

Summary
Membrane proteins are important pharmaceutical targets, but they pose significant challenges for fragment based drug discovery approaches. Here we present the first successful use of biophysical methods to screen for fragment ligands to an integral membrane protein. The E. coli inner membrane protein DsbB was solubilized in detergent micelles and lipid bilayer nanodiscs. The solubilized protein was immobilized with retention of functionality and used to screen 1,071 drug fragments for binding using Target Immobilized NMR Screening. Biochemical and biophysical validation of the 8 most potent hits revealed an IC50 range of 7 to 200 μM. The ability to insert a broad array of membrane proteins into nanodiscs, combined with the efficiency of TINS, demonstrates the feasibility of finding fragments targeting membrane proteins.
Keywords: Fragment-based drug discovery, immobilization, membrane proteins, DsbB, OmpA, Target Immobilized NMR Screening, Nanodisc
Introduction
With 60% of currently marketed drugs targeting membrane proteins1, it is clear that finding small molecules to modulate the function of such proteins is essential. High throughput screening (HTS) methods have been successful in identifying such compounds, but because the methods of detection rely on functional assays, they are generally only sensitive to submicromolar interactions. Such relatively tight interactions are generally only observed for larger compounds (300–500 Da). However, it has proved challenging to simultaneously optimize potency and absorption, distribution, metabolism, and excretion (ADME) properties of these “lead-like” or “drug-like” compounds. Furthermore, such large compounds inefficiently explore the binding sites of proteins2. Fragment-based drug discovery (FBDD) has become a powerful complementary approach to HTS for generating novel chemical modulators of pharmaceutical targets. FBDD screens small libraries (1,000–20,000 compounds) of so-called drug “fragments” that are often described by a “rule of threes”3 (Ro3, Mr < 300 Da, cLogP < 3, H-bond donors < 3, H-bond acceptors < 3, number of rotatable bonds < 3 and TPSA (total polar surface area) < 60 Å2) for binding to the target. Ro3 compliant compounds typically bind the target with KD greater than 10 μM. In order to detect such weak binding, sensitive biophysical techniques are typically required, particularly when the target is not an enzyme. Commonly used techniques for detecting fragment binding include NMR, X-ray crystallography and surface plasmon resonance (SPR)4.
Although biophysical methods have been successfully applied to an array of soluble protein targets5, they have failed in one way or another when applied to membrane proteins. There are two primary reasons for this failure: insufficient quantity of the target and problems related to the solubilization media. Many biophysical methods require tens or even hundreds of mg of purified, functional protein and most membrane proteins are difficult to produce in these quantities. However, recent advances have enabled the production of low mg quantities of a variety of MPs6–8. Membrane proteins that can be produced in sufficient quantity must then be solubilized in a surfactant while maintaining their functional state, which is also often challenging. Finally, non-specific partitioning of fragments into the surfactant has been a severe problem leading to high levels of false positives.
The use of detergent micelles to solubilise MPs has only met limited success in retaining the native function of the protein while at the same time the micelles often interfere with biophysical assays. A possible solution to this bottleneck would be to employ non-detergent media to functionally solubilize MPs. The Nanodisc (ND) has been developed as an alternative, surfactant free approach to solubilize MPs. NDs consist of a lipid bilayer that is surrounded by an amphiphilic α-helical membrane scaffold protein (MSP). A variety of proteins have been functionally solubilized in NDs9–11, which are much better mimics of the native membrane than detergent micelles. However, the suitability of NDs for biophysical assays of ligand binding to MPs has yet to be determined.
We have developed an NMR-based fragment screening approach which has proven capable of overcoming many of the challenges posed by membrane proteins12. The approach, called Target Immobilized NMR Screening (TINS)13, involves immobilizing a target and a reference in two compartments of a dual-cell sample holder14 and simultaneously injecting mixtures of fragments in an automated process. For each mixture a 1D 1H NMR spectrum is recorded while fragment binding to the target protein results in a decrease in peak amplitude. The reference, which is selected for minimal specific small molecule binding, serves to cancel out non-specific binding of fragments to protein surfaces. Hits can therefore be detected by comparing spectra of the compounds recorded in the presence of the target to those recorded in the presence of the reference. By repeatedly using the same sample to screen the entire fragment collection (>1,000 compounds), typically only ~25 nmol of protein is required, thus bringing many MPs within the requirements for TINS. Furthermore, the reference system is expected to account for non-specific binding of fragments to the media in which the membrane protein is solubilized.
We sought to apply TINS to a bona fide, integral membrane pharmaceutical target that could be functionally solubilized in detergent micelles and NDs. The inner membrane protein of Escherichia coli Disulphide bond forming protein B (DsbB), and its homologs in other gram-negative bacteria, is an oxidoreductase that is essential for protein disulfide bond formation in the periplasm15. Periplasmic DsbA functions as the catalyst for protein disulfide bond formation and is reoxidized by DsbB with concomitant reduction of bound ubiquinone or menaquinone. Since many bacterial virulence factors are secreted proteins that require disulfide bonds for proper folding and function, the DsbA/DsbB system is a potential antimicrobial drug target16–18. DsbB is an ideal candidate to test the TINS methodology since it can be produced in large quantities and solubilized in detergent micelles where it retains a robust enzymatic activity which is easily assayed. In addition, a wealth of biochemical data is available that describes the enzymatic activity of the wildtype as well as numerous relevant mutants15,19–21. Finally, the 3D structures of wildtype DsbB bound to its redox partner DsbA22 and of a mutant representing an enzymatic intermediate are available23. Selection of an appropriate reference system is critical to insure the robust performance of TINS. Our previous experience using the E. coli Outer membrane protein A (OmpA) transmembrane domain, which has native structure under the same detergent micelle conditions as DsbB 24, suggested that it had minimal small molecule binding and would therefore serve as a good reference (data not shown).
Here we report the first complete screen of a fragment library against an integral membrane protein. We have tested the applicability of TINS for fragment screening using both micelle and ND solubilized protein. Hits from the screen have been validated and characterized with respect to mode of action using an enzyme inhibition assay. Finally, the binding mode of two classes of inhibitors has been investigated by analysis of chemical shift perturbations induced upon fragment binding to 15N-labeled mutant DsbB.
Results
DsbB functional immobilization and enzymatic activity
Wildtype DsbB (containing endogenous quinone) has previously been solubilized in DPC micelles, which we refer to as DsbB/DPC, with retention of enzymatic function23. We prepared protein similarly and immobilized it on a Sepharose resin via a Schiff’s base intermediate. At the pH selected (7.4), this reaction is relatively specific for the free N-terminus. A final concentration of approximately 100 μM DsbB/DPC (nmol protein per ml settled bed volume) was achieved with an overall yield of 50%. The functionality of the immobilized enzyme was compared to non-immobilized, micelle solubilized enzyme. Table 1 shows that immobilized wildtype DsbB/DPC retained 90% activity in comparison to the non-immobilized protein and that the kcat of both forms of the protein was close to values previously reported25. The ready immobilization with retention of enzymatic activity suggests that the N-terminus of DsbB is accessible in the micelle solubilized protein. We used the same approach to immobilize OmpA which had also been solubilized in DPC micelles26 and shown to be natively folded. We observed a similar yield of OmpA immobilization. Since OmpA has no enzymatic activity, we had to assume that its structure was not grossly perturbed by the immobilization process. Independent experiments showed that immobilized samples of DPC solubilized DsbB were stable for at least one month after storage at 4 °C (data not shown).
Table 1.
Enzymatic activity of solubilized vs immobilized DsbB.
| Enzyme | kcat (M UQ1/M DsbB-min−1) |
|---|---|
| DsbB wt/DPC | 267 ± 14 |
| DsbB wt/DPC (immobilized) | 238 ± 27 |
| DsbB wt/DDM literature25 | 278 |
| DsbB wt/ND | 346 ± 13 |
| DsbB wt/ND (immobilized) | 329 ± 26 |
We next trapped DPC solubilized DsbB and OmpA in NDs. Gel filtration analysis of our preparations revealed Stokes diameters of 9.63, 9.68, and 9.52 nm for empty NDs (-/ND), NDs with embedded DsbB (DsbB/ND), and NDs with embedded OmpA (OmpA/ND), in accordance with literature values27 (See supplementary information). The DsbB/ND was immobilized using the same method as for DsbB/DPC with an overall yield of 75%. Non-immobilized and immobilized DsbB/ND were assayed for enzymatic activity for comparison to DsbB/DPC. As shown in Table 1, both preparations had a kcat that was somewhat greater than the micelle solubilized protein, indicating that they remained completely functional. The increased kcat for DsbB in NDs could possibly result from a more native functionality of the enzyme in the lipid bilayer environment of the ND.
Stability of the Immobilized Protein to Repeated Sample Application Cycles
In a method such as TINS where a single sample of the target is used to screen an entire compound collection, the integrity of the immobilized protein is clearly critical. Soluble proteins are routinely stable over more than 200 cycles of sample application and washing13. Solubilized MPs however, include two components, the surfactant and the protein itself, both of which must remain stable in order to ensure proper ligand screening. Preliminary studies of DsbB/DPC and OmpA/DPC clearly demonstrated that repeated cycles of compound application and washing in the absence of added detergent resulted in rapid degradation of DsbB activity12. Therefore deuterated DPC was included at a minimum concentration of 5 mM (approximately 3 x critical micellar concentration) in the buffer used to wash the compounds from the sample holder. The library, which consisted of 1,071 fragments at the time, was then screened in mixtures that averaged approximately 5 compounds each in the absence of DPC. Including control experiments designed to monitor the physical integrity of the target and reference samples, approximately 200 sample application/washing cycles were performed. To monitor the integrity of the DsbB sample during the screen, the binding of synthetic UQ1 was observed (Figure 1a). In TINS, binding of a fragment is best described by the T/R (target/reference) ratio, defined as the average ratio of the amplitude of peaks in the presence of the target, DsbB, to that in the presence of the reference, OmpA. It is clear from Figure 1a that binding of UQ1 to DsbB/DPC remained relatively constant throughout the screen which required 5.5 days to complete.
Figure 1.

Stability of the DsbB in Micelles and NDs. The stability of DPC solubilized (a) and ND solubilized (b) DsbB after multiple cycles of compound application and washing was assessed by binding of a known ligand. Binding is displayed as the average ratio of peak heights for the compound in the presence of DsbB over that in the presence of the reference (T/R ratio). The reference in (a) was DPC solubilized OmpA and in (b) -/ND. Note the difference in vertical scale between parts a and b.
Since it was not practical to rescreen the entire fragment collection multiple times, we selected a subset of 20 compound mixtures containing positive hits from the DsbB/DPC screen and 20 mixtures containing no positive hits from the DsbB/DPC screen (a total of 183 compounds) to assess the suitability of the ND system for ligand screening. The 40 mixes, along with the control experiments used for DsbB/DPC, were applied sequentially to the immobilized DsbB/ND using -/ND as the reference. We wanted to assess whether DsbB/ND and -/ND were stable in the absence of added phospholipid. As with DsbB/DPC, we monitored the integrity of DsbB/ND, as determined by binding of a known ligand (UQ1), during multiple cycles of compound application and washing in lipid free buffers. The T/R ratio for ligand binding to DsbB/ND vs -/ND is shown in Figure 1b. This data suggests a possible, initial small degradation in binding behaviour (although the variation is similar to that seen in Figure 1a), after which the ligand binding capacity of DsbB/ND remained constant. The constant T/R ratio during cycles 22 through 61 suggests that both DsbB/ND and -/ND remained intact.
Target immobilized NMR Screening (TINS) of DsbB/DPC
The fragment collection was screened for binding to DsbB at 500 μM each, in 182 mixtures. A spatially selective Hadamard NMR experiment28 was used to simultaneously acquire a 1D 1H spectrum of compounds in the presence of DsbB/DPC or OmpA/DPC. The data resulting from the screen could be analysed directly without deconvolution because fragments could be directly identified by comparing peaks from TINS spectra with reference spectra of the individual fragment (Figure 2). The screen resulted in 93 hits for DsbB, defined as fragments which had a T/R ratio less than 0.3, as shown by an example of a mix containing two hits in Figure 2. This particular cutoff was chosen by virtue of a step-like relationship between the observed TINS effect and the number of “hits” whereby even slightly raising the cut-off gave a large increase (more than two fold) in the number of compounds that were selected as hits (not shown). The resulting hit rate for DsbB was 8.7% which is well within the range we typically observe with soluble proteins using TINS (3–9.5%). Application of the same criteria to OmpA/DPC binding identified 7 compounds as hits for a hit rate of 0.6%, validating the earlier data suggesting that OmpA/DPC has minimal small molecule binding capacity.
Figure 2.

Detection of ligand binding to immobilized DsbB using TINS. The 1D 1H NMR spectrum of 3 different fragments in solution (a – c) is shown for reference. The 1H NMR spectrum of a mix of the 3 fragments in the presence of DsbB/DPC (red spectrum) or OmpA/DPC (blue spectrum) that have been immobilized on the sepharose support is shown in d. The spectra of the same mix recorded in the presence of DsbB/ND (green) or -/ND (magenta) is shown in e. The asterisk indicates the resonance from residual 1H DMSO and the bracket shows residual sugar 1H resonances from the sepharose media. The residual H2O resonance at 4.7 ppm has been filtered out.
Comparison of Micelle Solubilized vs ND Solubilized Protein for Ligand Binding Studies
The influence of detergent or ND on the quality of the NMR spectra of the fragments is shown in Figure 2(d and e). In both cases the compound whose spectrum is shown in 2c can be identified as specifically binding to DsbB. However, the signal-to-noise ratio of the compounds (Figure 2a and b) in 2e is nearly double that in 2d (most readily observed on the aromatic resonances, but see also the peak at 3.1 ppm). The improved quality of the spectra allows more reliable analysis of the peaks at 7.3 and 7.4 ppm, which are now clearly seen to indicate specific binding of this compound to DsbB/ND, consistent with the behaviour of the peaks at 2.8 and 2.2 ppm. The reduced signal in the presence of detergent solubilized protein is likely due to non-specific partioning of 30–40% of the compounds into the micelle.
The stability of the empty ND (-/ND) as shown in Figure 1b, affords the possibility to use NDs directly as a generic reference to account for non-specific ligand binding to the phospholipid bilayer and the scaffolding protein. To investigate this, we screened all 183 compounds for binding to DsbB/ND using either OmpA/ND or -/ND as a reference. By plotting the T/R for each compound from the screen using -/ND versus that using OmpA/ND we derive a two dimensional plot that gives an overview of the performance of the screen (Figure 3a). Overall there was a reasonable correlation in ligand binding to DsbB/ND using either empty NDs or OmpA/ND as the reference (R2 =0.78). In general however, the T/R ratio of fragments is lower with -/ND as a reference, indicating that specific binding to DsbB/ND is more pronounced. Since the NMR spectra of the fragments in the presence of DsbB/ND in the screen vs -/ND or OmpA/ND are similar, this suggests a higher level of non-specific binding of the fragments to OmpA/ND. We conclude therefore that -/ND is the preferred reference.
Figure 3.
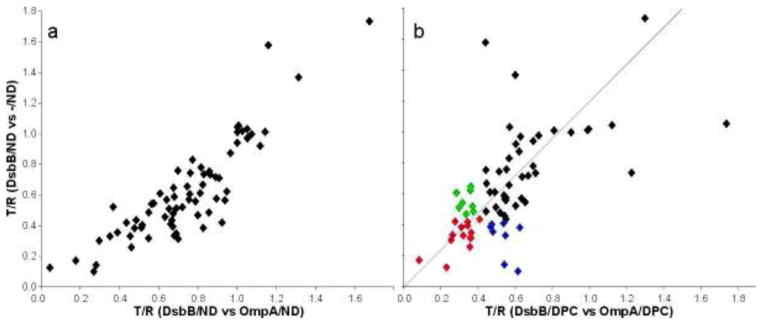
Comparison of TINS screening in micelles vs NDs. A total of 70 fragments were assayed for binding to DsbB solubilized in either detergent micelle or ND. a) The 70 fragments were screened for binding to DsbB/ND using either empty ND (-/ND) or OmpA/ND as a reference. The T/R (see text) for each compound is plotted for one screen versus the other. R2 = 0.78. b) The T/R for each compound in the DsbB/ND vs -/ND screen is plotted against the value from the DsbB/DPC vs OmpA/DPC screen. Hits common to both screens are show in red. Hits found only in the ND screen are shown in blue while those found only in tehe DPC screen are in green.
We then compared the ligand screening results from DsbB/DPC (OmpA/DPC as reference) to those from DsbB/ND (-/ND as reference). Upon inspection of the raw NMR data from the DPC screen we observed that although 183 compounds were present in the 40 mixes selected, only 127, about 2/3, gave observable NMR spectra. Presumably, those compounds missing from the NMR data had non-specifically adsorbed to the micelle. We compared the calculated Log of the octonal/water partition coefficient (cLogP)) for compounds that could be observed (median 0.9) and could not be observed (median 1.8) for evidence to support this assumption. Despite the inaccuracies of cLogP values, there is a clear trend towards more hydrophobic compounds in the group of compounds whose NMR spectra were unobservable in the presence of micelles. In contrast, 164 of 183 compounds gave observable spectra in the ND screen. The median cLogP for the observable compounds was 1.1 and 1.6 for the unobservable. However, tThe small size of the set of unobservable compounds in the presence of ND renders the median cLogP value meaningless. Of the 127 compounds with observable spectra in the DPC screen, 70 were of sufficiently high quality to allow a reliable comparison with the ND screen and we therefore focused our efforts on these. Inspection of Figure 3b clearly shows that the correlation between the micelles and NDs is much less pronounced than between the two ND references. Using the same criteria for hit selection for both, 22 hits were identified for DsbB/ND and 22 were identified for DsbB/DPC. Of these biophysically detected hits, 14 were common to both the micelle and ND (red) screen while 8 were unique to the ND screen (blue) and 8 unique to the micelle screen (green, see also Table 2). We analyzed the solubility of each of the 22 fragment hits using the cLogP. Interestingly, the hits specific for the ND screen are on average slightly more hydrophobic than the hits found in both screens, but the hits specific to the micelle screen are considerably more hydrophylic (Table 2). A possible explanation for this observation is that the more hydrophobic fragments exhibited greater non-specific binding to the micelle, thus masking specific binding to DsbB. This observation is consistent with the NMR data in Figure 2. Using the same criteria, 2 out of the 183 compounds assayed bound to -/ND (1.1% hit rate). Given the small sample this hit rate is similar to that for OmpA/DPC.
Table 2.
Fragment hits from the screen of DsbB in micelles and NDs.
| Hits | cLogP | BioAssay in ND | BioAssay in DPC | |
|---|---|---|---|---|
| Micelle | 8 | 1.34 | − | + |
| ND | 8 | 2.21 | ++ | ++ |
| Both | 14 | 2.13 | ++ | ++ |
Note: − poor correlation between enzyme inhibition and binding assay, + reasonable correlation (approximately 50% hits bioactive), ++ good correlation (80–90% bioactive), cLogP is the median value.
Hit validation using enzymatic assays
The TINS assay simply identifies compounds that bind to DsbB, but not necessarily in a biologically relevant manner. Therefore we felt it was critical to validate the hits in terms of biological activity. We used an enzymatic assay to assess the ability of the compounds to inhibit electron transfer mediated by DsbB. Each of the 93 fragments identified as TINS hits in the micelle screen was assayed for inhibition of DsbB-dependent reoxidation of DsbA at 250 μM (Figure 4). Eight compounds interfered with the assay when run in either fluorescence or absorbance mode and therefore were left out of the analysis. The remaining 85 hits exhibited a distribution of potencies against DsbB, including 60% with better than 30% enzymatic inhibition and 16% with either less than 20% inhibition or stimulation. This data confirms that a very high percentage of the hits found in the biophysical assay also modulate the enzymatic activity of DsbB and are functionally relevant.
Figure 4.

Distribution of biological activity of the hits found in the TINS fragment screen of DsbB. Each fragment was assayed singly at 250 μM. The percentage of inhibition of each category is provided in the legend. The height of each bar represents the percentage of all hits found in the TINS screen within the given range of potencies. The three ranges of inhibitors mentioned in the text (low, medium and high) are indicated.
We next used the bioassay to compare hits selected in the micelle screen to those selected in the ND screen (see Table 2). As expected, fragments common to both the micelle and ND screens yielded a strong correlation with biological activity with 12/14 exhibiting medium (30–70%) or high (>70%) inhibition of DsbB in both micelles and NDs. We observed a good correlation between ligands detected in the ND screen and biochemical activity against both micelle and ND solubilized DsbB where 6/7 compounds had medium inhibitory activity and the seventh was a mild stimulator. In contrast, while the micelle specific ligands correlated reasonably well with the bioassay using detergent solubilized DsbB where 5/8 were medium or strong inhibitors, none inhibited DsbB/ND.
To avoid the possibility of artifacts in the biochemical assay we selected the 13 fragments from the micelle screen showing strong inhibition in the single concentration point assay for further analysis. We first assayed these 13 fragments for potency (IC50) by dose-response experiments (Figure 5). Dose response experiments were carried out with increasing fragment concentrations, from 0.0001 to 10 mM, while both DsbA and UQ1 were kept in excess. Three of the 13 fragments indeed showed artefacts including signs of protein precipitation at higher compound concentration and/or steeper than expected Hill coefficients. The remaining 10 fragments titrated over 2 Log orders and exhibited a Hill coefficient close to unity. By these criteria the 10 fragments are reversible inhibitors with a 1:1 stoichiometry and are therefore well-behaved. The 8 most potent compounds had IC50 values between 7 and 170 μM (Table 3.) and consisted of a variety of scaffolds (see Figure 5). The calculated binding efficiency index29 (Table 3) indicates that these fragments are all very good or excellent starting points for hit elaboration projects.
Figure 5.

Potency determination of selected hits from the TINS screen. An example of an inhibition curve used to determine the IC50 for compound 2. The curve represents the mean ± S.E.M. of three independent experiments performed in triplicate. The structures of the 8 most potent compounds are shown and the IC50 values are provided in Table 3.
Table 3.
Biological characterization of fragment hits.
| Fragment | IC50 (μM) | Hill coefficient | BEI29 |
|---|---|---|---|
| 1 | 7 ± 1 | 0.80 ± 0.10 | 21.8 |
| 2 | 100 ± 10 | 1.40 ± 0.15 | 14.7 |
| 3 | 193 ± 11 | 1.20 ± 0.11 | 15.3 |
|
| |||
| 4 | 13 ± 1 | 0.80 ± 0.10 | 21.2 |
| 5 | 46 ± 12 | 0.80 ± 0.10 | 22.2 |
| 6 | 70 ± 10 | 1.00 ± 0.10 | 14.0 |
|
| |||
| 7 | 115 ± 11 | 1.15 ± 0.05 | 19.1 |
| 8 | 168 ± 10 | 1.40 ± 0.10 | 14.2 |
Note: BEI – Binding Efficiency Index = pIC50/MW
As a second validation step, we carried out a more detailed kinetic analysis of the mode of action of the 8 most potent fragments. Substrate-velocity experiments were performed in which either DsbA or UQ1 were titrated in the presence of saturating amounts of the other. The titrations were then repeated in the presence of increasing amounts of the inhibitory fragment (Figure 6, Table 4). In this analysis, fragments 1 – 3 behaved similarly (Supplementary data). This group is exemplified by fragment 2 where increasing concentrations result in moderate perturbation of the maximum enzymatic turn over rate (kcat) and apparent affinity of DsbA but a dramatic reduction (>6 fold) in the apparent affinity of UQ1. This data suggests that fragments 1 – 3 compete for the same binding site as UQ1. On the other hand, addition of fragments 4 – 8 simultaneously decreased both the apparent affinity and the kcat for UQ1 and DsbA as best exemplified by fragment 8 (Figure 6 and Supplementary data). This data suggests a mixed model of inhibition of DsbB by these fragments. We next sought biophysical confirmation of these two different modes of fragment interaction with DsbB.
Figure 6.

Mode of action determination for the most potent DsbB inhibitors. Fragment 2 was assayed in competition with synthetic UQ1 (a), the electron acceptor, or DsbA (b) the electron source. Fragment 8 was assayed in the same manner (panels c and d respectively). The kcat and Km apparent determined from the data are shown in Table 4 in the absence and presence of the indicated amount of each inhibitor. See supplementary information for data on the other 6 fragments.
Table 4.
Kinetic analysis of 2 fragment hits.
| Fragment | Substrate | kcat (M DsbA/M DsbB-s−1) | Km (μM) | |
|---|---|---|---|---|
| 2 | 0 μM | UQ1 | 3.7 ± 0.1 | 1.6 ± 0.1 |
| 75 μM | 3.1 ± 0.2 | 13.2 ± 1.7 | ||
|
| ||||
| 0 μM | DsbA | 4.3 ± 0.1 | 2.4 ± 0.2 | |
| 75 μM | 2.5 ± 0.1 | 2.8 ± 0.1 | ||
|
| ||||
| 8 | 0 μM | UQ1 | 4.0 ± 0.1 | 2.2 ± 0.0 |
| 75 μM | 2.3 ± 0.1 | 9.7 ± 1.5 | ||
|
| ||||
| 0 μM | DsbA | 4.3 ± 0.1 | 1.9 ± 0.1 | |
| 75 μM | 2.4 ± 0.2 | 4.1 ± 0.5 | ||
Confirmation of different modes of interaction with DsbB by NMR
Chemical shift perturbations of the protein NMR spectrum (typically 2D 15N-1H HSQC or 13C-1H HSQC) in the presence of compounds can both confirm binding to the target and localize the binding site on a protein when resonance assignments are available30. While the sequential assignment of wildtype DsbB is not available due to the poor quality of the NMR spectra, spectra of the DsbB[CSSC] double cysteine mutant are of high quality, resulting in a complete backbone resonance assignment for this form of the protein23. When purified from E. coli, DsbB[CSSC] contains the endogenous ubiquinone-831, thus compounds specific for this site must compete with UQ8 for binding. We first titrated the synthetic quinone UQ1 into a sample of 15N DsbB[CSSC]. Addition of UQ1 to 15N DsbB[CSSC] resulted in numerous chemical shift perturbations but two in particular afford a detailed analysis of the binding and allow a reliable comparison with the fragments found in TINS screening. As shown in Figure 7, the sidechain indole of Trp135 (in the vicinity of the quinone binding site, see Supplementary Information) and the backbone amide of Arg109 (close to the DsbA binding site) respond very differently to addition of UQ1. Titration of UQ1 resulted in the simultaneous disappearance of the Trp135ε-HN peak from DsbB[CSSC] bound to endogenous quinone and the appearance of a new peak close by in the spectrum. Due to its proximity and the unique chemical shift of the Trpε HN proton, in combination with the high level of conservation of this residue, the new peak is likely from the Trpε HN proton of the UQ1 bound DsbB[CSSC]. This pattern of peak changes is indicative of slow exchange on the NMR time scale (e.g. koff < 30 Hz Δδ in Figure 7a). In contrast, the backbone amide of Arg109 is essentially unchanged by the addition of UQ1. Mapping the chemical shift perturbations induced by UQ1 onto the surface of DsbB confirms that UQ1 binds at the UQ8 site (see Supplementary Information).
Figure 7.

NMR analysis of fragment binding to DsbB. The 8 most potent fragments were titrated into 15N DsbB[CSSC]. Data for the synthetic quinone UQ1 (a), competitive fragment 2 (b) and the mixed model fragment 8 (c) are shown. For each of these three compounds, the structure of the compound is shown in the left column and the characteristic peak perturbations in the [15N,1H] HSQC spectrum (green 0 mM fragment, blue 5 mM fragment and red 10 mM fragment) are shown in the middle (Arginine 109 backbone amide) and right columns (Tryptophan 135 sidechain indole).
Addition of all 8 fragments to 15N labelled DsbB[CSSC] resulted in detectable changes in chemical shifts, suggesting that the fragments selected by TINS screening and biochemical assays on wild type protein also bind the cysteine mutated form. The presence of chemical shift perturbations both in solvent exposed loops and in portions of the protein buried within the micelle (data not shown) suggests that the fragments are specifically binding to the protein and not non-specifically partitioning into the micelle. Fragment 2, which competitively inhibited ubiquinone binding, induced chemical shift perturbations in a variety of amino acids, including W135 and R109. The pattern of chemical shift perturbations induced by fragment 2 closely resembles that induced by UQ1. First, titration of 2 into 15N DsbB[CSSC] resulted in chemical shift changes in the W135ε-HN peak that were similar to those induced by UQ1 (i.e. slow exchange). Moreover, the resonance frequency of the new peak tentatively assigned to the DsbB[CSSC]-2 complex is similar to that of the DsbB[CSSC]-UQ1 complex. Similarly, R109HN, which is minimally affected by UQ1, undergoes only minor chemical shift perturbations in the presence of 2. Mapping the chemical shift perturbations induced by 2 onto the surface of DsbB confirms that binding is similar to UQ1 (see Supplementary Information). While 1 and 3 induce chemical shift perturbations in the spectrum of DsbB[CSSC], the characteristic ones observed at R109 and W135 are not seen, so the grouping of these compounds with 2 as UQ8 competitors is tentative, relying only on the kinetic data.
In contrast, the chemical shift changes induced by fragments 4 – 8 differ in both the overall pattern and the details from fragment 2 and UQ1 (Figure 7c and Supplementary Information). Addition of 8, for example, induced concentration dependent shifts in the Trp135ε-HN resonance to an entirely different chemical shift than did fragment 2 or UQ1. This concentration dependent shift is indicative of rapid exchange on the NMR time scale. There was no evidence for slow exchange for any of the fragments 4 – 8, although 4 and 7 show signs of line broadening of the backbone resonance of Q33 that may indicate intermediate exchange (not shown). In contrast, the backbone amide of R109, which is only mildly perturbed by UQ1 or 2, is very dramatically perturbed by the addition of fragment 8. This data suggests that fragments 4 – 8, which exhibit mixed mode DsbB inhibition, bind in either a different mode or different site to fragment 2 which is competitive with ubiquinone.
Discussion
The use of Ro3 compliant, “drug fragments” as a starting point for drug discovery has delivered a number of innovative compounds against soluble targets which are currently in clinical trials32. Membrane proteins have not made good targets for FBDD due to their challenging physicochemical properties. In particular, the difficulty of generating sufficient quantities of purified, functional protein and of detecting specific binding to the target, as opposed to non-specific partitioning into hydrophobic phases, have limited the applicability of biophysical ligand screening approaches. Here we have addressed these two issues by a) immobilizing the target and reusing a single sample to screen an entire fragment collection and b) using a reference sample to cancel out non-specific interaction of the fragments with the hydrophobic phase. Using TINS we have screened a collection of nearly 1,100 fragments with a single sample of less than 2 mg of protein and demonstrated that the protein was stable throughout the procedure. The stability of DsbB to repeated cycles of fragment application and washing depends on detergent micelles and the quinone cofactor. The detergent requirement could be overcome by including it in the buffer or using NDs to solubilize the protein. Endogenous UQ8 binds DsbB very tightly and is quite resistant to repeated detergent washing33.
Screening of the fragment library resulted in 93 ligands that were specific for DsbB. A number of observations suggest that most of these ligands are directly binding to DsbB and not indirectly via the micelle. First, the DsbB binding detected using TINS was relative to OmpA solubilized in identical conditions. Second, there is a range of potencies in the enzyme inhibition studies that includes a small number of non-inhibitors and activators. Third, and perhaps more critically, inhibition is saturable and occurs over 2 log orders, strongly suggesting a stoichiometric interaction. Fourth, titration of 8 different fragments into 15N labelled DsbB resulted in chemical shift perturbations at well defined sites in both solvent exposed and micelle buried portions of the protein. In particular, the similarity of the chemical shift perturbations induced by the synthetic quinone UQ1 and fragment 2 indicate the compounds are binding to the same or overlapping sites. An additional, likely important, factor contributing to the low false positive rate is that the fragments that make up the collection are highly soluble, with each having been tested at 500 μM in an aqueous buffer alone and in a mixture. Nonetheless, an appreciable fraction of these fragments exhibit sufficient non-specific binding to the micelle that they were only poorly or even not detected in the NMR spectra. This data suggests that ligand screening in the presence of micelle solubilized membrane proteins may bias the chemical nature of the fragment library. The median cLogP for observable compounds in the presence of ND solubilized proteins is slightly greater (0.9 vs 1.1) suggesting a reduced tendency of hydrophobic compounds to partition into the nanodisc.
The eight fragments with greatest potency in the single concentration enzyme inhibition assay were fully characterized for potency, mode of action, and binding site on DsbB. A simplistic analysis suggests that these fragments can be divided into two groups, one that competes only with quinone for DsbB binding and a second that perturbs the apparent affinity of DsbB for both quinone and DsbA. The different mode of action is best exhibited by the differing effect on the apparent Km for UQ1 or DsbA for each. Addition of fragment 2 reduced Km for UQ1 more than 8-fold, while it had only a marginal effect on the Km for DsbA (only 5% greater than experimental error). In contrast, addition of 8 reduced Km for UQ1 more than 4.4-fold and Km for DsbA more than 2-fold.
The different behaviour of the resonances of the backbone amide of R109 and the sidechain indole of W135 upon titration with the fragments provides further support for two modes of action. Titration of UQ1 DsbB[CSSC] results in slow exchange between the endogenous quinone bound form and a newly arising peak at a nearby position which we assign to the UQ1 complex. Similarly, addition of 2 resulted in slow exchange between the endogenous quinone bound form of W135ε NH and the appearance of a new peak with similar chemical shift as the UQ1 complex. An additional chemical shift perturbation indicating fast exchange with the endogenous complex was also observed. The fast exchange is likely due to competition between 2 and the quinone moiety of the bound UQ8, consistent with the competitive kinetics observed for this inhibitor. However, we have shown that the isoprenyl tail of UQ8 extends down the groove between TM1 and TM4, making extensive interactions with the protein23. Therefore, displacement of the quinone moiety apparently occurs on a slower timescale. Addition of 8 to DsbB[CSSC] also causes chemical shift perturbation of the W135ε NH but these suggest rapid exchange between a typical 2-state equilibrium rather than the more complex effects seen with 2. In addition, the bound state has a different resonance frequency than the bound state of UQ1 or 2. Additional large downfield chemicals shifts of the resonance of R109N, indicative of rapid exchange, are observed while UQ1 and 2 had no or only minor effects on this peak. From a drug discovery perspective, this data is exciting because it suggests, as with soluble enzymes, it is possible to find small molecule inhibitors with different modes of action and possibly non-overlapping or even different binding sites on membrane proteins.
We note that the concentration of the fragments required to induce chemical shift perturbations in DsbB[CSSC] is significantly higher than the IC50 values measured for the wild type protein but the same as required for UQ1. A likely explanation is that the conformation of the mutant differs slightly from the wild type protein, against which the fragments were selected. In addition, either the affinity for the quinone is higher for the DsbB[CSSC] mutant or more likely, the quinone binding site may be partially occluded. This latter possibility is clearly consistent with the reduced dynamic behaviour of DsbB[CSSC] with respect to the wild type protein, which results in the substantial improvement in the quality of the NMR spectra. This reduced dynamic behaviour of the disulfide mutant may be responsible for the slow exchange kinetics observed for UQ1 and 2 if release of the UQ8 from this binding site can only occur from a sparsely populated conformation.
Functional solubilization of membrane proteins in detergent micelles is a challenging process that must be individually optimized for each protein. NDs offer the potential to enable a more generic approach to handling membrane proteins since they can be used to functionally solubilise a variety of membrane proteins and can obviate the requirement for an intermediate micelle solubilized step10,27. Furthermore, immobilization can be made generic for all MPs with minimal effect on functionality by appropriate engineering of the MSP portion of the ND. Use of NDs as a generic solubilization/immobilization system for ligand screening is further enabled by the stability of the empty ND and the fact that it represents a high quality reference system to remove false positives. This conclusion is strongly supported by the observation that the 8 DsbB/DPC specific hits failed to inhibit DsbB/ND while 7 of 8 DsbB/ND hits also inhibited DsbB/DPC. Apparently, despite the reference sample, some compounds interact with DsbB in a micelle specific manner. This problem would be largely eliminated by using NDs.
Significance
Integral membrane proteins make up a significant portion of the human genome, carrying out numerous important normal as well as disease related functions. More than 50% of drugs currently on the market target a membrane protein, demonstrating the utility of targeting this class of proteins. Approaches for the development of drugs targeting this class of proteins have focused principally on the use of high throughput screening methods. Recently, fragment based drug discovery (FBDD) has emerged as a powerful additional screening approach. Because of the typically modest binding affinities of the small, functional group rich, i.e. high ligand efficiency, compounds utilized in FBDD, various biophysical techniques, including NMR, are typically used to detect binding. To date several compounds developed using FBDD have advanced to clinical trials. However, thus far FBDD has only been demonstrated for soluble proteins, not membrane proteins.
Herein we describe the first complete screen of a fragment collection against an integral membrane protein. The screen was performed using a detergent micelle solubilized protein using a simple and rapid 1D NMR method we described previously (TINS). The 93 hits were subsequently validated in an enzyme inhibition study. The use of a reference sample in the TINS experiment eliminated the well-documented problem of non-specific binding of compounds to the detergent. As membrane protein activity is enhanced in more bilayer like environments, we have also solubilised the protein in nanodiscs and shown that the screening approach is effective with this preparation as well. The use of nanodiscs further ameliorates issues with non-specific binding and should also extend the method to proteins which typically don’t behave well in detergents such as GPCRs.
Our results clearly establish the feasibility of using a fragment based approach for finding starting matter for subsequent development of compounds targeting membrane proteins, including the all-important GPCR class of proteins. In addition, increasing success in the preparation of membrane proteins in reasonable quantities should make many such proteins amenable to the use of TINS for fragment screening, thereby increasing its general utility.
Experimental Procedures
Protein Purification
DsbA, DsbB, and OmpA were expressed and purified as previously reported25,31,34. All proteins have a 6x-His tag at the N-terminus or C-terminus for affinity purification. Succesful refolding of OmpA from inclusion bodies was monitored by SDS-PAGE analysis34.
ND self-assembly
The ND self assembly procedure was repeated the same way for both OmpA and DsbB with slight adaptations from the previously reported procedures27. The reconstitution mixture contained Membrane Scaffold Protein MSP1D1(−) which lacked the His-tag, with mixed micelles of POPC and cholate at a ratio of 1:65:130. This reconstitution mixture was added to the OmpA or DsbB in detergent micelles (each with 10x the detergent CMC) in a volumetric ratio of 1:1 and incubated on ice for 4 hours. We always ensured a stoicheometry of MSP1D1(−) to OmpA or DsbB of 2:1. Upon addition of 0.7 g/ml of the hydrophobic adsorbent Bio-Beads SM-2 (Biorad, Hercules, CA) and gently mixing for 4 hours at 4 °C, the NDs underwent self-assembly, incorporating DsbB or OmpA in the lipid bilayer. This step was limiting, whereby detergent removal before 4 hours resulted in incomplete ND formation, but caused ND complex malformation if carried out for longer (i.e. 16h, data not shown). The His-tag of the embedded OmpA and DsbB was used to separate the empty non-tagged MSP1D1(−) complexes from the mixture by IMAC chromatography using Ni-NTA resin in a buffer containing 100 mM Tris-HCl (pH 7.5), 300 mM NaCl, and imidazole at 0 mM, 10 mM, and 100 mM for loading, washing, and elution respectively. The eluted fractions were applied to a gel filtration column (Superdex 200 10/300 from GE Healthcare) in order to remove the remaining aggregated, non-embedded OmpA and DsbB, and to exchange the ND-embedded proteins into phosphate buffered saline (PBS, pH 7.4) for compatibility with the immobilization step required for TINS. A set of standard proteins was used to calibrate the Stokes’ diameter vs. the retention time of the column.
Protein immobilization
Actigel ALD resin (Sterogene, Carlsbad, CA, USA) was used as a 50% slurry and all experiments were carried out at 4 °C when possible. The resin was washed with cold phosphate buffer (50 mM Na2HPO4, 50 mM KH2HPO4, 100 mM NaCl) at pH 7.5. 200 nmol of DPC solubilized DsbB was added to 1 ml bed volume of resin. The reductant sodium cyanoborohydride (NaCNBH3) was added to a final concentration of 0.1 M. After an overnight incubation at 4 °C, residual unreacted aldehydes were blocked by addition of 50 mM D11-Tris and fresh NaCNBH3 in the same buffer for another 2 hours. The same procedure was repeated for DPC solubilized OmpA. Quantification of immobilized protein was monitored by absorption of the supernatant at 280 nm before and after immobilization, and by SDS-page gel with a known standard curve and band volume analysis. This data indicated that a final concentration of 100 μM of both immobilized DsbB and OmpA was achieved, equating to a 50% yield. The procedure was repeated identically to immobilize DsbB and OmpA solubilized in ND (after pooling fractions containing particles ranging between 9.2 to 9.7 nm) and empty ND at a similar final concentration as the micelle solubilized protein. ND preparations could not be quantified by UV absorption, therefore they were loaded on SDS-page gels with a BSA standard curve for band volume quantification by Quantity One (Biorad), providing information on the concentration and ratio of ND molecules and incorporated proteins. The yield of immobilized, ND solubilized protein was 75%.
DsbB activity assays
DsbB activity was quantified by measuring the capacity of the enzyme to reoxidize the protein DsbA or reduce its cofactor Ubiquinone-5, also called Coenzyme Q1 (UQ1) at pH 6.2. DsbA was reduced with 10 mM DTT for 10 minutes on ice. DTT was subsequently removed by gel filtration on a PD-10 column pre-equilibrated with degassed, distilled water containing 0.1 mM EDTA. DsbA fluorescence (excitation at 295 nm and emission at 330 nm) was measured in the presence of DsbB and UQ1 in 50 mM sodium phosphate, 100 mM NaCl, 0.1 % detergent (DPC or DDM depending on which was used to solubilize DsbB) and 0.1mM EDTA) at 30 °C. Both UQ1 and DsbA were added at final concentrations of 30 μM. DsbB was added at a final concentration of 20 nM. The activity of DsbB in terms of moles ubiquinone reduced/moles DsbB min−1 could be calculated by using the initial slope of the fluorescence decrease upon DsbA oxidation, or by using the slope of absorption decrease at 275 nm upon reduction of UQ131.
To measure activity of immobilized DPC or ND solubilized DsbB, resin was aliquoted and diluted with degassed activity assay buffer to a final protein concentration of approximately 20 nM. For an appropriate baseline, an equivalent amount of resin without protein (blank resin) was prepared in the same manner. Quinone reduction was monitored in DPC samples after addition of 20 μM coenzyme Q1 and 20 μM DsbA and DsbA oxidation was measured for ND solubilized DsbB.
Target Immobilized NMR Screening
Immobilized, DPC solubilized DsbB and OmpA were each packed into a separate cell of a dual-cell sample holder14. Mixes of the 1,071 fragments were made by 200 fold dilution of a 100 mM stock of each compound in d6-DMSO such that the final DMSO concentration was never greater than 5%. Upon injection of each mix into the dual-cell sample holder, flow was stopped and spatially selective Hadamard spectroscopy28 was used to acquire a 1D 1H spectrum of each sample separately. A CPMG T2 filter of 80 ms was used to remove residual broad resonances from the sepharose resin. The cycle time was about 35 minutes, with 30 minutes required for the NMR experiment and 5 for sample handling, resulting in a total time of about 5.5 days to complete the screen. To maintain the proper fold of each protein, 5 mM deuterated DPC was included in the buffer (20 mM phosphate buffer in D2O, 100 mM NaCl, pH 7.6) used to wash the fragment mixes from the sample holder.
Biochemical hit validation
All fragments from the screen that were designated as positive for binding were assayed for DsbB inhibition at 250 μM. The amount of DMSO in all biochemical assay controls was adjusted to match the amount present when fragments were tested. Those compounds that showed more than 70% inhibition at 250 μM were further characterized by titration from 0.0001 mM to 10 mM to generate IC50 curves. The mode of action for the 8 most potent fragments was determined from competition enzyme assays. For this analysis either DsbA or UQ1 was titrated in from 0.2 to 40 μM, while the other was kept constant at 40 μM. For each titration point, slopes were measured in the presence of 5, 10, and 75 μM of the fragment. DsbB activity data was analyzed using the non-linear regression curve fitting routines in Graph Pad Prism v. 5.01 (Graph Pad, San Diego, CA, U.S.A.). Statistical significance was evaluated with the student’s T-test. Depending on the light absorbing properties of the fragments, they were used in either the fluorescence or UV-absorbance assay. Compounds which were not compatible with the assays due to high intrinsic fluorescence, high UV absorbance or irregular baselines were not included in the analysis.
Biophysical hit validation
Due to the poor quality of the NMR spectra of the wild-type DsbB, it was necessary to use a mutant that represents an intermediate in the disulfide oxidation pathway23. Validated hits from the screen were titrated at 1, 5, and 10 mM into 15N-labelled DsbB[CSSC] mutant (C44S, C104S). [15N,1H] HSQC experiments were acquired at 40 °C in a Bruker DRX 600 MHz spectrometer equipped with a cryoprobe. A reference titration of DMSO and a non-binding fragment from the screen were used to subtract chemical shift perturbations not related to fragment binding.
Supplementary Material
Highlights.
Use of NMR to detect weak binding of drug fragments to a membrane protein.
Comparison of different membrane protein solubilization media for ligand discovery.
Biological and biophysical characterization of ligands from library screening.
Acknowledgments
This research was supported in part by the Dutch Technology Foundation STW, applied science division of NWO and the Technology Program of the Ministry of Economic Affairs. This work was also supported by the US National Institutes of Health (R01 GM078296)
Footnotes
Conflict of Interest
G.S. acknowledges greater than 5% ownership in a company, ZoBio BV, whose goal is to commercialize TINS-based drug discovery.
Reference List
- 1.Zheng CJ, Han L, Yap CW, Xie B, Chen YZ. Progress and problems in the exploration of therapeutic targets (vol 11, pg 412, 2006) Drug Discovery Today. 2006;11:717. doi: 10.1016/j.drudis.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Carr RAE, Congreve M, Murray CW, Rees DC. Fragment-based lead discovery: leads by design. Drug Discovery Today. 2005;10:987–992. doi: 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- 3.Congreve M, Carr R, Murray C, Jhoti H. A rule of three for fragment-based lead discovery? Drug Discovery Today. 2003;8:876–877. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 4.Siegal G, ABE, Schultz J. Integration of fragment screening and library design. Drug Discovery Today. 2007;12:1032–1039. doi: 10.1016/j.drudis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Hajduk PJ, Meadows RP, Fesik SW. Drug design - Discovering high-affinity ligands for proteins. Science. 1997;278:497–499. doi: 10.1126/science.278.5337.497. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen SGF, et al. Crystal structure of the human bold beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 7.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the beta 1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci U S A. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahmane T, Damian M, Mary S, Popot JL, Baneres JL. Amphipol-Assisted in Vitro Folding of G Protein-Coupled Receptors. Biochem. 2009;48:6516–6521. doi: 10.1021/bi801729z. [DOI] [PubMed] [Google Scholar]
- 9.Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochem. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 10.Katzen F, et al. Insertion of membrane proteins into discoidal membranes using a cell-free protein expression approach. Journal of Proteome Research. 2008;7:3535–3542. doi: 10.1021/pr800265f. [DOI] [PubMed] [Google Scholar]
- 11.Leitz AJ, Bayburt TH, Barnakov AN, Springer BA, Sligar SG. Functional reconstitution of beta(2)-adrenergic receptors utilizing self-assembling Nanodisc technology. Biotechniques. 2006;40:601. doi: 10.2144/000112169. [DOI] [PubMed] [Google Scholar]
- 12.Früh V, Heetebrij RJ, Siegal G. In: Fragment-based Drug Discovery: A Practical Approach. Zartler ER, Shapiro M, editors. Wiley; Chichester, UK: 2008. pp. 135–158. [Google Scholar]
- 13.Vanwetswinkel S, et al. TINS, target immobilized NMR screening: An efficient and sensitive method for ligand discovery. Chem Biol. 2005;12:207–216. doi: 10.1016/j.chembiol.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Marquardsen T, et al. Development of a dual cell, flow-injection sample holder, and NMR probe for comparative ligand-binding studies. J Magn Reson. 2006;182:55–65. doi: 10.1016/j.jmr.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Bardwell JCA, et al. A Pathway for Disulfide Bond Formation Invivo. Proc Natl Acad Sci U S A. 1993;90:1038–1042. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inaba K, Ito K. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. EMBO J. 2002;21:2646–2654. doi: 10.1093/emboj/21.11.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stenson TH, Weiss AA. DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infection and Immunity. 2002;70:2297–2303. doi: 10.1128/IAI.70.5.2297-2303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagusztyn-Krynicka EK, Rybacki J, Lasica AM. Novel strategies for antibacterial drug discovery - antitoxin drugs. Postepy Mikrobiologii. 2009;48:93–104. [Google Scholar]
- 19.Jander G, Martin NL, Beckwith J. 2 Cysteines in Each Periplasmic Domain of the Membrane-Protein Dsbb Are Required for Its Function in Protein Disulfide Bond Formation. EMBO J. 1994;13:5121–5127. doi: 10.1002/j.1460-2075.1994.tb06841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regeimbal J, Bardwell JCA. DsbB catalyzes disulfide bond formation de novo. J Biol Chem. 2002;277:32706–32713. doi: 10.1074/jbc.M205433200. [DOI] [PubMed] [Google Scholar]
- 21.Kadokura H, Bader M, Tian HP, Bardwell JCA, Beckwith J. Roles of a conserved arginine residue of DsbB in linking protein disulfide-bond-formation pathway to the respiratory chain of Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:10884–10889. doi: 10.1073/pnas.97.20.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inaba K, et al. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 23.Zhou YP, et al. NMR solution structure of the integral membrane enzyme DsbB: Functional insights into DsbB-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat Struc Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 25.Bader MW, Xie T, Yu CA, Bardwell JCA. Disulfide bonds are generated by quinone reduction. J Biol Chem. 2000;275:26082–26088. doi: 10.1074/jbc.M003850200. [DOI] [PubMed] [Google Scholar]
- 26.Arora A, Abildgaard F, Bushweller JH, Tamm LK. NMR solution structure and dynamics of the outer membrane protein A transmembrane domain in dodecylphosphocholine micelles. Biophysical Journal. 2002;82:2512. [Google Scholar]
- 27.Civjan NR, Bayburt TH, Schuler MA, Sligar SG. Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers. Biotechniques. 2003;35:556. doi: 10.2144/03353rr02. [DOI] [PubMed] [Google Scholar]
- 28.Murali N, Miller WM, John BK, Avizonis DA, Smallcombe SH. Spectral unraveling by space-selective Hadamard spectroscopy. J Magn Reson. 2006;179:182–189. doi: 10.1016/j.jmr.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 29.Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discovery Today. 2005;10:464–469. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- 30.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 31.Bader M, Muse W, Ballou DP, Gassner C, Bardwell JCA. Oxidative protein folding is driven by the electron transport system. Cell. 1999;98:217–227. doi: 10.1016/s0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- 32.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discovery. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 33.Inaba K, Takahashi YH, Ito K. DsbB elicits a red-shift of bound ubiquinone during the catalysis of DsbA oxidation. J Biol Chem. 2004;279:6761–6768. doi: 10.1074/jbc.M310765200. [DOI] [PubMed] [Google Scholar]
- 34.Arora A, Rinehart D, Szabo G, Tamm LK. Refolded outer membrane protein A of Escherichia coli forms ion channels with two conductance states in planar lipid bilayers. J Biol Chem. 2000;275:1594–1600. doi: 10.1074/jbc.275.3.1594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.