cut and stained with anti–B220-FITC and anti-CD3 PE as previously described (9). Fluorescence microscopy with an Olympus BX60 epifluorescence microscope was coupled to a CCD camera and MagnaFire software (Optronics International, Chelmsford, MA) for image capture. Adobe Photoshop software (Adobe Systems, San Jose, CA) was used to optimize signal-to-noise ratio and to pseudocolor images, uniformly converting FITC staining to red and PE staining to blue for easier visualization. For analysis of islet organization in anti-CXCL13 treated versus isotype control-treated and untreated controls, images were made of all visible insulitis lesions on immunofluorescently stained slides of frozen sections of pancreata from three mice treated with anti-CXCL13, two isotype treated controls, and two untreated controls. Treated groups received 100 μg of anti-CXCL13 or isotype control injected i.p. three times a week from age 3–12 wk. Mice were sacrificed the day after the final injection. Each lesion was classified as disorganized, organized, or intermediate by an independent reviewer who was blinded to treatment group.
Flow cytometry
Lymphocytes suspended at 1 × 106/100 μl in a buffer solution were stained with Abs recognizing B220, IgM, and CXCR5 (BD Pharmingen). 7 Aminoactinomycin-D was used to exclude dead cells. Data were obtained using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (TreeStar, Ashland, OR). Lymphocyte gate by size and granularity is used for all plots.
CXCL13 blockade
Wild-type or VH125 transgenic NOD mice were i.p. injected with 100 μg monoclonal, rat-derived, anti-CXCL13 Ab (mAB470, R&D Systems, Minneapolis, MN) in sterile PBS three times weekly, from age 3 wk to the end of each study. Controls were treated with the same amount of isotype control (mAB006, R&D Systems), or were untreated.
BCR L chain analysis
VH125/NOD mice were treated with anti-CXCL13, three times a week as described previously, from ages 3 to 12 wk. Pancreatic RNA from these mice was reverse-transcribed and amplified using Vκ L chain (LC) primers, cloned, sequenced and analyzed for Vκ family and CDR mutations as previously described (9). Of note, NOD-specific comparisons were made for analysis of CDR mutations, to differentiate actual mutations from polymorphisms that are strain-specific for NOD κ genes (17, 18).
Mice and disease studies
NOD and C57BL/6 mice used in these studies are bred and maintained in our colony. VH125Tg/NOD mice were generated as previously described, are routinely backcrossed to wild-type NOD mice, and maintained as hemizygotes (19). Mice were housed in specific pathogen-free conditions. Disease studies were performed on VH125Tg/NOD mice injected with anti-CXCL13 blocking Abs (mAB470, R&D Systems), or isotype control Abs (mAB006, R&D Systems), or left untreated, as described previously. Blood glucose was monitored weekly and mice were considered diabetic at the first of two consecutive readings above 200 mg/dl. The Institutional Animal Care and Use Committee of Vanderbilt University has approved all procedures.
Statistics
Statistics for organization of islets and for LC distributions was performed using Stata Software, version 9 (College Station, TX) to determine p value by Fisher exact test, or Poisson regression, as noted. R software (www.r-project.org) was used to analyze incidence of SHM in islets using negative binomial regression.
Results
CXCL13 is expressed in NOD islets, and its receptor, CXCR5, is found on B lymphocytes in the pancreas
To identify chemotactic factors involved in B lymphocyte traffic to sites of inflammation in the pancreas, we initially used gene chip microarray as a screening tool, and found a 3-fold increase in transcript level for cxcl13 in pancreata from prediabetic NOD females compared with healthy pancreas from nonautoimmune C57BL/6 (not shown). To confirm these pilot studies, individual islets from prediabetic NOD and C57BL/6 females were isolated for examination by RT-PCR. Isolated islets were cultured overnight and then examined for extruded lymphocytes. Inflamed islets from NOD mice were selected for comparison with healthy islets from C57BL/6 mice. RNA encoding for CXCL13 was found to be expressed in inflamed islets isolated from three NOD mice, but not in islets from three C57BL/6 mice (Fig. 1A). Cxcl13 transcript was not detected in isolated NOD islets that were free of infiltrating lymphocytes (not shown).
FIGURE 1.

CXCL13 is expressed in inflamed islets of NOD mice. A, Gel electrophoresis showing RT-PCR products from isolated pancreatic islets using cxcl13-specific primers. First three lanes are from inflamed islets of three independent female NOD mice. Last three lanes are from islets of three independent female C57BL/6 mice. Lower lanes are controls using hprt primers from the same islets. B, Real-time PCR showing cxcl13 levels, normalized to hprt, from isolated islets. At least 20 islets were isolated from three mice per group, and pooled for each mouse. Normalized cxcl13 levels are significantly higher in islets from 12-wk-old NOD mice than from any other group. **p < 0.0001, compared with noninfiltrated NOR controls or 3-wk-old NOD mice; p < 0.001 for 6-wk-old NOD mice; p = 0.004 compared with 14-wk-old NOR mice. C, Immunohistochemical staining of frozen NOD pancreatic sections shows CXCL13+ (dark brown) cells among invading lymphocytes in early insulitis. Right panel shows control section processed with secondary reagents, but without primary anti-CXCL13 Ab (original magnification ×20).
These studies were extended using real-time PCR for comparisons of cxcl13 levels in islets at 3, 6, and 12 wk of age in NOD mice. Nonobese-resistant (NOR) mice, which are protected against disease, were used as controls. Islets were isolated from three female mice in each group, placed in overnight culture and then examined for extrusion of lymphocytes, prior to RNA extraction, to compare the levels of lymphocytic invasion in our samples. We found a little more than half the islets from 6-wk-old NOD mice extruded lymphocytes (average 54%, SD ± 17%); 12-wk-old NOD mice yielded lymphocytes from 62% (SD ± 9%) of islets and that contrasted with islets from 3-wk-old NOD mice, none of which extruded lymphocytes. NOR mice also showed lymphocyte extrusion in 26% (±14%) of islets, at 14 wk of age. Therefore, nonlymphocyte-extruding islets from additional NOR mice were obtained to provide another control sample. Islets were harvested together with any lymphocytes extruded, and pooled for each mouse, taking all islets, without selecting for inflammation (n = at least 20 islets per mouse). mRNA was obtained from pooled islets, translated to cDNA, and real-time PCR was performed for cxcl13 levels and normalized to hprt levels for each sample. Cxcl13 expression in islets from 12-wk-old female NOD mice was significantly higher than in any other group, as shown in Fig. 1B (p < 0.0001, compared with noninfiltrated NOR controls). Cxcl13 expression in islets from 3-wk-old NOD mice was comparable to that of noninfiltrated NOR controls (p = 0.76). Islets from 6-wk-old NOD mice and from 14-wk-old NOR (26% infiltrated) mice had higher levels of CXCL13 expression compared with noninfiltrated NOR islets, but not to a statistically significant degree (p = 0.33 and 0.20, respectively).
Immunohistochemical staining of frozen sections from the pancreata of 10- to 12-wk-old NOD mice further confirmed the presence of CXCL13 in lymphocytic infiltrates (Fig. 1C). CXCL13 was limited to areas of the islets that contained invading lymphocytes, and was not found in healthy islets, suggesting its presence does not precede lymphocytic invasion.
B lymphocytes in the spleen express CXCR5, the receptor for CXCL13. Lymphocytes were extracted from inflamed pancreata of 10-wk-old female prediabetic NOD mice and analyzed by flow cytometry for the presence of this receptor (Fig. 2). B cells in inflamed pancreata were found to express CXCR5, although at slightly lower levels than in the spleen (mean fluorescence intensity on B cells from spleen 46.15 ± 1.4, versus 14.81 ± 2.9 on B cells from pancreas, n = 3). Thus, both CXCL13 and its receptor, CXCR5, are present within inflamed pancreatic islets of NOD mice.
FIGURE 2.
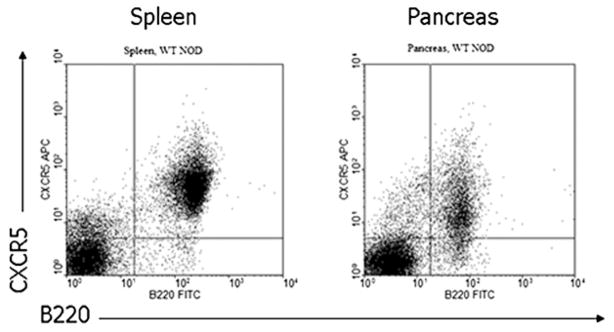
CXCL13 receptor is expressed on B cells in pancreas and spleen. Flow cytometry shows CXCR5 expression on B220+ B cells from pancreas and spleen of the same female NOD mouse. Image is representative of data from six mice.
B cell organization in islet tertiary lymphoid structures is CXCL13 dependent
Previous studies in our laboratory indicated some B cell-related therapies that are disease-protective are associated with loss of B cells from inflamed islets (20). We hypothesized that CXCL13 drives B lymphocyte recruitment to inflamed islets, and that CXCL13 blockade could reduce their presence there. NOD mice were treated with 100 μg anti-CXCL13 Ab three times weekly, beginning at age 3 wk, a time point at which few, if any, lymphocytes are present in islets, to 12 wk, when insulitis is prominent (21). Pancreata were then harvested and analyzed for alterations in lymphocyte populations by flow cytometry, H&E staining, and immunofluorescent staining of frozen sections. Insulitis development was not impaired. However, immunofluorescent staining showed the architecture of ectopic lymphocytic structures that develop in the islets was strikingly altered. Fig. 3A (and Supplemental Fig. 1) shows normal arrangement of B cells (pink) surrounding T cells (blue) in an islet from a frozen section of the pancreas of an untreated NOD mouse, whereas Fig. 3B (and Supplemental Fig. 1) shows loss of organization, with B and T cells intermingled in the islet of an NOD mouse treated with anti-CXCL13. Quantification of multiple islets by a blinded, independent scorer indicated significant differences between anti-CXCL13–treated and isotype-treated or untreated controls, with 68% of insulitis lesions from anti-CXCL13–treated mice disorganized, compared with 11% of those from isotype-treated controls, and 5.5% of untreated controls (Fig. 3D). In contrast, 66% of lesions from isotype-treated controls and 72% of those from untreated mice contained well-organized structures, compared with only 12% of those from anti-CXCL13–treated mice. Intermediate levels of organization, in which small areas of T or B cells could be seen grouped together (Supplemental Fig. 1), were seen in 21–22% of insulitis lesions from all groups (CXCL13 versus isotype control p < 0.001; CXCL13 versus untreated control p < 0.001, comparing pooled, independent, islets from: anti-CXCL13 n = 34, isotype control n = 27, untreated n = 18; anti-CXCL13–treated mice n = 3; isotype control mice n = 2; untreated controls mice n = 2).
FIGURE 3.

CXCL13 blockade does not stop B lymphocyte invasion of pancreatic islets, but scrambles tertiary lymphoid organization. A, Immunofluorescent staining of a typical, untreated, inflamed islet from a 12-wk-old female NOD mouse, with B220+ B cells (pseudocolored pink) surrounding CD3+ T cells (pseudocolored blue) (original magnification ×20). B, Immunofluorescent staining of a typical inflamed islet from a 12-wk-old female NOD mouse treated with CXCL13-blocking Ab from age 3 wk (original magnification ×20). C, Bar chart showing B lymphocytes as proportion of total lymphocytes extracted from pancreata of 12-wk-old female NOD mice treated with anti-CXCL13, isotype control, or not treated, analyzed by flow cytometry. Bar height shows average for three mice per group, with SD as indicated. D, Bar chart showing percent of inflamed islets classified as organized, disorganized, or intermediate from 12-wk-old female NOD mice treated with anti-CXCL13, isotype control Abs, or not treated. p < 0.001 for CXCL13 versus isotype-treated controls; p < 0.001 for CXCL13 versus untreated controls; p = 0.9 for isotype-treated controls versus untreated controls. Independent islets from anti-CXCL13, n = 34; isotype control, n = 27; untreated, n = 18; anti-CXCL13–treated mice, n = 3; isotype control mice, n = 2; untreated controls mice, n = 2.
However, flow cytometry analysis of lymphocytes from pancreata indicates that CXCL13 blockade did not reduce B lymphocyte proportions in pancreatic infiltrates (Fig. 3C). Thus, CXCL13 blockade curtails lymphocytic organization within insulitis lesions, without significantly impeding B lymphocyte presence there.
Disease-associated B lymphocyte selection into islets occurs despite CXCL13-blockade induced disruption of their structural organization
NOD mice that harbored an anti-insulin H chain Tg have a skewed repertoire of Vκ genes when recovered from islets. Our previous work showed that the Vκ4 LC family predominates in the pancreata of VH125/NOD mice, differing notably from spleen and draining pancreatic lymph nodes (PLNs), where, as expected for primary repertoires, the Vκ1 and Vκ9 families predominate (9, 17). To determine whether CXCL13-driven cellular organization contributes to this selection, VH125/NOD mice were treated with Ab blockade, and the Vκ composition of islet cDNA libraries was determined, as previously reported for untreated mice (9). Immunofluorescent staining on sections of the same pancreata confirmed that B lymphocyte disorganization also occurred in these mice (not shown). The pattern of LC families expressed in the pancreas (black bars) and draining pancreatic lymph nodes (gray bars) of anti-CXCL13–treated VH125/NOD mice is shown in Fig. 4A. The Vκ4 family predominated in the pancreata of CXCL13-treated mice, as previously shown in the pancreata of untreated VH125/NOD mice (9). The Vκ4 family accounted for 70% of LCs identified in this experiment, compared with 26% in draining PLNs from the same mice, a 2.7-fold difference. Vκ1 and Vκ9 family representations were lower in pancreas, relative to PLNs, 0.38-fold and 0.34-fold, respectively (overall repertoire shift, p = 0.002 by Fisher exact test, n = 53). The fold changes in the predominant Vκ families in the pancreata, relative to draining PLNs, do not significantly differ between untreated and CXCL13-treated VH125/NOD mice (Fig. 4B). The proportion of Vκ4 family genes is 2.7-fold higher in treated mice, and 3.5-fold higher in untreated mice, whereas Vκ1 and Vκ9 families are found expressed in pancreata <0.5-fold relative to PLNs, in treated and untreated mice (p = 0.066, 95% CI 0.77–1.01, by Poisson regression; mice n = 8, independent clones n = 104). Thus, the repertoire shift in the inflamed tissue relative to the secondary lymphoid organs is maintained, despite loss of the tertiary lymphoid, B cell-related morphology, indicating that intact B cell organization in TLOs is not essential for selection of the Vκ4-predominant LC repertoire found to be paired with the VH125 BCR H chain in B cells from the pancreas.
FIGURE 4.
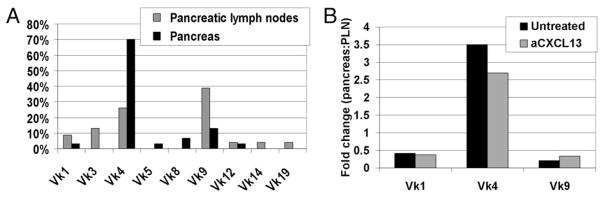
CXCL13 blockade fails to alter selection of the B cell repertoire found in pancreatic islets. A, LC gene families expressed by B cells from pancreata (black bars) and draining PLNs (gray bars) of 12-wk-old prediabetic female VH125/NOD mice treated with anti-CXCL13 from the age of 3 wk. p = 0.002 by Fisher exact test; mice, n = 3; independent clones, n = 53. B, The ratio of predominant B lymphocyte Vκ families found in pancreata, relative to those from PLNs, in untreated VH125/NOD (black bars) and anti-CXCL13–treated VH125/NOD mice (gray bars). p = 0.066; 95% CI 0.77–1.01 by Poisson regression; mice, n = 8; independent clones, n = 104.
CDR mutations in islet-derived BCRs, indicative of SHM, occur despite disrupted B lymphocyte organization in islets
B lymphocytes undergo SHM in response to activation by T cells. Our previously published analyses showed that GCs, the site of SHM, are present within well-organized TLOs in inflamed NOD islets, and that CDR mutations indicative of SHM are present in LC sequences from the pancreas (9). As GL7 staining of frozen pancreatic sections in anti-CXCL13–treated mice did not show GCs in islet infiltrates (not shown), we evaluated individual LC sequences from amplified Vκ cDNA, as described previously, for CDR mutations. Unexpectedly, 34% of LC sequences examined showed evidence of mutation by nucleotide replacement in the CDRs (Fig. 5B). Fourteen percent had one CDR mutation, 14% had two or three CDR mutations, and 7% had either four or five mutations. Seventy-one percent of these mutations resulted in amino acid replacements. This is similar to findings in untreated controls, in which 35% of LC sequences were found to contain mutations in the CDRs, with number of CDR mutations per LC ranging from one to eight as shown in Fig. 5A. The findings indicate that T–B interactions in the islets that result in SHM do not heavily depend on the structural support of the follicle-like B cell arrangement classically found in well-formed TLOs.
FIGURE 5.
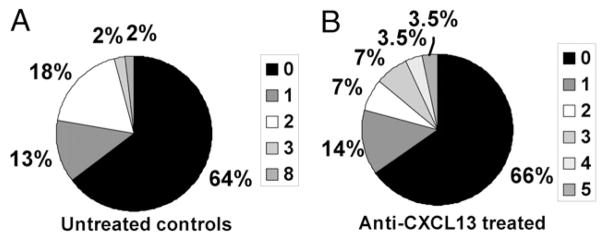
CXCL13 blockade does not significantly decrease CDR mutations found in BCR LCs from pancreata. Gene segments encoding BCR κ LCs cloned from pancreata of VH125/NOD mice were examined for evidence of somatic hypermutation as indicated by mutations in the CDRs. A, A total of 35% of BCR LC gene segments from pancreata of untreated controls have mutations in the CDR-encoding region. B, A total of 34% of BCR LC gene segments from anti-CXCL13–treated mice have mutations in the CDR-encoding regions. Keys show shading corresponding to number of mutations. Untreated, n = 45 clones; anti-CXCL13, n = 29 clones, from at least three mice per group. p = 0.861 by negative binomial regression.
Diabetes progresses despite B lymphocyte disorganization in tertiary lymphoid structures
Disease studies were performed on VH125/NOD mice treated with anti-CXCL13, isotype control, or left untreated. Female mice were injected with 100 μg monoclonal anti-CXCL13, or isotype control, in sterile PBS i.p., three times a week from the age of 3 wk. Blood glucose levels were checked weekly, and mice were considered diabetic when levels exceeded 200 mg/dl, confirmed by a second reading. The study was stopped for lack of efficacy when at least 50% of anti-CXCL13–treated mice had developed diabetes by the age of 17 wk (Fig. 6), a rate consistent with our VH125Tg/NOD colony, and no better than controls in this study (log rank p = 0.94 anti-CXCL13 versus isotype-treated controls; p = 0.27 anti-CXCL13 versus untreated controls, n = 30 mice). Pancreata from these mice harvested at the time of sacrifice and imaged using immunofluorescent staining showed disrupted TLO architecture, indicating that this dose of treatment was adequate to prevent B lymphocyte organization in islets, despite the lack of disease effect.
FIGURE 6.

CXCL13 blockade does not protect against development of diabetes. Female VH125/NOD mice were administered monoclonal anti-CXCL13 (◆) or isotype control (■), or were untreated (▲). Blood glucose levels were checked weekly, and mice diagnosed with diabetes at >200 mg/dl. Study was stopped for lack of efficacy when at least 50% of anti-CXCL13–treated mice had become diabetic. Mice, n = 10 per group. Log rank p = 0.94 CXCL13 versus isotype control treated; log rank p = 0.27 anti-CXCL13 versus untreated controls.
Discussion
This study addresses the role of B lymphocyte organization within TLOs in the autoimmune β cell attack that leads to T1D in the NOD mouse model. Organized tertiary lymphoid structures are found at the site of inflammation in multiple autoimmune diseases. TLOs are typical for fully invaded islets in the NOD mouse model of T1D, but their role in disease has not been defined. Because TLOs have been associated with selection of B cells (9) and recruitment and activation of naive T cells (6), as well as with disease progression in NOD mice, it has been hypothesized that these structures contribute to the disease process. Previous interventions that resulted in disease protection, such as lymphotoxin β receptor blockade, effectively eliminated lymphocytes from islets altogether, but could not address the role of their organization (10). The studies presented in this article use CXCL13 blockade, downstream of lymphotoxin β, to independently analyze the effects of structural organization of B lymphocytes in inflamed islets. The data provide evidence that the majority of B lymphocytes within pancreatic islets need not be organized into follicle-like structures to encourage the selection of the distinct BCR repertoire found there, or for SHM at the site, or for disease to occur.
CXCL 13 is a B lymphocyte chemoattractant critical to recruitment and retention of naive B cells to splenic follicles and B cell areas of lymph nodes, as well as to the light zones of GCs (11, 22). Because TLOs in islets mimic the architecture of secondary lymphoid organs, it was expected that CXCL13 blockade would affect B lymphocyte recruitment and activity within the islets. The discovery that neither B cell presence nor repertoire found within the islets were altered by CXCL13 blockade suggests that B cells populating the islets are not dominated by the naive recirculating population that typically homes to this chemokine in secondary lymphoid tissues. Our previous finding that B cells in the islets have a repertoire that is distinct from those in secondary lymphoid tissue, coupled with these new data showing they are not deterred from the islets by blockade of the primary homing signal for naive B cells, suggests the B cells in TLOs of T1D may not be analogous to ectopic lymph node follicles. Rather, despite their organized appearance, the B cells in this setting are more likely to have been selected in an Ag-specific way, and may home there, or remain there, in response to inflammatory chemokines, with CXCL13 serving primarily in an organizational capacity. The caveat to that would be the possibility that the dosage of Ab used, although high enough to alter cellular organization, may still have allowed some homing of naive cells to occur.
We previously discovered GCs within islet TLOs, along with molecular evidence of SHM. In the current study, we failed to locate GCs in the scrambled insulitis lesions of anti-CXCL13–treated mice, but CDR mutations indicative of SHM were still plentiful. Although SHM typically occurs in GCs, previous findings in a mouse model of rheumatoid arthritis indicate GC-independent SHM can occur (23, 24). Our findings are consistent with this concept, and suggest the T–B interactions that drive SHM within islets do not necessarily depend on the structural organization provided by ectopic B lymphocyte aggregates. The fact that we have shown oligoclonality of B cells in each islet (9), and others have shown T cell oligoclonality (25) may indicate cognate T and B clonal expansion in each islet, producing large collections of cognate pairs in close proximity to each other, obviating the need for T–B zone-specific homing signals.
In conclusion, we show in this study that CXCL13 underlies the B lymphocyte organization within TLOs in pancreatic islets, but this microarchitecture does not appear to contribute significantly to the recruitment of the skewed B cell repertoire found there, for SHM of B cell receptors indicative of T–B cellular interactions at the site, or for diabetes development. Additional questions yet to be explored include: the ways in which cellular organization affects dendritic cell and T cell pathogenic function in islet TLOs, and whether these cellular clusters may have other functions not explored in this study, such as downregulation or modulation of the autoimmune response.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants K08 DK070924 and T32 HL069765, The Vanderbilt Diabetes Center, The Vanderbilt Physician-Scientist Development Award, and the National Institutes of Health Loan Repayment Program. The Vanderbilt DNA Sequencing Facility is supported by National Institutes of Health Grants CA68485 (to the Vanderbilt-Ingram Cancer Center), DK20593 (to the Vanderbilt Diabetes Research and Training Center), and HL65962 (to the Vanderbilt Pharmacogenomics Research Center). The Vanderbilt Medical Center Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (Grant P30 CA68485) and the Vanderbilt Digestive Disease Research Center (Grant DK058404).
We are grateful to Matt Luther and Vivian Siegel for insightful discussion and critical manuscript review, and to Guowu Yu, Liping Liu, and Allison Sullivan for technical assistance. We also thank the Biostatistics Clinic, Vanderbilt School of Medicine Department of Biostatistics, and the Vanderbilt Ingram Cancer Center Microarray Shared Resource. Flow cytometry experiments were performed in the Veterans’ Administration Flow Cytometry Core and the Vanderbilt Medical Center Flow Cytometry Shared Resource.
Abbreviations used in this paper
- GC
germinal center
- LC
Ig L chain
- NOR
non-obese-resistant
- PLN
pancreatic lymph node
- SHM
somatic hypermutation
- T1D
type 1 diabetes
- TLO
tertiary lymphoid organ
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6:205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 2.Manzo A, Vitolo B, Humby F, Caporali R, Jarrossay D, Dell’accio F, Ciardelli L, Uguccioni M, Montecucco C, Pitzalis C. Mature antigen-experienced T helper cells synthesize and secrete the B cell chemoattractant CXCL13 in the inflammatory environment of the rheumatoid joint. Arthritis Rheum. 2008;58:3377–3387. doi: 10.1002/art.23966. [DOI] [PubMed] [Google Scholar]
- 3.Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. 2006;176:7676–7685. doi: 10.4049/jimmunol.176.12.7676. [DOI] [PubMed] [Google Scholar]
- 4.Manzo A, Paoletti S, Carulli M, Blades MC, Barone F, Yanni G, Fitzgerald O, Bresnihan B, Caporali R, Montecucco C, et al. Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis. Eur J Immunol. 2005;35:1347–1359. doi: 10.1002/eji.200425830. [DOI] [PubMed] [Google Scholar]
- 5.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 6.Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu YX. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 7.Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Th1-biased tertiary lymphoid tissue supported by CXC chemokine ligand 13-producing stromal network in chronic lesions of autoimmune gastritis. J Immunol. 2003;171:4359–4368. doi: 10.4049/jimmunol.171.8.4359. [DOI] [PubMed] [Google Scholar]
- 8.Barone F, Bombardieri M, Manzo A, Blades MC, Morgan PR, Challacombe SJ, Valesini G, Pitzalis C. Association of CXCL13 and CCL21 expression with the progressive organization of lymphoid-like structures in Sjögren’s syndrome. Arthritis Rheum. 2005;52:1773–1784. doi: 10.1002/art.21062. [DOI] [PubMed] [Google Scholar]
- 9.Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol. 2007;178:5643–5651. doi: 10.4049/jimmunol.178.9.5643. [DOI] [PubMed] [Google Scholar]
- 10.Wu Q, Salomon B, Chen M, Wang Y, Hoffman LM, Bluestone JA, Fu YX. Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J Exp Med. 2001;193:1327–1332. doi: 10.1084/jem.193.11.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansel KM, V, Ngo N, Hyman PL, Luther SA, Förster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 12.Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, Hamburger J, Ainsworth J, Mathews J, Salmon M, et al. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjögren’s syndrome. Arthritis Rheum. 2001;44:2633–2641. doi: 10.1002/1529-0131(200111)44:11<2633::aid-art443>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 13.Carlsen HS, Baekkevold ES, Johansen FE, Haraldsen G, Brandtzaeg P. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut. 2002;51:364–371. doi: 10.1136/gut.51.3.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148:11–23. doi: 10.1016/j.jneuroim.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 15.Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- 16.Ettinger R, Munson SH, Chao CC, Vadeboncoeur M, Toma J, McDevitt HO. A critical role for lymphotoxin-beta receptor in the development of diabetes in nonobese diabetic mice. J Exp Med. 2001;193:1333–1340. doi: 10.1084/jem.193.11.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodward EJ, Thomas JW. Multiple germline kappa light chains generate anti-insulin B cells in nonobese diabetic mice. J Immunol. 2005;175:1073–1079. doi: 10.4049/jimmunol.175.2.1073. [DOI] [PubMed] [Google Scholar]
- 18.Henry RA, Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Vκ polymorphisms in NOD mice are spread throughout the entire immunoglobulin kappa locus and are shared by other autoimmune strains. Immunogenetics. 2010 doi: 10.1007/s00251-010-0457-9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hulbert C, Riseili B, Rojas M, Thomas JW. B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol. 2001;167:5535–5538. doi: 10.4049/jimmunol.167.10.5535. [DOI] [PubMed] [Google Scholar]
- 20.Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Peritoneal B cells govern the outcome of diabetes in non-obese diabetic mice. Eur J Immunol. 2004;34:2387–2395. doi: 10.1002/eji.200324744. [DOI] [PubMed] [Google Scholar]
- 21.Miyazaki A, Hanafusa T, Yamada K, Miyagawa J, Fujino-Kurihara H, Nakajima H, Nonaka K, Tarui S. Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal study. Clin Exp Immunol. 1985;60:622–630. [PMC free article] [PubMed] [Google Scholar]
- 22.Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 23.Shlomchik MJ, Euler CW, Christensen SC, William J. Activation of rheumatoid factor (RF) B cells and somatic hypermutation outside of germinal centers in autoimmune-prone MRL/lpr mice. Ann N Y Acad Sci. 2003;987:38–50. doi: 10.1111/j.1749-6632.2003.tb06031.x. [DOI] [PubMed] [Google Scholar]
- 24.Herlands RA, William J, Hershberg U, Shlomchik MJ. Anti-chromatin antibodies drive in vivo antigen-specific activation and somatic hypermutation of rheumatoid factor B cells at extrafollicular sites. Eur J Immunol. 2007;37:3339–3351. doi: 10.1002/eji.200737752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker FJ, Lee M, Chien YH, Davis MM. Restricted islet-cell reactive T cell repertoire of early pancreatic islet infiltrates in NOD mice. Proc Natl Acad Sci USA. 2002;99:9374–9379. doi: 10.1073/pnas.142284899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.