

Summary
Oxygen is essential for multicellular existence. Its reduction to water by the mitochondrial electron transport chain forms the cornerstone of aerobic metabolism. Conditions in which oxygen is limiting for electron transport result in bioenergetic collapse in metazoans. However, compared with postnatal existence, all of mammalian development occurs in a hypoxic environment in utero. Not just an epiphenomenon, this ‘physiological hypoxia’ is required for the activation of a transcriptional response mediated by the hypoxia-inducible factor (HIF) family of transcriptional regulators that coordinates the expression of hundreds of genes, many with developmentally critical functions. Oxygen tension, therefore, is a morphogen. Understanding the physiological significance of hypoxia responses during human development and the role of the HIF family of transcriptional regulators will have important consequences for the care of preterm neonates. Defining clinical care guidelines for the proper oxygenation of critically ill neonates that take account of these observations is therefore of paramount importance. The pharmacological stabilization of HIF family members may therefore have clinical utility in premature infants in whom this important morphogen has been inactivated by exposure to supraphysiological oxygen levels.
Keywords: Hypoxia; Hypoxia-inducible factor; Necrotizing enterocolitis; Neonate, retinopathy of prematurity; Prolyl hydroxylase inhibitor; Respiratory distress syndrome
Introduction
As photosynthesizing organisms increased ambient oxygen levels in the Earth’s early atmosphere, life forms evolved that could utilize this newly oxygen-rich environment via aerobic metabolism.1 The reduction of oxygen to water by the mitochondrial electron transport chain (ETC) is able to generate the ATP needed to meet the bioenergetic demands of increasingly complex multicellular existence. However, this process is not 100% efficient, and the incomplete reduction of oxygen results in the formation of toxic reactive oxygen species (ROS) that can cause oxidative damage to cellular macromolecules. This dual nature of oxygen as an essential but potentially harmful molecule required the evolution of oxygen-sensing mechanisms to respond to high as well as to low levels of oxygen, respectively known as hyperoxia and hypoxia. However, intermediate levels of these ROS appear to have been co-opted for signaling purposes, paradoxically to signal low oxygen levels.2
In mammals, one of the most important regulators of oxygen homeostasis is the hypoxia-inducible factor (HIF) complex. HIF coordinates the myriad responses to decreased oxygen tension by promoting compensatory mechanisms acting at the cellular as well as organismal level that include enhancing the oxygen-carrying capacity of blood, decreasing cellular oxygen demand, and increasing glycolysis, to name just a few.3,4 This response is critical for many facets of normal mammalian development.4
What is HIF?
HIF is a heterodimeric protein complex composed of an α and β subunit that forms a sequence-specific DNA-binding transcription factor.5,6 Both subunits contain basic helix-loop-helix (bHLH) as well as PAS (PER, AHR, ARNT, and SIM) domains that mediate dimerization and DNA binding at the hypoxia response elements (HREs) of target genes.7–10 The identification of an HRE in the enhancer region of the erythropoietin (Epo) gene led to the discovery of HIF-1, composed of HIF-1α and ARNT. HIF-1 binding to the HRE either promotes or represses the transcription of a broad range of genes involved in maintaining biological homeostasis in response to changing oxygen levels. There are three structurally similar HIF-α proteins: HIF-1α, HIF-2α, and HIF-3α these are strictly regulated in an oxygen-dependent manner and confer oxygen regulation to the complex overall.3 HIF-β subunits, also known as aryl hydrocarbon receptor nuclear translocator (ARNT 1, 2, 3) are constitutively expressed nuclear proteins.5 Although each of the three α-subunits is known to contribute in vivo to hypoxia responses, only two of the β-subunits are thought to participate.
Normoxic degradation of HIF-α subunits
HIF-1α is constitutively transcribed and translated, but its stability and transcriptional activity are regulated by several post-translational modifications. For example, under normoxic conditions, it is hydroxylated on critical proline residues.11–13 Post-translational hydroxylation within the oxygen-dependent degradation domain (ODDD) located at the N-terminal transactivation domain (NTAD) occurs at one or both of two highly conserved proline residues. The molecular basis for this oxygen-dependent hydroxylation has been the subject of intense study over the past decade and is now known to be accomplished by members of the prolyl hydroxylase domain (PHD) family of proteins. PHD enzymes belong to a non-heme, Fe2+ and 2-oxoglutarate-dependent (a Krebs cycle intermediate) dioxygenase superfamily whose activity is dependent on oxygen availability. PHD uses both oxygen and a-ketoglutarate as substrates for the dioxygenase reaction that splits oxygen.14 One oxygen atom is transferred to hydroxylate the proline residue and the other reacts with 2-oxoglutarate to form succinate and carbon dioxide. The enzymes therefore require molecular oxygen for their activity, but whether they represent the elusive cellular oxygen sensors is not clear, as oxygen concentrations within the physiologic range are unlikely to directly affect their activities.
Prolyl-4-hydroxylation of the ODDD creates a binding site for the von Hippel–Lindau tumor suppressor protein (pVHL), which is a component of an E3 ubiquitin ligase complex.15 As a result, HIF-1α is polyubiquitinated at several sites and rapidly targeted for proteosomal degradation. Mutation of both proline residues disrupts interaction of HIF-1α with pVHL and increases its stability in normoxic conditions whereas mutating one proline residue only partially stabilizes HIF-1α.16 Mutations or deficiencies of PHD or pVHL lead to accumulation of high levels of HIF-α proteins during normoxia. 2-Oxoglutarate analogs, which inactivate PHDs, also increase the half-life of HIF-1α11–13 which is normally <5 min in normoxic conditions.
Additionally, factor inhibiting HIF (FIH) hydroxylates an asparaginyl moiety at the C-terminus of the HIF-1α and HIF-2α subunits. Hydroxylation of Asp803 blocks interaction of the C-terminal transactivation domain (CTAD) with the coactivator p300 and its paralogue CREB-binding protein (CBP).17,18 FIH-1, like PHD family members, is an Fe2+- and 2-oxoglutarate-dependent dioxygenase. Upon availability of oxygen and Fe2+, FIH-1 hydroxylates the conserved asparaginyl residue within the CTAD and prevents interaction with coactivator p300 and CBP due to steric interference.19
PHD isoforms
The PHD family consists of three members, PHD1, PHD2 and PHD3 (also known as HIF prolyl hydroxylase (HPH) or Egg-laying Nine (EGLN), HPH3/EGLN2, HPH2/EGLN1 and HPH1/EGLN3, respectively. The various PHD isoforms differ with regard to their tissue distribution, protein structure, cellular localization, protein interactions and hydroxylation of HIF-α isoforms.20 PHDs are regulated by the presence of oxygen, ferrous iron, and 2-oxoglutarate. Ferrous iron (Fe2+) is found in the active site of the enzyme’s catalytic triad comprised of two histidines and one aspartate residue. When diatomic molecular oxygen is cleaved, one oxygen atom is transferred to the proline residue. At the same time, the second oxygen atom interacts with 2-oxoglutarate which decarboxylates to form succinate and carbon dioxide.14 These enzymes can therefore be inhibited via iron chelation. Structural analogs of 2-oxoglutarate such as dimethyl-oxalyl-glycine (DMOG) have also been shown to inhibit PHD activity.
PHD1 is a 43.6 kDa enzyme highly expressed in the testis, with lower levels reported in the kidney, heart, and liver.21 Unlike PHD2 or PHD3, PHD1 is constitutively expressed and is not induced by hypoxia or other hypoxia mimetics (DFO and CoCl2) but is responsive towards estrogen. PHD1 hydroxylates HIF-1α at both the N-terminal ODDD at Pro564 and C-terminal ODDD at Pro402. PHD2 is known as the dominant prolyl-4-hydroxylase.20 It is ubiquitously expressed but shows highest expression in the heart with moderate expression in the brain. A majority of PHD2 is localized in the cytoplasm with a smaller population found in the nucleus. The 46 kDa enzyme preferentially regulates HIF-1α over HIF-2α and hydroxylates both the N- and C-terminus ODDD. Aditionally, the HRE that is recognized by HIF-1 is also found in gene regulatory regions of Phd2 and Phd3, thereby conferring oxygen sensitivity to their expression. In-vivo studies confirm that PHD2 is the most important PHD family member during development. PHD2−/− embryos all died between E12.5 and E14 whereas Phd1−/− and Phd3−/− embryos were viable.22 Somatic Phd2−/− mouse models reveal elevated EPO levels in the kidney due to increased HIF-α levels.23 Phd1−/−/Phd3−/− double knock-out mice do not exhibit erythropoiesis phenotypes.
By contrast with PHD2, PHD3 is distributed uniformly between the nucleus and cytoplasm. It is upregulated by hypoxia and other hypoxia mimetics, but still shows considerable hydroxylase activity under hypoxic conditions for an oxygen-dependent enzyme.24 Similar to PHD1, the 27.3 kDa enzyme displays greater activity towards HIF-2α over HIF-1α. PHD2 preferentially hydroxylates the C-terminal ODDD at Pro564 in a sequence-specific manner, whereas PHD1 and PHD3 recognize the ODDD based on their spatial conformations.
HIF-α family members
There are three main HIF-α subunits. HIF-1α is ubiquitously expressed whereas the related HIF-2α exhibits a much more tissue-restricted expression pattern.25 HIF-2α, also known as endothelial PAS domain protein 1(EPAS1), is structurally similar and can regulate many of the same target genes as HIF-1α, although HIF-α family members can also act in different ways.26 In cells that express both HIF-1α and HIF-2α, each appears to regulate overlapping as well as distinct functions. In endothelial cells, for example, HIF-1α controls proliferation, metabolism, and survival whereas HIF-2α controls cell migration, adhesion, and vessel integrity.27
In murine embryos, HIF-2α, but not HIF-1α, induces Oct-4, a stem cell factor important in the self-renewal of undifferentiated embryonic stem cells.28 These discovery of specific HIF-2α target genes that promote growth and repress differentiation is consistent with possible HIF-2α involvement in tumorigenesis. pVHL-defective renal carcinomas overproduce HIF-1α and HIF-2α or just HIF-2α alone. Inhibition of HIF-2α, but not HIF-1α, can override the tumor suppressor activity of pVHL.29 HIF-1α and HIF-2α can also perform opposite roles. For instance, HIF-1α inhibits c-Myc activity by interacting through its PAS B domain and displaces Myc from regulatory sequences.30 As a result, the c-Myc-repressed cyclin-dependent kinase inhibitors p21 and p27 are upregulated, and c-Myc-activated cyclin D2 and E2F are downregulated. HIF-2α, therefore, potentiates c-Myc activity to enhance cell growth. Not much is known about the HIF-3α subunit other than that it can act in a dominant negative fashion as an inhibitory PAS domain protein by repressing HIF-1α transcriptional activity.31
Oxygen-sensing mechanisms
A large body of evidence suggests that reactive oxygen species can modulate HIF activity. Many intracellular sources of ROS such as the endoplasmic reticulum, peroxisomes, and plasma membrane NADPH oxidases have been cited, but the most significant contribution comes from the mitochondrial electron transfer chain (ETC). The ETC consists of five multiprotein complexes that are embedded within the inner mitochondrial membrane.2 Complexes I and II oxidize the energy-rich molecules NADH and FADH2, respectively, and transfer the resultant electrons to ubiquinol that then shuttles them to complex III. Complex III ferries these electrons across the inner mitochondrial membrane to cytochrome c, which carries them on to complex IV. Complex IV then uses these electrons to reduce oxygen to water. Each of these steps is associated with the pumping of protons into the intermembrane space, generating a proton gradient, the dissipation of which is coupled to complex V to drive the energy-costly phosphorylation of ADP to ATP. Early studies with ETC inhibitors, or cells depleted of mitochondrial DNA and hence critical electron transport chain components, laid the groundwork for more recent studies supporting a role for mitochondrial electron transport in hypoxic stabilization of HIF-1α.32 These experiments showed that hypoxia could paradoxically trigger an increase in mitochondrial ROS production, and that antioxidant treatment could inhibit hypoxic HIF activation. Subsequently, it was shown that exogenous hydrogen peroxide could stabilize HIF-1α during normoxia33 and cells depleted of mitochondrial DNA or critical ETC components failed to produce ROS during hypoxia.34–36
Though there are many possible mechanisms by which mitochondrial ROS could regulate HIF-α subunit stability, direct inhibition of PHD activity would be quite attractive. In fact, one report showed that elevated ROS levels led to enzyme inactivation of PHD2 by catalyzing a ferrous oxidation in its active domain.37 It has been proposed that ROS alter PHD2 at important disulfide bonds to inhibit its activity, a regulatory mechanism similarly seen in protein tyrosine phosphatases.38,39 Alternatively, ROS might initiate a signal transduction cascade that post-translationally modifies and inhibits PHD2 via the p38 mitogen-activated protein kinase family. In support of this, mouse embryonic fibroblasts lacking p38 or its upstream effector MKK3/6 failed to stabilize HIF-1α in response to hypoxia-generated ROS.40
HIF target genes and clinical implications of HIF activity in neonates
Hypoxia is defined by oxygen tensions below normal physiological levels that trigger a compensatory cellular response. Hypoxia, therefore, is context dependent. At term, following the first breath of a newborn infant, oxygen transport mechanisms in the lung enable a rapid increase in blood oxygen content. Prior to this point, from conception through birth, all of mammalian development occurs under oxygen tensions that are significantly more hypoxic than postnatal existence.41 Partial pressures of oxygen within the fetal circulation, as well as within the uterus at the time of implantation, rarely exceed 20 mmHg. This reflects a ‘physiological hypoxia’ that is normoxic for the developing human. And although the elevated hematocrit and enhanced oxygen affinity of fetal hemoglobin enable similar total oxygen contents between the maternal and fetal circulations, the partial pressure of oxygen within fetal tissues – the freely diffusible component able to interact with the mitochondrial ETC within developing tissues – is usually in the teens. Thus, following delivery of a preterm neonate and the institution of pulmonary oxygenation strategies, a profound change in the cellular oxygen environment of infants ensues. Understanding the consequences of this environmental insult is of paramount importance for improving the care of preterm infants in the intensive care nursery.
Under hypoxic conditions, oxygen-dependent PHDs and FIHs are inactivated, HIF-1α is no longer hydroxylated and can translocate to the nucleus, where it heterodimerizes with ARNT to activate the transcription of many target genes.5 HIF-1 can activate the transcription of many genes that regulate diverse functions including erythropoiesis and iron transport, angiogenesis, and glucose metabolism, to name just a few.3 Following the transition to postnatal existence and the associated exposure to increased oxygen levels, many of these pathways are prematurely inactivated in preterm infants.
Erythropoiesis, iron transport and anemia
Low oxygen conditions increase blood oxygen-carrying capacity by upregulating the expression of genes involved in erythropoiesis and iron metabolism. EPO is a glycoprotein hormone initially produced by the fetal liver and then by specialized kidney cells that is necessary for red blood cell survival.42 Hypoxia orchestrates the transcription of Epo and Epo receptor (EpoR) genes43 as well as genes involved in hemoglobin synthesis such as transferrin (Tf) and transferrin receptor (Tfr).44,45 Tfr is a cell-membrane-associated glycoprotein that regulates cellular uptake of iron from transferrin. Tfr transports ferric iron (Fe3+) bound to transferrin into cells that can then be incorporated into the heme group of hemoglobin in erythrocytes. Hypoxic induction of these pathways is one of the main mechanisms contributing to elevated fetal hematocrits in utero.1 The developmental requirement for HIF activity in regulating erythropoiesis was demonstrated in Arnt-null embryos that showed erythropoietic defects as well as abnormal expression of iron metabolism proteins. HIF deficiency affected erythropoiesis during embryo and yolk sac development.46 Interestingly, postnatal deletion of HIF-2α showed that it is the key HIF-α subunit responsible for regulating adult erythropoeisis, especially under stress conditions.47 Therefore, in addition to the clinically indicated blood sampling that results in significant anemia requiring blood transfusions in preterm neonates, exogenous oxygen administration, by suppressing HIF activity, likely also plays a contributory role in promoting clinically significant anemia in the ICN.
Angiogenesis and retinopathy of prematurity
In addition to residing in a globally hypoxic environment, proliferating tissues in utero also experience localized gradients of oxygen tension due to an increase in cell mass and the resultant cell distances from nearby vasculature. The globally hypoxic nature of the in-utero environment ensures that small changes in cellular oxygen tension are met with rapid and compensatory activation of HIF-dependent gene expression. This is because HIF-α stability begins to increase at approximately 6% O2, representing the high end of in-utero O2 tensions. Between 0.5% and 6%, small decreases in O2 tension result in immediate and at times exponential increases in HIF-α subunit stability6, ensuring rapid and well-coordinated hypoxia responses. Local tissue responses to decreased oxygenation induce HIF-1 activity and trigger expression of angiogenic factors such as vascular endothelial growth factor (VEGF). VEGF promotes migration of endothelial cells in a process called angiogenesis to promote blood vessel growth to increase oxygen delivery. Importantly, Hif-1α−/− or Arnt−/− embryos exhibit failed vascularization and embryonic lethality due to impaired VEGF production.48–51
The eye develops rapidly during gestation with blood supply to the optic nerve starting at about 16 weeks and blood vessel growth towards the retina continuing until birth. In premature neonates, this normal blood vessel growth is disrupted and ultimately results in dysregulated compensatory retinal neovascularization, which contributes to retinopathy of prematurity (ROP). ROP is a disease that occurs in premature babies and is the leading cause of blindness among children in the USA. In the retina, expression of HIF-α proteins and Vegf mRNA patterns are temporally and spatially correlated.52 Both HIF-1α and -2α appear to play contributory roles. In addition to VEGF, the VEGF receptor FLT-1 is also induced when endothelial cells are exposed to hypoxic conditions.53 Hif-2α+/+ mice were indistinguishable from Hif-2α+/− mice under normoxia. Interestingly, on a particular genetic background, a subset of Hif-2α−/− embryos were viable and resulted only in minor vascular phenotypic changes, implying that HIF-2α does not play a major role in developmental vascularization. However, Hif-2α+/− and Hif-2α−/− mice54 had reduced retinal neovascularization and absent or diminished levels of angiogenic factors when subjected to an oxygen-induced retinopathy protocol. Conditional knockdown of Hif-2α expression in the retina suggested that Epo was the critical target gene responsible for the effect of HIF-2α, not Vegf.55
Glucose metabolism and diabetes
Metabolic adaptations are induced by hypoxia as individual cells reprogram their glucose metabolism from oxidative to glycolytic pathways. The oxygen-dependent tricarboxylic acid (TCA) or Krebs cycle produces 38 ATP molecules per glucose molecule whereas anaerobic metabolism provides only two ATP molecules. As a result, hypoxic cells increase their glucose uptake and reduce oxygen consumption in an attempt to maintain energy homeostasis. HIF-1 plays a key role by up-regulating the expression of glycolytic enzymes and glucose transporters such as GLUT1 and GLUT3.56 HIF-1 mediates transcription of the mitochondrial enzyme pyruvate dehydrogenase kinase 1 (PDK-1) to shunt pyruvate away from mitochondria and reducing cellular oxygen consumption. ARNT activity in the pancreas has been shown to be important for the maintenance of euglycemia,57 as has hepatic ARNT activity.58 Therefore ARNT and the ARNT-like BMAL1 have been proposed as genetic loci associated with diabetes in adults.59–61 Whether dysregulated HIF activity contributes to the frequently observed glucose instability in premature neonates is currently unclear.
Lung development and respiratory distress syndrome
Lung disease is another common problem facing neonatologists due to arrested pulmonary vascular development and angiogenesis. Affecting about 1% of all births and 60% of premature infants born at <32 weeks of gestation and weighing <1000 g, respiratory distress syndrome (RDS) is a major cause of neonatal morbidity and mortality.62 In addition to its role in tissue angiogenesis, the three VEGF also stimulate vascularization, branching morphogenesis, and alveolar development in the lung.63 VEGF is a major factor regulating fetal lung maturation and is tightly regulated by HIF activity. HIF-1α expression levels decreased by almost 80% and HIF-2α levels decreased by 55% in the lungs of RDS lambs in high oxygen concentrations following premature birth along with concomitant decreases in VEGF mRNA. The inhibition of PHDs using pharmological inhibitors DMOG and FG-4095 show parallel increases of HIF-1α, -2α and VEGF.64 Gene deletion studies show that HIF-1α and HIF-2α are critical for normal pulmonary development; however, similar to what was found in retinal development, HIF-1α seems to act as the primary player in fetal lung development whereas HIF-2α functions in the fine-tuning of vascularization. Surprisingly, HIF-2α-deficient mice had reduced VEGF levels in alveolar cells that caused fatal RDS in neonatal mice due to insufficient surfactant production,65 suggesting that HIF-2α has a greater role than just promoting lung vascularization. In these studies, mice that were delivered prematurely exhibited improved alveolar maturation following intratracheal VEGF administration. Inhibition of VEGF also impairs normal pulmonary alveolar and vascular development in a rat model.66
Inflammation and necrotizing enterocolitis
Necrotizing enterocolitis (NEC) is a disease of prematurity wherein the lining of the intestinal wall undergoes necrosis and results in tissue death.67 This serious life-threatening gastrointestinal disorder has no definitive etiology but it is generally believed that decreased blood flow due to unknown causes damages the bowels and increases intestinal permeability and susceptibility to bacterial invasion. However, changes in the oxygen environment of the developing intestinal epithelium may also be playing contributory roles. For example, in mouse models of experimental colitis, HIF activity has been shown to be protective.68 HIF-deficient intestines exhibited more severe features of intestinal colitis compared with their wild-type counterparts and HIF-overexpressing animals were protected. The intestinal epithelial barrier-protecting agents Mucin 3 and Trefoil Factor are induced by hypoxia in an HIF-dependent manner,69,70 suggesting a direct link between HIF activity and intestinal barrier function. Additionally, HIF family members have been shown to play important roles in innate immunity.71 Conditional gene targeting in myeloid lineages has revealed important roles for HIF activity in monocytes/macrophages as well as neutrophils. HIF promotes the bactericidal properties of phagocytic cells and enhances the innate immune functions of dendritic cells, mast cells and epithelial cells. Dynamic changes in HIF levels are seen in the context of various infectious states. This effect appears at least in part to be mediated via cross-talk between HIF and nuclear factor (NF)-κB-dependent transcriptional pathways.72 NF-κB, critical regulator of the innate immune response, has been shown to be critical for hypoxic HIF activity in the innate immune system, formerly linking ancient immune responses with ancient oxygen-sensing pathways. Taken together, these studies suggest that the in-utero hypoxic environment may be protective for intestinal epithelial integrity and barrier function and that premature exposure to elevated oxygen tensions in the setting of preterm birth may predispose these infants to NEC.
PHD inhibitors in the ICN
Inhibition of developmentally critical HIF activity due to exogenous oxygen administration in preterm neonates contributes to their various comorbidities. It is therefore reasonable to speculate that, in addition to decreasing oxygen use, pharmacological stabilization of HIF family members should produce significant benefits for this at-risk population. In an oxygen-induced retinopathy mouse model, for example, PHD inhibition significantly attenuated the degree of disease.73 In fact, a single dose of PHD inhibitor produced a more significant protective effect than multiple rounds of EPO or VEGF administration, highlighting the pleiotropic effects of oxygen exposure and HIF activity in developing neonates. PHD inhibition also enhances lung angiogenesis in a primate model of bronchopulmonary dysplasia, likely due to increased VEGF production.74,75 Development of lung microvasculature is critical for distal airway formation and therefore PHD inhibitors may play a protective role in promoting normal lung development in premature neonates. Therefore, the time to seriously consider PHD inhibitors in the intensive care nursery is here. While decreasing the oxygen exposure of premature neonates should still be the primary goal, PHD inhibition could have a significant impact on outcomes for premature infants.
Practice points
Oxygen is a morphogen.
Human development occurs under a physiological hypoxia and preterm delivery results in premature exposure to supraphysiological oxygen levels.
Inactivation of HIF by exogenous oxygen administration impairs retinal vascularization, lung development and hematopoiesis and possibly contributes to the etiology of NEC.
Research directions
Test the utility of PHD inhibitors in the neonatal intensive care setting to activate HIF-dependent gene expression in premature neonates.
Develop biomarkers of lung disease and NEC using genomics as well as proteomics and mass spectrometry-based approaches to track disease severity, progression and response to PHD inhibition.
Figure 1.
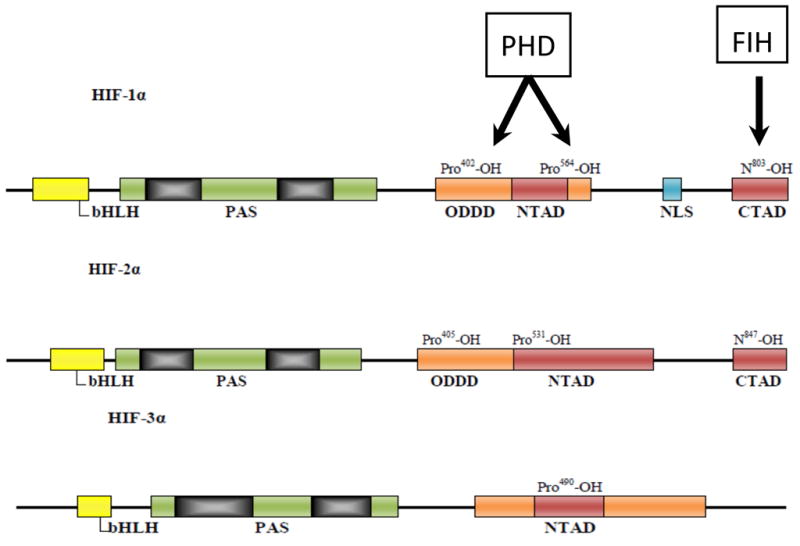
Schematic diagram of hypoxia-inducible factor (HIF)-α subunit molecular structure and interacting regulatory hydroxylases. HIF-1α belongs to the basic helix-loop-helix (bHLH) and PSA (PER–ARNT–SIM) protein family. All HIF-α subunits contain an N-terminal transactivation domain (NTAD) within the oxygen-dependent degradation domain (ODDD) that mediates oxygen stability through the hydroxylation of proline residues via prolyl hydroxylase domains (PHD). The C-terminal transactivation domain (CTAD) region found only in HIF-1α and HIF-2α contains an asparagine residue that is hydroxylated by factor inhibiting HIF (FIH) and prevents recruitment of p300/CBP transcriptional activators.
Figure 2.

Outline of hypoxia-inducible factor (HIF)-α regulation. During normoxia, prolyl hydroxylase domains (PHD) hydroxylate HIF-α on proline residues in the presence of Fe2+ and 2-oxoglutarate. Hydroxylated HIF-α is recognized by the E3 ubiquitin ligase von Hippel–Lindau tumor suppressor protein (pVHL) and is ubiquinated and targeted for proteasomal degradation. During hypoxia, mitochondria-generated reactive oxygen species (ROS) can lead to the inhibition of PHD activity via unknown mechanisms, allowing HIF-1α to translocate to the nucleus, dimerize with HIF-1β, bind to the HRE, and induce expression of target genes such as Vegf, Glut-1 and Pdk-1. Hypoxia mimetics such as dimethyl-oxalyl-glycine (DMOG) display similar effects of blocking PHD hydroxylation.
Acknowledgments
Funding sources
None.
Footnotes
Conflict of interest statement
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maltepe E, Saugstad OD. Oxygen in health and disease: regulation of oxygen homeostasis – clinical implications. Pediatr Res. 2009;65:261–8. doi: 10.1203/PDR.0b013e31818fc83f. [DOI] [PubMed] [Google Scholar]
- 2.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–6. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 4.Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev Cell. 2009;17:755–73. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 7.Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3″ to the human erythropoietin gene. Proc Natl Acad Sci USA. 1991;88:5680–4. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol. 2000;59:47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- 9.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 10.Pugh CW, Ebert BL, Ebrahim O, Maxwell PH, Ratcliffe PJ. Analysis of cis-acting sequences required for operation of the erythropoietin 3′ enhancer in different cell lines. Ann NY Acad Sci. 1994;718:31–9. doi: 10.1111/j.1749-6632.1994.tb55701.x. discussion 9–40. [DOI] [PubMed] [Google Scholar]
- 11.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 12.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 13.Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 14.Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun. 2005;338:617–26. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 15.Kim W, Kaelin WG., Jr The von Hippel–Lindau tumor suppressor protein: new insights into oxygen sensing and cancer. Curr Opin Genet Dev. 2003;13:55–60. doi: 10.1016/s0959-437x(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 16.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lando D, Peet DJ, Gorman JJ, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain – a hypoxic switch. Science. 2002;295(5556):858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molec Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–28. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- 21.Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80:421–6. doi: 10.1139/o02-115. [DOI] [PubMed] [Google Scholar]
- 22.Takeda K, Ho VC, Takeda H, et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor a levels in mice lacking prolyl hydroxylase domain protein 2. Molec Cell Biol. 2006;26:8336–46. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minamishima YA, Moslehi J, Bardeesy N, et al. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–44. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong GH, Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008;15:635–41. doi: 10.1038/cdd.2008.10. [DOI] [PubMed] [Google Scholar]
- 25.Jain S, Maltepe E, Lu MM, Simon C, Bradfield CA. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech Dev. 1998;73:117–23. doi: 10.1016/s0925-4773(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 26.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Molec Cell Biol. 2003;23:9361–74. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skuli N, Liu L, Runge A, et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood. 2009;114:469–77. doi: 10.1182/blood-2008-12-193581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Covello KL, Kehler J, Yu H, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–70. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel–Lindau-associated renal cell carcinoma. Molec Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414(6863):550–4. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 32.Chandel NS, Maltepe E, Goldwasser E, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 34.Brunelle JK, Bell EL, Quesada NM, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–14. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Mansfield KD, Guzy RD, Pan Y, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–9. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–94. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–7. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 39.Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7:560–77. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 40.Emerling BM, Platanias LC, Black E, et al. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Molec Cell Biol. 2005;25:4853–62. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okazaki K, Maltepe E. Oxygen, epigenetics and stem cell fate. Regen Med. 2006;1:71–83. doi: 10.2217/17460751.1.1.71. [DOI] [PubMed] [Google Scholar]
- 42.Goldwasser E. From protein to gene to protein: the molecular biology of erythropoietin. Am J Kidney Dis. 1991;18(4 Suppl 1):10–3. [PubMed] [Google Scholar]
- 43.Yoon D, Pastore YD, Divoky V, et al. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem. 2006;281:25703–11. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- 44.Rolfs A, Kvietikova I, Gassmann M, Wenger RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. 1997;272:20055–62. doi: 10.1074/jbc.272.32.20055. [DOI] [PubMed] [Google Scholar]
- 45.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274:24142–6. doi: 10.1074/jbc.274.34.24142. [DOI] [PubMed] [Google Scholar]
- 46.Adelman DM, Maltepe E, Simon MC. Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 1999;13:2478–83. doi: 10.1101/gad.13.19.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gruber M, Hu CJ, Johnson RS, et al. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. 2007;104:2301–6. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386(6623):403–7. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 49.Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Dev Biol. 1997;191:297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- 51.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozaki H, Yu AY, Della N, et al. Hypoxia inducible factor-1alpha is increased in ischemic retina: temporal and spatial correlation with VEGF expression. Invest Ophthalmol Vis Sci. 1999;40:182–9. [PubMed] [Google Scholar]
- 53.Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem. 1997;272:23659–67. doi: 10.1074/jbc.272.38.23659. [DOI] [PubMed] [Google Scholar]
- 54.Ding K, Scortegagna M, Seaman R, Birch DG, Garcia JA. Retinal disease in mice lacking hypoxia-inducible transcription factor-2alpha. Invest Ophthalmol Vis Sci. 2005;46:1010–6. doi: 10.1167/iovs.04-0788. [DOI] [PubMed] [Google Scholar]
- 55.Morita M, Ohneda O, Yamashita T, et al. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003;22:1134–46. doi: 10.1093/emboj/cdg117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–25. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 57.Gunton JE, Kulkarni RN, Yim S, et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell. 2005;122:337–49. doi: 10.1016/j.cell.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 58.Wang XL, Suzuki R, Lee K, et al. Ablation of ARNT/HIF1beta in liver alters gluconeogenesis, lipogenic gene expression, and serum ketones. Cell Metab. 2009;9:428–39. doi: 10.1016/j.cmet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hung MS, Avner P, Rogner UC. Identification of the transcription factor ARNTL2 as a candidate gene for the type 1 diabetes locus Idd6. Hum Mol Genet. 2006;15:2732–42. doi: 10.1093/hmg/ddl209. [DOI] [PubMed] [Google Scholar]
- 60.Woon PY, Kaisaki PJ, Braganca J, et al. Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci USA. 2007;104:14412–7. doi: 10.1073/pnas.0703247104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das SK, Sharma NK, Chu WS, Wang H, Elbein SC. Aryl hydrocarbon receptor nuclear translocator (ARNT) gene as a positional and functional candidate for type 2 diabetes and prediabetic intermediate traits: mutation detection, case–control studies, and gene expression analysis. BMC Med Genet. 2008;9:16. doi: 10.1186/1471-2350-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eichenwald EC, Stark AR. Management and outcomes of very low birth weight. N Engl J Med. 2008;358:1700–11. doi: 10.1056/NEJMra0707601. [DOI] [PubMed] [Google Scholar]
- 63.Carpenter T, Schomberg S, Steudel W, et al. Endothelin B receptor deficiency predisposes to pulmonary edema formation via increased lung vascular endothelial cell growth factor expression. Circ Res. 2003;93:456–63. doi: 10.1161/01.RES.0000090994.15442.42. [DOI] [PubMed] [Google Scholar]
- 64.Asikainen TM, Ahmad A, Schneider BK, White CW. Effect of preterm birth on hypoxia-inducible factors and vascular endothelial growth factor in primate lungs. Pediatr Pulmonol. 2005;40:538–46. doi: 10.1002/ppul.20321. [DOI] [PubMed] [Google Scholar]
- 65.Compernolle V, Brusselmans K, Acker T, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–10. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 66.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol. 2002;283:L555–62. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 67.Henry MC, Moss RL. Necrotizing enterocolitis. Annu Rev Med. 2009;60:111–24. doi: 10.1146/annurev.med.60.050207.092824. [DOI] [PubMed] [Google Scholar]
- 68.Karhausen J, Furuta GT, Tomaszewski JE, et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Louis NA, Hamilton KE, Canny G, et al. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–27. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 70.Hernandez C, Santamatilde E, McCreath KJ, et al. Induction of trefoil factor (TFF)1, TFF2 and TFF3 by hypoxia is mediated by hypoxia inducible factor-1: implications for gastric mucosal healing. Br J Pharmacol. 2009;156:262–72. doi: 10.1111/j.1476-5381.2008.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–17. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sears JE, Hoppe G, Ebrahem Q, Anand-Apte B. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc Natl Acad Sci USA. 2008;105:19898–903. doi: 10.1073/pnas.0805817105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Asikainen TM, Chang LY, Coalson JJ, et al. Improved lung growth and function through hypoxia-inducible factor in primate chronic lung disease of prematurity. FASEB J. 2006;20:1698–700. doi: 10.1096/fj.06-5887fje. [DOI] [PubMed] [Google Scholar]
- 75.Asikainen TM, Waleh NS, Schneider BK, Clyman RI, White CW. Enhancement of angiogenic effectors through hypoxia-inducible factor in preterm primate lung in vivo. Am J Physiol Lung Cell Mol Physiol. 2006;291:L588–95. doi: 10.1152/ajplung.00098.2006. [DOI] [PubMed] [Google Scholar]