

Abstract
Described in this report is an enantioselective route toward the chamigrene natural product family. The key disconnections in our synthetic approach include sequential enantioselective decarboxylative allylation and ring-closing olefin metathesis to form the all-carbon quaternary stereocenter and spirocyclic core present in all members of this class of compounds. The generality of this strategy is demonstrated by the first total syntheses of elatol and the proposed structure of laurencenone B, as well as the first enantioselective total syntheses of laurencenone C and α-chamigrene. A brief exploration of the substrate scope of the enantioselective decarboxylative allylation/ring-closing metathesis sequence with fully substituted vinyl chlorides is also presented.

1. Introduction
The chamigrene subclass of sesquiterpenes is an abundant and continually expanding family of natural products, with over one hundred members discovered to date (Figure 1).1,2 Isolated from a variety of sources including terrestrial and marine plants, these compounds have demonstrated a diverse array of biological activity and have been suggested to play a chemical defense role in the marine alga genus Laurencia.3 For example, elatol (1), originally isolated from Laurencia eletata,2a has displayed antifeedant activity,3a antifouling activity,4a antibacterial activity (including human pathogenic bacteria),3b–c,4b antifungal activity,3d and cytotoxicity against HeLa and Hep-2 human carcinoma cell lines.4c As a consequence, significant effort has been devoted to the total synthesis of several members of this class;5 nevertheless, no general strategy for their preparation has yet been developed. Inspired by both this lack of a common approach and the overall synthetic challenge posed by these molecules, we sought to generate a new route toward the chamigrene family that would allow ready access to a wide range of its constituents in enantioenriched form. Herein we report in full our efforts toward this goal, culminating in the first enantioselective total syntheses of elatol (1), α-chamigrene (5), laurencenone C (8), and the proposed structure of laurencenone B (7).6
Figure 1.

Examples of chamigrene natural products.
Structurally, chamigrenes are characterized by a spiro[5.5]undecane core bearing an all-carbon quaternary stereocenter at the spirocycle junction. Moreover, this stereocenter is found to occur in both enantiomeric series in nature. The moderately-to-densely functionalized A ring also contains a second, non-stereogenic quaternary carbon that is vicinal to the spirocycle junction, while the B ring often possesses a tri- or tetrasubstituted olefin, or functionality readily derived from such an alkene. We envisioned a strategy toward these challenging motifs based on methodological advances recently reported by our laboratories. Specifically, enantioselective decarboxylative allylation7 could be used to generate either enantiomer of the all-carbon quaternary stereocenter, while ring-closing metathesis (RCM)8 could be employed to concomitantly provide the B-ring tri- or tetrasubstituted olefin and the spirocyclic core of 10 (Eq. 1).
 |
(1) |
Through proper functionalization, we anticipated that intermediate 11 could thus serve as a common precursor to a variety of chamigrene natural products.
2. Results and discussion
2.1. Synthesis of the proposed structure of laurencenone B (7)
From the outset, we set as the final goal of our studies the preparation of elatol (1), one of the more biologically interesting and synthetically demanding chamigrenes. Initially, however, we chose to focus on a simpler target in order to validate our synthetic approach. The proposed structure of laurencenone B (7) possesses features identical to those of elatol (1) with respect to our key disconnections, namely the all-carbon quaternary stereocenter and B-ring tetrasubstituted olefin, but contains a less elaborately functionalized A ring. Also like 1,9 no total synthesis of 7 had yet been reported.10 Scheme 1 outlines our retrosynthetic analysis.11 Laurencenone B (7) could ultimately arise from spirocycle 12 through Stork-Danheiser alkylation using an appropriate methyl anion source. Compound 12 in turn could be obtained by RCM of α,ω-diene 13. Although unprecedented in the literature,12,13 we anticipated formation of a fully substituted chlorinated olefin could be achieved with the enhanced reactivity of catalyst 49 (vide infra).8 α,ω-Diene 13 itself could be the product of an enantioselective decarboxylative allylation of racemic β-ketoester (±)-14 using the Pd(0) complex of a phosphinooxazoline (PHOX) ligand.14,15 Finally, intermediate 14 could be prepared from commercially available dimedone (15) via vinylogous ester 11.16
Scheme 1.

We began our synthetic efforts with the condensation of dimedone (15) and isobutyl alcohol to afford known vinylogous ester 11 as shown in Scheme 2.16 Acylation of 11 with carbonate 16 then provided racemic β-ketoester (±)-17, albeit in low yield. Unfortunately, direct alkylation of 17 with 4-iodo-2-methyl-1-butene (18)17 did not yield any of the desired product (±)-14 even under forcing conditions. Alternative routes to (±)-14 were briefly examined; however, concurrent model studies indicated that the proposed β-ketoester (±)-14 would likely not be a viable substrate in the enantioselective decarboxylative allylation step. Racemic β-ketoester analog (±)-22, readily prepared due to the absence of β-dimethyl substitution, surprisingly provided less than 10% conversion to allylated product 24 under our standard reaction conditions at 65 °C after 22 h (Scheme 3). Although a rigorous explanation of this unexpectedly low reactivity awaits further study, we speculated that the presence of the tethered olefin substituent at the α-position may interact with the Pd(0)-PHOX complex, thus interfering with oxidative addition to the allyl ester moiety. Similar retardation of reaction rates has been observed with analogous substrates in our laboratories.18
Scheme 2.

Scheme 3a.

a dmdba = bis(3,5-dimethoxybenzylidene)acetone.
To circumvent the anticipated poor performance of proposed β-ketoester (±)-14 in the enantioselective decarboxylative allylation, we attempted to take advantage of the ability to effect this transformation through one of two other structurally discreet substrates: namely a trimethylsilyl enol ether (25) or enol carbonate (26) (Eq. 2).
 |
(2) |
Not only would this approach provide the necessary reactivity to produce α,ω-diene 13, it would also alleviate the need to preform vicinal quaternary carbon centers, allowing such functionality to be installed in the allylation step. To this end, we prepared vinylogous ester (±)-27, the immediate precursor to both 25 and 26 (Scheme 4). Direct conversion of 11 to (±)-27 by alkylation with 4-iodo-2-methyl-1-butene (18) proceeded in only low conversion;19 alternatively, 1,4-addition to methyl vinyl ketone (MVK)20 followed by Wittig methylenation of the crude product mixture led to (±)-27 in good yield over two steps. While an attempt to capture the corresponding lithium enolate of (±)-27 with trimethylsilyl chloride did not generate an isolatable product,21 treatment of the same enolate with TMEDA and chloroformate 20 afforded stable enol carbonate 26 in 73% yield. Gratifyingly, exposure of 26 to our standard allylation conditions22 at 40 °C now provided α,ω-diene (+)-13 in 52% yield by 1H NMR and 78% ee. Slightly higher enantioselectivity (81% ee) could be obtained by reaction at 24 °C,23 but at a significant expense of yield (28% by 1H NMR, 23% isolated).
Scheme 4.

Having achieved base reactivity using enol carbonate 26, we sought to further optimize the enantioselective decarboxylative allylation in order to generate a synthetically viable reaction. Based on the temperature dependence of enantioselectivity in our initial screening, we reasoned that increasing catalyst reactivity should allow access to lower reaction temperatures and thus improved ee. Careful scrutiny of the proposed catalytic pathway24 (Scheme 5) led us to hypothesize that the low reactivity of 26 was likely derived from one of three scenarios: (1) slow oxidative addition of Pd(0)-PHOX complex 29 to allyl enol carbonate 26; (2) slow decarboxylation of Pd(II)-enol carbonate 31 to provide Pd(II)-enolate 32; or (3), slow alkylation of 32 to yield the desired product (+)-13. To differentiate between these possibilities, we conducted the set of control reactions depicted in Scheme 6. Subjection of enol carbonate 26 to conditions developed in our laboratories for enantioselective decarboxylative protonation25 readily afforded olefin 27,26 demonstrating that oxidative addition and decarboxylation proceed as desired. Furthermore, less sterically-hindered deschloro analog 33 underwent facile decarboxylative allylation to yield bis(olefin) 34. From these studies, slow alkylation was determined to be the problematic step in this transformation.
Scheme 5.

Scheme 6a.

a Conditions: (a) HCO2H, Pd(OAc)2 (10 mol%), (−)-23 (12.5 mol%), MS 4Å, benzene, 40 °C, 3.1 h; (b) Pd(dmdba)2 (10 mol%), (−)-23 (13 mol%), benzene, 40 °C, 1.1 h.
In order to enhance the sluggish rate of alkylation identified above, we attempted to increase the electrophilicity of the π-allyl Pd(II) intermediate by incorporating electron-withdrawing substituents into the PHOX ligand framework.27 As shown in Table 1, moving from electron-rich to electron-deficient ligands resulted in a dramatic improvement in conversion from <20% to >99%, with good selectivity for the desired allylation product (+)-13 over the undesired protonation byproduct 27. A solvent screen employing one of the most active PHOX analogs, ligand (−)-40, revealed higher enantioselectivity with aromatic solvents compared to ethereal solvents (Table 2), while a temperature screen using the two most active ligands, (−)-40 and (−)-41, demonstrated the latter to exhibit the sharpest dependence of enantioselectivity on temperature (Table 3). Ultimately, the best compromise between reactivity and selectivity was provided by ligand (−)-41 in benzene at 13 °C (Table 3, entry 5), affording bis(olefin) (+)-13 in 90% isolated yield and 87% ee (Table 4, entry 2). A brief examination of closely related analogs of enol carbonate 26 gave similarly good yields and enantioselectivities for the corresponding α,ω-dienes (Table 4, entries 1, 3–4).
Table 1.
Effect of ligand electronics on enantioselective decarboxylative allylation.

| entry | ligand | conversiona (%) | allylationa,b (%) | eec,d (%) |
|---|---|---|---|---|
| 1 |

|
11 | 6 | n.d. |
| 2 |

|
18 | 14 | n.d. |
| 3 |

|
12 | 6 | n.d. |
| 4e |

|
59 | 52 | 78 |
| 5 |

|
75 | 60 | 76 |
| 6 |

|
98 | 87 | 83 |
| 7 |

|
>99 | 92 | 75 |
| 8 |

|
>99 | >99 | 78 |
Determined by 1H NMR spectroscopic analysis of the crude product mixture.
The remaining mass balance was comprised of protonation byproduct 27.
Determined by chiral HPLC analysis.
n.d. = not determined.
7 h reaction time.
Table 2.
Effect of solvent on enantioselective decarboxylative allylation with ligand (−)-40.

| entry | solvent | conversiona (%) | allylationa,b (%) | eec,d (%) |
|---|---|---|---|---|
| 1 | TEA | 7 | 3 | n.d. |
| 2 | Et2O | >99 | 88 | 67 |
| 3 | THF | >99 | 90 | 71 |
| 4 | 1,4-dioxane | >99 | 87 | 72 |
| 5 | toluene | >99 | 90 | 76 |
| 6 | benzene | >99 | 93 | 77 |
Determined by 1H NMR spectroscopic analysis of the crude product mixture.
The remaining mass balance was comprised of protonation byproduct 27.
Determined by chiral HPLC analysis.
n.d. = not determined.
Table 3.
Effect of temperature on enantioselective decarboxylative allylation with ligands (−)-40 and (−)-41.

| entry | ligand | solvent | T (°C) | t (h) | conversiona (%) | allylationa,b (%) | eec (%) |
|---|---|---|---|---|---|---|---|
| 1 | (−)-40 | benzene | 40 | 13 | >99 | 92 | 75 |
| 2 | (−)-40 | benzene | 30 | 13 | >99 | 93 | 77 |
| 3 | (−)-41 | benzene | 40 | 13 | >99 | >99 | 78 |
| 4 | (−)-41 | benzene | 30 | 15 | >99 | 97 | 83 |
| 5 | (−)-41 | benzene | 13 | 24 | >99 | 95 | 87 |
| 6 | (−)-41 | toluene | 0 | 17 | 72 | 63 | 89 |
Determined by 1H NMR spectroscopic analysis of the crude product mixture.
The remaining mass balance was comprised of protonation byproduct 27.
Determined by chiral HPLC analysis.
Table 4.
Scope of enantioselective decarboxylative allylation with ligand (−)-41.

| entry | enol carbonate | R | n | α,ω-diene | yielda (%) | eeb (%) |
|---|---|---|---|---|---|---|
| 1 | 42 | Me | 1 | (+)-45 | 92 | 91 |
| 2 | 26 | Me | 2 | (+)-13 | 90 | 87 |
| 3 | 43 | Me | 3 | (−)-46 | 91 | 90 |
| 4 | 44 | H | 2 | (+)-24 | 80 | 94 |
Isolated yield.
Determined by chiral HPLC analysis.
We next turned our attention to the completion of the chamigrene spirocyclic core via RCM of bis(olefin) (+)-13. In a limited survey of catalysts, second generation complex 47 provided moderate conversion to the desired tetrasubstituted olefinic product (−)-12 and minor unidentified byproducts, while complex 48 yielded comparable reactivity and substantial unknown impurities (Table 5, entries 1 and 2). Catalyst 49, however, afforded rapid and clean conversion under our standard conditions (Table 5, entry 3), furnishing spirocycle (−)-12 in 97% isolated yield (Table 6, entry 2). Interestingly, this catalytic system proved ineffective when applied to the RCM of the α,ω-diene analogs synthesized above. Removal of the dimethyl substitution vicinal to the spirocycle junction led to significantly diminished conversion (Table 6, entry 4), while extension or contraction of the homoallyl chain by a single methylene unit resulted in complete loss of reactivity (Table 6, entries 1 and 3). Similarly, a simplified acyclic substrate (53) produced little of the ring-closed product using catalyst 49 (Eq. 3).
 |
(3) |
This seemingly unique reactivity of intermediate (+)-13 was unanticipated, and efforts to understand the basis of these effects are ongoing.
Table 5.
Catalyst comparison in RCM of α,ω-diene (+)-13.




Determined by 1H NMR spectroscopic analysis using 1,4-bis(trimethylsilyl)benzene as an internal standard.
Conversion values are approximate due to overlapping of significant amounts of unknown impurities with (+)-13 in the 1H NMR spectrum.
Table 6.
Scope of RCM with catalyst 49.

| entry | α,ω-diene | R | n | spirocycle | Result |
|---|---|---|---|---|---|
| 1a | (+)-45 | Me | 1 | 50 | no conversionc |
| 2b | (+)-13 | Me | 2 | (−)-12 | 97% yieldd |
| 3a | (−)-46 | Me | 3 | 51 | no conversionc |
| 4b | (+)-24 | H | 2 | 52 | 19% conversionc,e |
C6D6.
Benzene.
Determined by 1H NMR spectroscopic analysis of the crude product mixture.
Isolated yield.
Did not determine optical rotation.
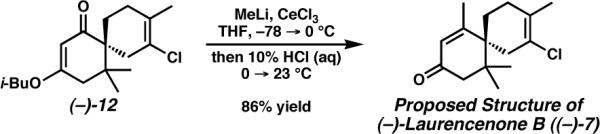
With spirocycle (−)-12 in hand, all that remained to complete Laurencenone B (7) was Stork-Danheiser alkylation using methyl anion. While methyl lithium alone added only very slowly to vinylogous ester (−)-12,28 inclusion of CeCl329 provided a significant increase in reaction rate,30 generating (−)-Laurencenone B ((−)-7) in 86% isolated yield after acid-mediated hydrolysis and elimination (Scheme 7). Overall, the first total synthesis of the proposed structure of (−)-Laurencenone B ((−)-7) was carried out in seven steps and 37% yield from dimedone (15). Unfortunately, discrepancies existed between the published 1H NMR data for the natural product2g and that of the synthetic material, although the single IR peak reported did match. Furthermore, no 13C NMR data or natural sample was available for comparison. 1H NMR, 13C NMR, and IR data for semisynthetic (+)-laurencenone B ((+)-7)10 did however match that of the synthetic material, while the opposite sign of optical rotation was obtained.31 Based on these observations,32 we conclude that the structure of the natural product designated as laurencenone B (7) has been incorrectly assigned.
Scheme 7.
2.2. Syntheses of laurencenone C (8) and α-chamigrene (5)
Having established a viable synthetic route to the chamigrene natural product family, we chose to demonstrate the generality of this strategy by preparing the related compounds laurencenone C (8)2g–h and α-chamigrene (5).2e Although four racemic total syntheses of (±)-laurencenone C ((±)-8)5a–c,e and ten of (±)-α-chamigrene ((±)-5)5a–c,33 have been previously reported, no enantioselective approaches have yet been devised.34,35 Conversion of (±)-8 to (±)-5 has been described by both Takeshita5a and Chen,5b and retrosynthetically we planned the same final two-step sequence to access α-chamigrene (5) (Scheme 8). Laurencenone C (8) could then be traced back to vinylogous ester (±)-27, the same intermediate used for (−)-Laurencenone B ((−)-7), through an analogous set of transformations. In the forward direction, acylation of compound (±)-27 with allyl chloroformate (55) proceeded smoothly to provide enol carbonate 33 in 70% yield (Scheme 9). Now in the absence of a 2-chloro substituent on the allyl moiety (cf. Table 3, entry 6), enol carbonate 33 displayed excellent reactivity at 0 °C in the enantioselective decarboxylative allylation, affording α,ω-diene (+)-34 in 87% yield and 87% ee. RCM, followed by methyl cerium addition29 31, 30 32 and acid-mediated hydrolysis/elimination, then yielded (−)-laurencenone C (8) in seven steps and 31% overall yield from dimedone (15).36 Following a modified variant of the reported procedures,37 (−)-8 was subsequently condensed with ethanedithiol (57) to afford dithiolane (−)-58 in 92% yield. Finally, reduction with Na0 5b provided (−)-α-chamigrene (5) in nine steps and 13% overall yield from 15,38 completing the first enantioselective total syntheses of both (−)-α-chamigrene (5) and (−)-laurencenone C (8).
Scheme 8.

Scheme 9.

2.3. Synthesis of elatol (1)
Following the completion of the syntheses of three simpler chamigrenes, we returned to the preparation of elatol (1) in order to demonstrate the ability of our approach to access a more complex member of this natural product class. Comparison of 1 to the proposed structure of (+)-laurencenone B ((+)-7) revealed the two molecules to be separated by three formal chemical reactions: (1) diastereoselective introduction of the 10-bromide; (2) diastereoselective reduction of the 9-ketone; and (3) transposition of the 7,8-olefin to the 7,14-position. We envisioned a simple two-step procedure to effect these transformations as shown in Scheme 10. Elatol (1) could ultimately be derived from sequential directed reductive olefin transposition and diastereoselective reduction of 10,14-dibromolaurencenone B (59) in a single pot.39 Dibromide 59 in turn could be the product of concomitant allylic and α'-bromination of (+)-laurencenone B ((+)-7).40 Substitution of PHOX ligand (+)-41 for (−)-41 in the initial synthesis of (−)-laurencenone B ((−)-7) would then provide the necessary starting material (+)-7.
Scheme 10.

Repetition of the (−)-laurencenone B ((−)-7) synthesis in the opposite enantiomeric series afforded (+)-7 in comparable yield and 87% ee as expected (Scheme 11). After surveying several electrophilic bromine sources41 and reaction conditions, conversion of (+)-7 to 10,14-dibromolaurencenone B (59) was effected in ≥8:1 dr42 using Br2 in the presence of HBr (aq)/AcOH,40c 42 presumably under thermodynamic control. Double reduction of α-bromoketone 59 with DIBAL39b 41 then provided (+)-elatol (1) in 32% isolated yield over two steps (3.9:1 syn:anti,43 11:1 SN2′:SN2).44 In comparison, reduction with LAH45 or Red-Al®46 provided ≤2:1 SN2′:SN2 for the reductive olefin transposition. The first total synthesis of (+)-elatol (1) was thus completed in only nine steps and 11% overall yield from dimedone (15).
Scheme 11.

3. Conclusion
We have successfully developed a concise and general enantioselective route to the chamigrene natural product family, resulting in the first total syntheses of (+)-elatol (1) and the proposed structure of laurencenone B (7), the first asymmetric total syntheses of (−)-α-chamigrene (5) and (−)-laurencenone C (8), and the first preparation of a fully substituted chlorinated olefin via RCM. Moreover, we have demonstrated the ability of our enantioselective decarboxylative allylation reaction to readily access enantioenriched vinylogous esters bearing vicinal quaternary carbon centers. Efforts to expand the scope of both the enantioselective decarboxylative allylation with vinylogous esters and the RCM with vinyl halides are the focus of ongoing studies.
4. Experimental
4.1. General
Unless stated otherwise, reactions were conducted under an ambient atmosphere. Anhydrous solvents were prepared by passing the solvents through activated alumina columns. Commercially obtained reagents were used as received, unless specified otherwise. Anhydrous diisopropylamine, triethylamine, N,N,N′,N′-tetramethylethylenediamine, and N,N′-dimethylethylenediamine (79) were obtained from distillation over CaH2. Methyl vinyl ketone, boron trifluoride diethyl etherate, and hexamethylphosphoramide were distilled prior to use. 4-Iodo-2-methyl-1-butene (18) was prepared according to the method of Hiersemann.17 Phosphinooxazoline ligands (−)-23 and (−)-35–41 were prepared according to the previously published method.27 28 Anhydrous CeCl3 was prepared according to the method of Imamoto.47 5-Iodo-2-methyl-1-pentene (67) was prepared according to the method of Boyer.48 Olefin 70 was prepared according to the previously published method.49 Salt 75 was prepared according to the method of Drauz.50 Phosphine 7851 was prepared according to the method of Bussaca.52 Thin-layer chromatography (TLC) was conducted with E. Merck silica gel 60 F254 pre-coated plates [0.25 mm (analytical) or 0.5 mm (preparative)] and visualized using a combination of UV light (254 nm), p-anisaldehyde staining, and potassium permanganate staining. TLC data include Rf and eluent (% by volume). ICN silica gel (particle size 0.032–0.063 mm), SiliCycle® SiliaFlash® P60 Academic Silica Gel (particle size 40–63 μm; pore diameter 60 Å), or Florisil® (100–200 mesh) were used for flash column chromatography. All flash column chromatographic purification steps are reported as follows: size of immobile phase column (length × diameter) and eluent (% by volume). Analytical chiral HPLC analyses were performed with an Agilent 1100 Series HPLC instrument. Preparative HPLC purifications were performed with a Beckman System Gold® or an Agilent 1200 Series HPLC instrument. 1H NMR spectra were recorded on a Varian Mercury 300 (at 300 MHz) or a Varian Inova 500 (at 500 MHz) instrument and are reported relative to the residual solvent peak (δ 7.26 for CDCl3, 7.16 for C6D6, and δ 2.05 for acetone-d6) or Me4Si (δ 0.00) in the case of CCl4. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. 13C NMR spectra were recorded on a Varian Mercury 300 (at 75 MHz), or a Varian Inova 500 (at 126 MHz) instrument and are reported relative the residual solvent peak (δ 77.0 for CDCl3 and 128.06 for C6D6,). Data for 13C NMR spectra are reported in terms of chemical shift (δ ppm), as well as multiplicity and coupling constant (Hz) where applicable. 19F NMR spectra were recorded on a Varian Inova 500 (at 470 MHz) instrument and are reported in terms of chemical shift (δ ppm) without the use of a reference peak. IR spectra were recorded on a Perkin Elmer Spectrum BXII spectrometer and are reported in frequency of absorption (cm−1). Optical rotations were measured with a Jasco P-1010 polarimeter, using a 100 mm path-length cell. High-resolution mass spectra were obtained from the California Institute of Technology Mass Spectral Facility.
4.2. Procedures for the synthesis of the proposed structure of (+)-laurencenone B ((+)-7)
4.2.1. Vinylogous ester 1116
To a 500 mL round-bottomed flask equipped with a magnetic stir bar, a Dean-Stark trap, and a condenser was added dimedone (15) (10.0 g, 71.3 mmol), p-toluenesulfonic acid monohydrate (269 mg, 1.41 mmol), anhydrous benzene (120 mL), and isobutanol (33.0 mL, 358 mmol). The resulting solution was then heated to reflux in an oil bath and stirred for 21 h. After cooling to 23 °C, the reaction was washed with saturated NaHCO3 (aq) (100 mL), and the aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (2 × 100 mL) and dried over MgSO4. Solvent was removed under reduced pressure, and the crude product mixture was azeotroped with toluene (4×) to remove excess isobutyl alcohol. The product was then distilled at 109–110 °C and 1.3 torr to provide vinylogous ester 1116 (12.561 g, 90%) as a slightly yellow liquid. 1H NMR (300 MHz, CDCl3) δ 5.33 (br s, 1H), 3.60 (d, J = 6.6 Hz, 2H), 2.28 (s, 2H), 2.21 (s, 2H), 2.03 (app. septet, J = 6.7 Hz, 1H), 1.07 (s, 6H), 0.97 (d, J = 6.7 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 199.6, 176.4, 101.4, 74.7, 50.7, 42.8, 32.4, 28.3, 27.7, 19.0; IR (neat film, NaCl) ν 2961, 2876, 1660, 1608, 1471, 1404, 1382, 1367, 1320, 1221, 1162, 1145, 1014, 992, 822 cm−1; HRMS (EI+) m/z calcd. for C12H20O2 [M]+: 196.1463, found 196.1463.
4.2.2. Olefin (±)-27
Part 1: Alkylation20

A 250 mL two-neck round-bottomed flask equipped with a 25 mL addition funnel, a stir bar, and two septa was flame dried under vacuum. After refilling with Ar, anhydrous THF (95 mL) and anhydrous diisopropylamine (2.0 mL, 14.3 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.6 M in hexanes, 5.4 mL, 14.0 mmol) was added dropwise via syringe and the solution stirred at 0 °C for 1 h. The resulting LDA solution was then cooled to −78 °C over 16 min. Vinylogous ester 11 (2.50 g, 12.7 mmol) in anhydrous THF (10 mL) was then added dropwise over 16 min via the addition funnel, washing the funnel with THF (5 mL) upon complete addition. A yellow solution was obtained. After stirring at −78 °C for 1 h, methyl vinyl ketone (1.05 mL, 12.9 mmol) was added dropwise quickly via syringe. A slight exotherm was observed. The reaction was stirred for an additional 3 h at −78 °C, whereupon H2O (20 mL) was added. The mixture was warmed to 23 °C, at which time the organic layer was separated. The aqueous layer was extracted with Et2O (2 × 100 mL) and EtOAc (2 × 100 mL), at which point it was completely incorporated into the organic layers. The combined organic layers were washed with saturated NaHCO3 (aq) (300 mL) and brine (2 × 300 mL), respectively, and then dried over MgSO4. Solvent was removed under reduced pressure to provide 3.469 g of ketone (±)-61 as a reddish-orange liquid containing 12.9% by mass starting vinylogous ester 11, 4.1% by mass EtOAc, and 1% by mass Et2O as determined by 1H NMR spectroscopy. The crude yield corrected for impurities was 84%. The crude material was carried directly to Part 2. An aliquot from a separate run was purified by preparative thin-layer chromatography (0.25 mm thickness, eluted twice with 30% EtOAc/hexanes eluent). Rf 0.40 (eluting twice with 30% EtOAc/hexanes eluent); 1H NMR (500 MHz, CDCl3) δ 5.20 (s, 1H), 3.57 (dd, J = 9.4, 6.7 Hz, 1H), 3.54 (dd, J = 9.5, 6.6 Hz, 1H), 2.64 (ddd, J = 18.2, 9.0, 5.2 Hz, 1H), 2.51 (ddd, J = 18.3, 9.0, 6.6 Hz, 1H), 2.36 (d, J = 17.8, 1H), 2.19 (d, J = 17.6 Hz, 1H), 2.13 (s, 3H), 2.01 (app. septet, J = 6.7 Hz, 1H), 1.82–1.92 (m, 2H), 1.53–1.64 (m, 1H), 1.08 (s, 3H), 1.00 (s, 3H), 0.96 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 209.1, 202.6, 175.0, 100.4, 74.6, 55.9, 42.2, 41.3, 35.0, 30.2, 28.5, 27.7, 24.6, 20.0, 19.1; IR (neat film, NaCl) ν 2962, 2934, 2897, 2876, 1716, 1652, 1611, 1471, 1424, 1406, 1382, 1366, 1324, 1298, 1276, 1221, 1199, 1179, 1155, 1131, 1067, 1056, 1008, 994, 968, 952, 912, 904, 851, 831, 745, 710, 693, 627 cm−1; HRMS (EI+) m/z calcd. for C16H26O3 [M]+: 266.1882, found 266.1928.
Part 2: Wittig olefination

To a flame-dried 500 mL round-bottomed flask equipped with a magnetic stir bar was added methyltriphenylphosphonium bromide (8.00 g, 22.4 mmol). After attaching an oven-dried condenser and sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous THF (255 mL) was added via syringe, and the mixture was cooled to 0 °C in an ice bath. Potassium tert-butoxide (2.39 g, 21.5 mmol) was added under positive Ar pressure to give a yellow mixture, and the system was flushed with Ar while being allowed to warm to 23 °C over 25 min. The crude ketone (±)-61 from Part 1 (3.461 g, 82% pure by mass, 10.7 mmol) in anhydrous THF (5 mL) was then added via syringe, washing the original flask with THF (5 mL). The reaction was placed in a 70 °C oil bath and stirred for 2 h. After cooling to 23 °C, the reaction mixture was filtered through a pad of silica gel (2.5 × 10 cm), eluting with THF (1 L). Solvent was removed under reduced pressure. Flash chromatography over silica gel (9 × 5 cm, 20% Et2O/pentane eluent) then provided olefin (±)-27 (2.489 g, 74% over two steps) as a slightly yellow liquid. The isolated yield for Part 2 corrected for impurities in crude ketone (±)-61 was 88%. Rf 0.33 (20% Et2O/pentane eluent); 1H NMR (500 MHz, CDCl3) δ 5.23 (s, 1H), 4.69–4.73 (m, 1H), 4.66–4.69 (m, 1H), 3.57 (d, J = 6.4 Hz, 2H), 2.33 (d, J = 17.6 Hz, 1H), 2.20 (d, J = 17.6 Hz, 1H), 2.08–2.19 (m, 1H), 1.960–2.07 (m, 2H), 1.90–1.96 (m, 1H), 1.72 (s, 3H), 1.55–1.69 (m, 2H), 1.07 (s, 3H), 0.98 (s, 3H), 0.96 (d, J = 6.8 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 202.3, 174.3, 145.9, 109.9, 100.6, 74.6, 56.3, 41.7, 36.6, 35.1, 28.6, 27.7, 24.6, 24.1, 22.6, 19.1; IR (neat film, NaCl) ν 2961, 2933, 2875, 1656, 1613, 1471, 1424, 1404, 1382, 1365, 1297, 1220, 1191, 1153, 1007, 885, 832 cm−1; HRMS (EI+) m/z calcd. for C17H28O2 [M]+: 264.2089, found 264.2081.
4.2.3.Chloroformate 2053

A 500 mL three-neck round-bottomed flask equipped with a 250 mL addition funnel, a stir bar, and three septa was flame dried under vacuum. After refilling with Ar, anhydrous Et2O (30 mL) was added via syringe, and the system was cooled to approx. −10 °C. Diphosgene (63) (7.7 mL, 63.8 mmol) was added dropwise via syringe and the solution was cooled to approx. −20 °C. 2-Chloroallyl alcohol (62) (5.0 mL, 62.8 mmol) in anhydrous Et2O (75 mL) was added dropwise via the addition funnel, and the solution was stirred at approx. −20 °C for an additional 15 min. The reaction was cooled to approx. −30 °C, and anhydrous triethylamine (8.8 mL, 63.1 mmol) in anhydrous Et2O (75 mL) was added dropwise via the addition funnel, generating a white precipitate. The reaction was then cooled to approx. −38 °C and stirred for 3 h, at which time it was allowed to warm to 23 °C while stirring for an additional 15 h. During this time, the reaction mixture became very thick, and Et2O (50 mL) was added to obtain a stirable suspension. Ar was bubbled through the suspension for 2 h to remove excess phosgene, employing a saturated NaHCO3 (aq)/triethylamine trap. The reaction mixture was filtered via vacuum filtration through a fritted funnel, and the filtrate was concentrated by rotary evaporation under reduced pressure. The product was then distilled at 50 °C and 20 torr to provide chloroformate 20 (4.987 g, 51%) as a colorless liquid containing a minor unidentified impurity (1% by area) by 1H NMR spectroscopy; 1H NMR (500 MHz, CDCl3) δ 5.58 (dt, J = 1.0, 2.0 Hz, 1H), 5.53 (d, J = 2.2 Hz, 1H), 4.84 (d, J = 0.7 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 150.4, 133.5, 117.4, 72.2; IR (CHCl3, KBr) ν 1775 cm−1; HRMS (EI+) m/z calcd. for C4H4Cl2O2 [M]+: 153.9588, found 153.9593.
4.2.4 Enol carbonate 26
A 250 mL two-neck round-bottomed flask equipped with a 50 mL addition funnel, a stir bar, and two septa was flame dried under vacuum. After refilling with Ar, anhydrous THF (55 mL) and anhydrous diisopropyl amine (1.45 mL, 10.3 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.5 M in hexanes, 3.8 mL, 9.5 mmol) was added dropwise via syringe, and the solution was stirred at 0 °C for 30 min. The resulting LDA solution was then cooled to −78 °C over 15 min. Olefin (±)-27 (2.282 g, 8.63 mmol) in anhydrous THF (10 mL) was then added dropwise over 26 min via the addition funnel, washing the funnel with THF (5 mL) upon complete addition. A yellow solution was obtained. After stirring at −78 °C for 1 h, anhydrous N,N,N′,N′-tetramethylethylenediamine (1.55 mL, 10.3 mmol) was added dropwise over 2 min via syringe. After stirring at −78 °C for an additional 1 h, chloroformate 20 (1.471 g, 9.49 mmol) in anhydrous THF (10 mL) was then added dropwise over 22 min via the addition funnel, washing the funnel with THF (5 mL) upon complete addition. A visible exotherm was observed, and the solution changed from yellow to brownish-red in color. The reaction was stirred for an additional 3 h at −78 °C, whereupon saturated NaHCO3 (aq) (35 mL) and H2O (35 mL) were added. The mixture was allowed to warm to 23 °C, at which time the organic layer was immediately separated. The aqueous layer was extracted with Et2O (2 × 200 mL), and the combined organic layers were washed with brine (2 × 200 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over Florisil® (12 × 5 cm, 2.5% Et2O/petroleum ether eluent) then provided enol carbonate 26 (2.412 g, 73%) as a colorless liquid containing minor unidentified impurities (2% by area) by 1H NMR spectroscopy. Rf 0.17 (2.5% Et2O/petroleum ether eluent); 1H NMR (300 MHz, CDCl3) δ 5.52–5.56 (m, 1H), 5.43–5.46 (m, 1H), 4.76 (s, 1 H), 4.71–4.74 (m, 2H), 4.66–4.72 (br m, 2H), 3.48 (d, J = 6.6 Hz, 2H), 2.19 (s, 2H), 2.01–2.19 (m, 4H), 1.97 (app. septet, J = 6.7 Hz, 1H), 1.74 (br s, 3H), 1.09, (s, 6H), 0.94 (d, J = 6.7 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 157.7, 153.0, 145.9, 142.0, 135.2, 123.3, 115.2, 109.8, 91.1, 73.9, 69.1, 43.4, 37.9, 35.4, 27.9, 26.0, 24.2, 22.4, 19.2; IR (neat film, NaCl) ν 3074, 2960, 2873, 1762, 1662, 1622, 1470, 1382, 1365, 1329, 1276, 1240, 1183, 1160, 1100, 1046, 1024, 972, 952, 922, 888, 782, 639 cm−1; HRMS (EI+) m/z calcd. for C21H31ClO4 [M]+: 382.1911, found 382.1912.
4.2.5. α,ω-Diene (−)-13
To a flame-dried 500 mL round-bottomed flask equipped with a magnetic stir bar was added phosphinooxazoline ligand (+)-41 (0.36 g, 0.61 mmol) and bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (0.44 g, 0.54 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (150 mL, deoxygenated prior to use by sparging with N2 in a flame-dried flask for 1.9 h) was added via syringe, and the resulting solution was stirred for 30 min at 23 °C during which time it turned dark yellow-brown. The solution was then cooled in an ice bath until solution just begins to freeze (7 min) and transfered to an 11 °C cooling bath. Concurrently, a second flame-dried round bottomed flask was charged with enol carbonate 26 (2.056 g, 5.37 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Deoxygenated anhydrous benzene (6.5 mL) was added via syringe, and the resulting solution was cooled in an ice bath, becoming very viscous at this time. The enol carbonate solution was then transferred via syringe to the catalyst solution at 11 °C, washing the original flask with deoxygenated anhydrous benzene (6.5 mL, allowed to cool briefly before transferring). The reaction, which turned green, was then stirred at 11 °C for 24 h. Solvent was removed under reduced pressure. The residue was suspended in Et2O and filtered through filter paper to remove precipitated Pd. The filtrate was then concentrated under reduced pressure. The residue was suspended in 5% Et2O/petroleum ether (some solid precipitates) and applied to a silica gel column (12 × 5 cm). Flash chromatography (5% Et2O/petroleum ether eluent) then provided α,ω-diene (−)-13 (1.488 g, 82%) as a highly viscous, slightly yellow oil. Observed 87% ee as determined by chiral HPLC analysis (Chiralpak® AD, 1% EtOH/hexanes, 1 mL/min, 254 nm, tR (minor) = 6.1 min; tR (major) = 6.7 min). Rf 0.06 (5% Et2O/petroleum ether eluent); [α]24D −10.55° (c 0.995, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.30 (br s, 1H), 5.28 (s, 1H), 5.23 (s, 1H), 4.67–4.71 (m, 1H), 4.64–4.68 (m, 1H), 3.59 (dd, J = 9.5, 6.6 Hz, 1H), 3.54 (dd, J = 9.3, 6.8 Hz, 1H), 2.94–3.18 (br m, 1H), 2.46–2.83 (br m, 1H), 2.57 (br d, J = 15.6 Hz, 1H), 2.12–2.38 (br m, 1H), 2.12–2.26 (m, 1H), 1.88–2.12 (br m, 1H), 2.01 (app. septet, J = 6.7 Hz, 1H), 1.71 (s, 3H), 1.50–1.82 (br m, 2H), 1.21 (br s, 3H), 1.11 (s, 3H), 0.97 (d, J = 6.8 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 202.6, 172.9, 146.2, 141.7 (br), 116.7, 109.5, 100.2 (br), 74.5, 54.6, 43.1, 38.9, 37.4 (br), 32.9, 28.3, 27.7, 24.8, 24.6, 22.8, 19.0; IR (neat film, NaCl) ν 3073, 2963, 2939, 2877, 1651, 1622, 1471, 1449, 1431, 1406, 1383, 1367, 1298, 1222, 1206, 1172, 1157, 1134, 1069, 1009, 967, 956, 884, 845, 767, 752, 692, 656, 624 cm−1; HRMS (EI+) m/z calcd. for C20H31ClO2 [M]+: 338.2013, found 338.2009.
4.2.6. Chloroalkene (+)-12
To a flame-dried 100 mL round-bottomed flask equipped with a magnetic stir bar was added catalyst 49 (124.9 mg, 0.219 mmol). After attaching an oven-dried condenser and sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (26 mL, deoxygenated prior to use by sparging with N2 in a flame-dried flask for ≥1 h) was added via syringe. To the resulting solution was added α,ω-diene (−)-13 (1.483 g, 4.38 mmol) in deoxygenated anhydrous benzene (6 mL) via syringe, washing the original flask with deoxygenated anhydrous benzene (2 × 6 mL). The reaction was placed in a 60 °C oil bath and stirred for 18 h. After cooling to 23 °C, solvent was removed under reduced pressure. Flash chromatography over silica gel (12 × 5 cm, 20% Et2O/petroleum ether eluent until product begins to elute, then 30% Et2O/petroleum ether eluent) then provided chloroalkene (+)-12 (1.313 g, 97%) as an off-white waxy powder. Rf 0.38 (20% Et2O/petroleum ether eluent); [α]24D +99.27° (c 1.005, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.19–5.23 (m, 1H), 3.59 (dd, J = 9.3, 6.3 Hz, 1H), 3.52 (dd, J = 9.5, 6.6 Hz, 1H), 2.50–2.70 (br m, 1H), 2.47 (br d, J = 17.1 Hz, 1H), 2.19–2.33 (br m, 1H), 1.61–2.14 (br m, 5 H), 2.01 (app. septet, J = 6.7 Hz, 1H), 1.76 (br s, 3H), 1.06 (s, 3H), 0.97 (d, J = 6.8 Hz, 6H), 0.95 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 202.3, 172.5, 127.3, 125.5 (br), 99.7, 74.5, 52.6, 41.5, 37.5, 34.4 (br), 29.5, 27.7, 25.7 (br), 24.1, 23.8, 19.7, 19.1; IR (neat film, NaCl) ν 2961, 2931, 2876, 2838, 1657, 1618, 1470, 1434, 1404, 1383, 1365, 1344, 1297, 1220, 1169, 1137, 1045, 1008, 965, 908, 858, 830, 817, 742, 685, 621 cm−1; HRMS (FAB+) m/z calcd. for C18H28ClO2 [M + H]+: 311.1778, found 311.1789.
4.2.7. Proposed structure of (+)-laurencenone B ((+)-7)29 31
In a glove box, anhydrous CeCl3 (3.973 g, 16.1 mmol) was added to a flame-dried 250 mL round-bottomed flask equipped with a magnetic stir bar. The flask was sealed with a septum and removed from the glove box. The flask was placed under high vacuum and refilled with Ar. Anhydrous THF (39 mL) was added via syringe, and the suspension was stirred for 3 h at 23 °C. The suspension was cooled to −78 °C over 15 min, and MeLi (1.37 M in Et2O, 9.0 mL, 12.3 mmol) was added dropwise over 10 min via syringe. An exotherm was observed, and the liquid phase turned yellow. After stirring for 35 min at −78 °C, chloroalkene (+)-12 (1.286 g, 4.14 mmol) in anhydrous THF (5 mL) was added dropwise over 3 min via syringe, washing the original flask with THF (2 × 4 mL). The reaction was stirred for 30 min at −78 °C and then placed in an ice bath. After stirring an additional 68 min at 0 °C, 10 wt% HCl (aq) (52 mL) was added, during which time a significant exotherm was observed. The mixture was then allowed to warm to 23 °C as it was stirred for 66 min. The reaction mixture was extracted with Et2O (3 × 150 mL), and the combined organic layers were washed with brine (2 × 250 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over silica gel (10 × 4 cm, 15% Et2O/pentane eluent until product begins to elute, then 30% Et2O/pentane eluent) then provided (+)-laurencenone B ((+)-7) (929 mg, 89%) as a white powder. Discrepancies existed between the published 1H NMR data reported in the original isolation paper2g and that of the synthetic material. IR data matched that reported for the natural product. No 13C NMR or optical rotation data were reported. 1H NMR, 13C NMR, and IR data matched that reported for semisynthetic material obtained from the degradation of elatol.10 The optical rotation matched in sign, but not in magnitude. These comparisons are outlined in the supplementary data. Rf 0.26 (30% Et2O/petroleum ether eluent); [α]24D +47.08° (c 0.36, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.89 (s, 1H), 2.50–2.69 (br m, 2H), 2.26 (br d, J = 18.3 Hz, 1H), 2.13–2.23 (br m, 1H), 1.99–2.14 (br m, 2H), 1.98 (d, J = 0.98 Hz, 3H), 1.93 (ddd, J = 12.3, 12.3, 5.4, Hz, 1H), 1.81 (br s, 3H), 1.72–1.80 (m, 1H), 1.06 (s, 3H), 0.97(s, 3H); 1H NMR (500 MHz, acetone-d6) δ 5.80 (s, 1H), 2.52–2.69 (br m, 2H), 2.31 (dd, J = 18.1, 0.98 Hz, 1H), 2.19–2.27 (br m, 1H), 2.02–2.16 (br m, 1H), 1.88–2.02 (m, 2H), 1.98 (d, J = 1.2 Hz, 3H), 1.76–1.88 (br m, 1H), 1.80 (br s, 3H), 1.09 (s, 3H), 0.96 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 198.2, 168.6, 129.6, 127.5, 126.2 (br), 48.8, 46.3, 40.4, 36.3, 30.4 (br), 30.1, 24.8, 23.9 (two overlapping CH3 peaks as determined by gHSQC and DEPT NMR spectroscopy; second peak appears as an upfield shoulder), 19.7; IR (neat film, NaCl) ν 3025, 2963, 2933, 2855, 1667, 1612, 1462, 1439, 1417, 1392, 1375, 1350, 1331, 1319, 1306, 1282, 1257, 1207, 1192, 1171, 1150, 1137, 1125, 1106, 1070, 1048, 1023, 1010, 988, 972, 954, 930, 913, 869, 834, 818, 684 cm−1; HRMS (EI+) m/z calcd. for C15H21ClO [M]+: 252.1281, found 252.1270.
4.3. Procedures for the synthesis of the proposed structure of (−)-laurencenone B ((−)-7)
4.3.1. α,ω-Diene (+)-13
Following a similar procedure to that for the preparation of α,ω-diene (−)-13, enol carbonate 26 (299.9 mg, 0.783 mmol) was converted to α,ω-diene (+)-13 (240.2 mg, 90%) using phosphinooxazoline ligand (−)- 41 (52.3 mg, 0.0884 mmol), bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (63.7 mg, 0.0781 mmol), and deoxygenated anhydrous benzene (24 mL total) at 12 °C. Observed 87% ee as determined by chiral HPLC analysis (Chiralpak® AD, 1% EtOH/hexanes, 1 mL/min, 254 nm, tR (major) = 6.0 min; tR (minor) = 6.6 min). 1H NMR data matched that of α,ω-diene (−)-13. [α]25D +10.46° (c 1.005, CHCl3).
4.3.2. Chloroalkene (−)-12
Following a similar procedure to that for the preparation of chloroalkene (+)-12, α,ω-diene (+)-13 (229.7 mg, 0.678 mmol) was converted to chloroalkene (−)-12 (204.7 mg, 97%) using catalyst 49 (19.3 mg, 0.0338 mmol) and deoxygenated anhydrous benzene (7 mL total). 1H NMR data matched that of chloroalkene (+)-12. [α]26D −101.03° (c 1.0, CHCl3).
4.3.3. Proposed structure of (−)-laurencenone B ((#x2212;)-7
Following a similar procedure to that for the preparation of the proposed structure of (+)-laurencenone B ((+)-7), chloroalkene (−)-12 (99.9 mg, 0.321 mmol) was converted to the proposed structure of (−)-laurencenone B ((−)-7) (69.6 mg, 86%) using anhydrous CeCl3 (312 mg, 1.27 mmol), MeLi (1.3 M in Et2O, 0.74 mL, 0.962 mmol), anhydrous THF (4 mL total), and 10 wt% HCl (aq) (4 mL). A minor unidentified impurity was observed by 1H NMR spectroscopy (≤3 mol% as determined by DEPT NMR spectroscopy). 1H NMR data matched that of (+)-laurencenone B ((+)-7). [α]25D −42.01° (c 0.357, CHCl3).
4.4. Procedures for the synthesis of (−)-laurencenone C (8)
4.4.1. Enol carbonate 33
A 250 mL two-neck round-bottomed flask equipped with a 50 mL addition funnel, a stir bar, and two septa was flame dried under vacuum. After refilling with Ar, anhydrous THF (40 mL) and anhydrous diisopropylamine (1.45 mL, 10.3 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.51 M in hexanes, 3.8 mL, 9.54 mmol) was added dropwise via the addition funnel over 22 min, washing the funnel sequentially with anhydrous hexanes (5 mL) and anhydrous THF (5 mL) upon complete addition. The reaction was then stirred at 0 °C for 30 min. The resulting LDA solution was cooled to −78 °C over 30 min. Olefin (±)-27 (2.285 g, 8.64 mmol) in anhydrous THF (10 mL) was then added dropwise over 11 min via the addition funnel, washing the funnel with anhydrous THF (5 mL) upon complete addition. A yellow solution was obtained. After stirring at −78 °C for 66 min, anhydrous N,N,N′,N′-tetramethylethylenediamine (1.55 mL, 10.3 mmol) was added dropwise over 2 min via syringe. After stirring at −78 °C for an additional 69 min, allyl chloroformate (55) (1.0 mL, 9.41 mmol) in anhydrous THF (10 mL) was then added dropwise over 21 min via the addition funnel, washing the funnel with THF (5 mL) upon complete addition. The reaction was stirred for an additional 3 h at −78 °C, at which time saturated NaHCO3 (aq) (35 mL) and H2O (35 mL) were added. The mixture was allowed to warm to 23 °C with the aid of a 23 °C water bath, at which time the organic layer was immediately separated. The aqueous layer was extracted with Et2O (2 × 200 mL), and the combined organic layers were washed with brine (2 × 200 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over Florisil® (12.5 × 5 cm, 2.5% Et2O/petroleum ether eluent) then provided enol carbonate 33 (2.101 g, 70%) as a slightly pale yellow liquid containing residual Et2O (<1% by mass) and minor unidentified impurities (5% by area) by 1H NMR spectroscopy. Rf 0.21 (2.5% Et2O/petroleum ether eluent); 1H NMR (500 MHz, CDCl3) δ 5.96 (ddt, J = 17.1, 10.5, 5.7 Hz, 1H), 5.39 (ddt, J = 7.1, 1.5, 1.5 Hz, 1H), 5.28 (ddt, J = 10.5, 1.2, 1.2 Hz, 1H), 4.76 (s, 1H), 4.66–4.71 (m, 2H), 4.66 (ddd, J = 5.7, 1.4, 1.4 Hz, 2H), 3.47 (d, J = 6.6 Hz, 2H), 2.19 (s, 2H), 2.09–2.17 (m, 2H), 2.01–2.09 (m, 2H), 1.97 (app. septet, J = 6.7 Hz, 1H), 1.74 (br s, 3H), 1.08 (s, 6H), 0.94 (d, J = 6.8 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 157.5, 153.5, 146.0, 141.9, 131.5, 123.1, 119.0, 109.7, 91.3, 73.8, 68.6, 43.4, 37.9, 35.4, 27.8, 26.0, 24.1, 22.4, 19.2; IR (neat film, NaCl) ν 2960, 2874, 1758, 1662, 1622, 1470, 1422, 1382, 1363, 1330, 1292, 1241, 1160, 1146, 1109, 1041, 1024, 994, 970, 952, 887, 784 cm−1; HRMS (EI+) m/z calcd. for C21H32O4 [M]+: 348.2301, found 348.2307.
4.4.2. α,ω-Diene (+)-34
To a flame-dried 250 mL round-bottomed flask equipped with a magnetic stir bar was added phosphinooxazoline ligand (−)-41 (192 mg, 0.325 mmol) and bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (234 mg, 0.287 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous toluene (77 mL) was added via syringe, and the resulting solution was stirred for 31 min at 23 °C. The solution was then cooled to 0 °C over 15 min in an ice bath. Concurrently, a second flame-dried round bottomed flask was charged with enol carbonate 33 (1.003 g, 2.88 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous toluene (5 mL) was added via syringe, and the resulting solution was cooled to 0 °C in an ice bath. The enol carbonate solution was then transferred via cannula under positive N2 pressure to the catalyst solution, washing the original flask with anhydrous toluene (5 mL, allowed to cool briefly before transferring). The reaction was then stirred at 0 °C for 9.5 h, at which point solvent was removed under reduced pressure. Flash chromatography over silica gel (12.5 × 5 cm, 5% Et2O/petroleum ether eluent) then provided α,ω-diene (+)-34 (760 mg, 87%) as a yellow liquid. Observed 87% ee as determined by chiral HPLC analysis (Chiralcel® OD-H, 0.25% EtOH/hexanes, 1 mL/min, 254 nm, tR (minor) = 9.0 min; tR (major) = 10.3 min). Rf 0.07 (5% Et2O/petroleum ether eluent); [α]24D +25.52° (c 0.995, CHCl3);1H NMR (500 MHz, CDCl3) δ 5.85–6.04 (br m, 1H), 5.24 (d, J = 0.97 Hz, 1H), 5.03–5.10 (m, 1H), 4.96–5.01 (m, 1H), 4.65–4.69 (m, 1H), 4.64 (br s, 1H), 3.58 (dd, J = 9.5, 6.6 Hz, 1H), 3.54 (dd, J = 9.3, 6.8 Hz, 1H), 2.35–2.90 (br m, 2H), 2.28 (dd, J = 15.4, 8.1 Hz, 1H), 1.94–2.10 (br m, 2H), 2.01 (app. septet, J = 6.7 Hz, 1H), 1.62–1.89 (br m, 3H), 1.69 (s, 3H), 1.10 (s, 3H), 1.07 (s, 3H), 0.96 (d, J = 6.8, 6H); 13C NMR (75 MHz, CDCl3) δ 203.2, 173.0, 146.5, 137.1 (br), 116.1, 109.3, 100.7, 74.5, 53.5, 42.7, 38.7, 33.7 (br), 32.8, 28.7, 27.7, 25.5, 25.3, 22.8, 19.1; IR (neat film, NaCl) ν 3072, 2961, 2937, 2876, 1653, 1621, 1471, 1449, 1405, 1383, 1366, 1297, 1221, 1207, 1172, 1149, 1009, 995, 967, 955, 907, 885, 846, 833, 768, 755, 654 cm−1; HRMS (EI+) m/z calcd. for C20H32O2 [M]+: 304.2402, found 304.2388.
4.4.3. Olefin (−)-56
To a flame-dried 50 mL round-bottomed flask equipped with a magnetic stir bar was added catalyst 49 (70.6 mg, 0.124 mmol). After attaching an oven-dried condenser and sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (20 mL, deoxygenated prior to use by sparging with N2 in a flame-dried flask for 1 h) was added via syringe. To the resulting solution was added α,ω-diene (+)-34 (750 mg, 2.46 mmol) in deoxygenated anhydrous benzene (3 mL) via syringe, washing the original flask with deoxygenated anhydrous benzene (2 × 1 mL). The reaction was placed in a 60 °C oil bath and stirred for 11.5 h. After cooling to 23 °C, solvent was removed under reduced pressure. Flash chromatography over silica gel (9 × 3 cm, 20% Et2O/petroleum ether eluent until product begins to elute, then 30% Et2O/petroleum ether eluent) then provided olefin (−)-56 (663 mg, 97%) as a white powder. Rf 0.23 (20% Et2O/petroleum ether eluent); [α]25D −91.53° (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.38–5.50 (br m, 1H), 5.17–5.24 (m, 1H), 3.58 (dd, J = 9.3, 6.8 Hz, 1H), 3.52 (dd, J = 9.5, 6.6 Hz, 1H), 2.40–2.82 (br m, 1H), 2.13–2.23 (br m, 1H), 1.87–2.13 (br m, 2H), 2.00 (app. septet, J = 6.7 Hz, 1H), 1.58–1.87 (br m, 4H), 1.64 (d, J = 1.5 Hz, 3H), 1.03 (s, 3H), 0.96 (d, J = 6.3 Hz, 6H), 0.94 (s, 3H); 13C NMR (75 MHz, CDCl3, overlapping of resonances was observed) δ 203.7, 172.3, 132.1 (br), 120.4 (br), 99.9, 74.4, 49.9, 41.5, 37.6, 27.72, 27.68, 26.1 (br), 24.2, 23.9, 23.4, 19.1; IR (neat film, NaCl) ν 3066, 3017, 2960, 2923, 2901, 2876, 2835, 1657, 1620, 1470, 1445, 1403, 1383, 1364, 1294, 1262, 1218, 1201, 1169, 1133, 1077, 1033, 1009, 968, 944, 906, 860, 830, 797, 765, 730, 714, 656 cm−1; HRMS (FAB+) m/z calcd. for C18H29O2 [M + H]+: 277.2168, found 277.2155.
4.4.4. (−)-Laurencenone C (8)
In a glove box, anhydrous CeCl3 (689 mg, 2.80 mmol) was added to a flame-dried 50 mL round-bottomed flask equipped with a magnetic stir bar. The flask was sealed with a septum and removed from the glove box. The flask was placed under high vacuum and refilled with Ar. Anhydrous THF (7 mL) was added via syringe, and the suspension was stirred for 2 h at 23 °C. The suspension was cooled to −78 °C over 15 min, and MeLi (1.30 M in Et2O, 1.7 mL, 2.21 mmol) was added dropwise over 5 min via syringe. An exotherm was observed, and the liquid phase turned yellow. After stirring for 30 min at −78 °C, olefin (−)-56 (200.3 mg, 0.725 mmol) in anhydrous THF (1 mL) was added dropwise over 7 min via syringe, washing the original flask with THF (1 mL). The reaction was stirred for 35 min at −78 °C and then placed in an ice bath. After stirring an additional 65 min at 0 °C, 10 wt% HCl (aq) (9 mL) was added, during which time a significant exotherm was observed. The mixture was then allowed to warm to 23 °C as it was stirred for 1.5 h. The reaction mixture was extracted with Et2O (3 × 50 mL), and the combined organic layers were washed with brine (2 × 150 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over silica gel (10 × 3 cm, 10% Et2O/pentae eluent until product begins to elute, then 30% Et2O/pentane eluent) then provided (−)-laurencenone C (8) (127 mg, 80%) as a colorless oil containing a minor unidentified impurity (2% by area) by 1H NMR spectroscopy. Discrepancies existed between the published 1H NMR data reported in the original isolation paper2g and that of the synthetic material. IR data matched that reported for the natural product. No 13C NMR or optical rotation data were reported. 1H NMR, 13C NMR, and IR data matched that reported for isolation of the natural product from a second source,2h with the exception of one 13C NMR resonance. The optical rotation matched in sign, but not in magnitude. These comparisons are outlined in the supplementary data. Rf 0.28 (30% Et2O/petroleum ether eluent); [α]26D −87.98° (c = 1.00, CHCl3);1H NMR (500 MHz, CDCl3) δ 5.87 (br s, 1H), 5.47–5.53 (br m, 1H), 2.48–2.76 (br m, 1H), 2.19–2.28 (br m, 1H), 1.82–2.14 (br m, 5H), 1.97 (d, J = 1.5 Hz, 3H), 1.71–1.82 (br m, 1H), 1.68 (br s, 3H), 1.03 (s, 3H), 0.95 (s, 3H);1H NMR (500 MHz, acetone-d6) δ 5.76 (br s, 1H), 5.49–5.55 (br m, 1H), 2.46–2.73 (br m, 1H), 2.21–2.31 (br m, 1H), 1.74–2.12 (br m, 6H), 1.97 (d, J = 1.5 Hz, 3H), 1.65–1.69 (br m, 3H), 1.03 (s, 3H), 0.93 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 198.6, 170.4, 134.1, 127.0, 121.6, 48.9, 43.4, 40.4, 30.6 (br), 28.2, 27.9, 24.8, 24.2, 23.9 (br), 23.3; IR (neat film, NaCl) ν 3016, 2963, 2928, 2907, 2879, 2853, 1667, 1610, 1447, 1437, 1418, 1390, 1376, 1332, 1320, 1282, 1257, 1209, 1190, 1170, 1146, 1122, 1074, 1036, 1022, 977, 944, 912, 870, 834, 818, 800, 767 cm−1; HRMS (EI+) m/z calcd. for C15H22O [M]+: 218.1671, found 218.1673.
4.5. Procedures for the synthesis of (−)-α-chamigrene (5)
4.5.1. Dithiolane (−)-5837 39
To a flame-dried 1 dram screw-cap vial equipped with a stir bar and a septum-bearing cap was added laurencenone C (8) (88.3 mg, 0.404 mmol). The flask was placed under high vacuum and refilled with Ar. Anhydrous MeOH (3 mL) followed by ethanedithiol (57) (60 μL, 0.715 mmol) were added via syringe. Boron trifluoride diethyl etherate (60 μL, 0.486 mmol) was then added via syringe in two portions (41 and 19 μL, respectively) 20 min apart, and the reaction was stirred for a total of 27 h at 23 °C. During this time, a fine dark precipitate was observed. MeOH was removed under reduced pressure, and the residue was dissolved in EtOAc (50 mL). The solution was washed sequentially with saturated NaHCO3 (aq) (2 × 25 mL), H2O (2 × 25 mL), and brine (2 × 25 mL) and then dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over silica gel (7.5 × 2 cm, 2.5% Et2O/pentane eluent) then provided dithiolane (−)-58 (109.6 mg, 92%) as a colorless oil containing minor unidentified impurities (3% by area by 1H NMR spectroscopy) which were present in the starting ethanedithiol. 1H NMR data matched that of the reported racemic compound.5a–b R f 0.29 (2.5% Et2O/petroleum ether eluent); [α]25D −51.47° (c 1.015, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.53–5.56 (m, 1H), 5.42–5.46 (m, 1H), 3.26–3.44 (m, 4H), 2.63 (d, J = 14.7 Hz, 1H), 2.06–2.16 (m, 2H), 1.81–1.95 (m, 3H), 1.60–1.71 (m, 2H), 1.69 (d, J = 0.98 Hz, 3H), 1.64 (bs, 3H), 0.97 (s, 3H), 0.91 (s, 3H).
4.5.2. (−)-α-Chamigrene (5)5b
A 100 ml three-neck round-bottomed flask equipped with a cold finger, a stir bar, and three septa was flame-dried under vacuum. After refilling with Ar, the flask was placed in a dry ice/CHCl3 bath which was used to maintain a reaction temperature between −60 and −45 °C, while the cold finger was maintained at −78 °C with dry ice and acetone. NH3 (g) was condensed within the flask to give a solvent volume of approx. 10 mL. A solution of dithiolane (−)-58 (110 mg, 0.373 mmol) in anhydrous Et2O (14 mL) cooled to −60 °C was then added via cannula under positive N2 pressure, washing the original flask with anhydrous Et2O (6 mL, allowed to cool briefly before transferring). Na0 (approx. 146 mg, 6.35 mmol) was added portion-wise under positive Ar pressure in small pieces, during which time the solution turned a persistent purple color. After complete addition, the reaction was stirred for 15 min, and the cold bath was then removed giving a gentle reflux while stirring for an additional 55 min. MeOH (1.5 mL) was added portion-wise via syringe to minimize any exotherm, and a mixture of a colorless solution and a white precipitate was obtained. The cold finger was then allowed to warm to 23 °C while the NH3 (l) and Et2O evaporated overnight. H2O (40 mL) was added to dissolve most solids, and the reaction was extracted with Et2O (3 × 50 mL). The organic layer was washed with brine (2 × 150 mL) and dried over MgSO4. Solvent was removed under reduced pressure. The crude product was then split into two approximately equal portions, each of which was subjected to preparative thin-layer chromatography (0.5 mm thickness, 100% hexanes eluent). The product was extracted from the silica with hexanes, and solvent was removed under reduced pressure to provide a combined yield of 33.6 mg (44%) of (−)-α-chamigrene (5) as a colorless oil. 1H NMR and IR data matched that reported in the original isolation paper,2e and these comparisons are outlined in the supplementary data. Rf 0.80 (100% hexanes eluent); [α]25D −64.60° (c 0.21, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.44–5.48 (m, 1H), 5.31–5.35 (br m, 1H) 2.12 (dddd, J = 18.1, 5.1, 2.4, 2.4 Hz, 1H), 1.80–2.01 (br m, 5H), 1.60–1.79 (m, 9H), 1.19 (ddd, J = 13.7, 6.1, 4.39 Hz, 1H), 0.89 (s, 3H), 0.83 (s, 3H); 1H NMR (300 MHz, CCl4) δ 5.36–5.43 (m, 1H), 5.23–5.29 (m, 1H), 2.09 (dddd, J = 18.1, 4.9, 2.4, 2.4 Hz, 1H), 1.55–2.02 (m, 14H), 1.17 (ddd, J = 13.3, 4.9, 4.9 Hz, 1H), 0.89 (s, 3H), 0.82 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 140.3, 133.8, 122.8, 122.5, 40.5, 35.9, 32.8, 30.7, 29.1, 28.9, 25.1, 23.47, 23.46, 23.3 (br), 23.0; IR (neat film, NaCl) ν 3055, 3015, 2958, 2923, 2841, 2729, 2675, 1683, 1655, 1640, 1473, 1463, 1448, 1436, 1386, 1380, 1366, 1342, 1320, 1312, 1280, 1258, 1248, 1220, 1188, 1163, 1148, 1129, 1088, 1072, 1029, 1020, 1006, 978, 954, 943, 901, 832, 810, 800, 760, 688, 661, 605 cm−1; HRMS (EI+) m/z calcd. for C15H24 [M]+: 204.1878, found 204.1883.
4.6. Procedures for the synthesis of (+)-elatol (1)
4.6.1. (+)-Elatol (1)
Part 1: Bromination40c 42
A half-dram screw-cap vial equipped with a stir bar and a septum-bearing cap was charged with (+)-laurencenone B ((+)-7) (9.9 mg, 0.0392 mmol). A solution of 48% HBr (aq) in glacial acetic acid [250:1 v/v AcOH:48% HBr (aq), 0.2 mL] was then added. To the resulting solution was added in one portion a stock solution of bromine in 250:1 v/v AcOH:48% HBr (aq) (0.501 M, 157.5 μL, 0.0789 mmol), resulting in a deep red color. After stirring for approx. 10 min, the reaction turned an orange-red color. After stirring for a total of 31 min, the solution was added to ice water (5 mL). The resulting mixture was extracted with Et2O (4 × 10 mL), and the combined organic layers were washed with saturated NaHCO3 (aq) (2 × 20 mL) and brine (2 × 20 mL). After drying over MgSO4, solvent was removed under reduced pressure, ultimately isolating the crude dibromide 59 in a 1 dram screw-cap vial. The crude material was carried directly to Part 2. A scalemic mixture from additional runs was partially purified (approx. 93% pure by area by 1H NMR spectroscopy) by preparative HPLC (Phenomenex® Gemini™, 25®100% CH3CN/0.1% TFA (aq), 8 mL/min, 250 nm, tR = 70–72 min). 1H NMR (500 MHz, C6D6) δ 6.05 (s, 1H), 4.55 (s, 1H), 3.67 (d, J = 10.7 Hz, 1H), 3.36–3.41 (m, 1H), 2.56–2.67 (m, 1H), 2.22–2.32 (br m, 1H), 1.52–1.56 (br m, 3H), 1.35–1.44 (br m, 1H), 1.14–1.25 (br m, 1H), 1.08 (ddd, J = 12.5, 12.5, 5.1 Hz, 1H), 0.80 (s, 3H), 0.68–0.76 (br m, 1H), 0.74 (s, 3H);13C NMR (126 MHz, CDCl3) δ 190.1, 164.1, 129.9, 129.8, 126.1, 65.7, 49.1, 47.1, 36.5, 30.9, 30.3, 29.7, 24.3, 19.8, 18.7; IR (neat film, NaCl) ν 2979, 2932, 1678, 1438, 1396, 1376, 1284, 1251, 1216, 1170, 1133, 1007, 911, 868, 818, 760, 733 cm−1; HRMS (EI+) m/z calcd. for C15H19ClOBr81Br [M]+: 409.9471, found 409.9482. A minor diastereomer was not isolated. The dr was determined to be ≥8:1 by 1H NMR spectroscopic analysis of the crude product of a third run. This minimal value was based upon resonances that could not be ruled out as corresponding to a minor diastereomer based on the preparative HPLC separation above.
Part 2: Reduction39b 41
The 1 dram screw-cap vial containing the crude dibromide 59 from Part 1 was equipped with a stir bar and a septum-bearing cap and placed under high vacuum. After refilling with Ar, anhydrous THF (0.3 mL) was added via syringe, and the solution was cooled to −78 °C. Concurrently, a second 1 dram screw-cap vial equipped with a septum-bearing cap was flame-dried under high vacuum and refilled with Ar. A stock solution of diisobutylaluminum hydride in anhydrous THF (1.59 M, 0.15 mL, 0.239 mmol) was added via syringe, and the solution was cooled to −78 °C. The precooled stock solution was then added to the crude dibromide 59 solution at −78 °C in one portion via cannula under positive N2 pressure, whereupon gas evolution was observed. The colorless solution was stirred for 2 h at −78 °C, at which point the reaction was placed in a 60 °C heating block, and the Ar inlet was removed. After stirring for an additional 2 h at 60 °C, the reaction was cooled to 23 °C with the aid of a 23 °C water bath. Et2O (0.5 mL) was added, and the reaction was quenched with a saturated solution of potassium sodium tartrate (aq) (0.5 mL), whereupon gas evolution was observed. After stirring vigorously for 1 h, the opaque biphasic mixture was transferred to a 25 mL round-bottomed flask with Et2O (5 mL) and a saturated solution of potassium sodium tartrate (aq) (5 mL). The mixture was then stirred vigorously until both layers were transparent. The organic layer was separated, and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic layers were washed with brine (2 × 20 mL) and dried over MgSO4. Solvent was removed under reduced pressure. The ratio of SN2′:SN2 reduction was determined to be 11:1 by 1H NMR spectroscopic analysis of the crude reaction mixture. The crude product was azeotroped with hexanes (4×) and then subjected to preparative thin-layer chromatography (0.5 mm thickness, eluted four times with 15% Et2O/petroleum ether eluent, then once with 15% EtOAc/hexanes eluent). The product was extracted from the silica with EtOAc and azeotroped with hexanes (3×). Finally, flash chromatography over silica gel (5.3 × 0.5 cm, 5% EtOAc/hexanes eluent until product begins to elute, then 30% EtOAc/hexanes eluent) then provided (+)-elatol (1) (4.4 mg) as a colorless oil containing 6% of the undesired SN2 reduction product by 1H NMR spectroscopy. The corrected yield was 32% over two steps. The dr of the initial ketone reduction was determined to be 3.9:1 syn:anti (relative to the bromide) by 1H NMR spectroscopic analysis of the crude product of separate run quenched at −78 °C after 2 h. 1H NMR data matched that reported in the original isolation paper,2a and this comparison is outlined in the supplementary data. Rf 0.64 (eluted four times with 15% Et2O/petroleum ether eluent, then once with 15% EtOAc/hexanes eluent); [α]23D +92.09° (c 0.22, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.13 (s, 1H), 4.80 (s, 1H), 4.61 (d, J = 2.9 Hz, 1H), 4.13–4.17 (m, 1H), 2.55–2.66 (br m, 2H), 2.51 (dd, J = 14.6, 2.7 Hz, 1H), 2.33–2.41 (br m, 1H), 2.16–2.20 (m, 1H), 1.92–2.00 (br m, 1H), 1.75–1.86 (br m, 2H), 1.70 (br s, 3H), 1.58–1.67 (m, 1H), 1.08 (s, 3H), 1.07 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 140.7, 128.1, 124.1, 115.9, 72.2, 70.9, 49.1, 43.1, 38.6, 37.9, 29.3, 25.6, 24.2, 20.8, 19.4; IR (neat film, NaCl) ν 3564, 3475, 3086, 2975, 2949, 2913, 2831, 1808, 1724, 1681, 1643, 1470, 1453, 1430, 1394, 1372, 1354, 1340, 1319, 1292, 1254, 1225, 1207, 1188, 1165, 1141, 1130, 1106, 1086, 1073, 1031, 1014, 982, 956, 943, 897, 879, 826, 811, 761, 737, 685, 706, 667, 642, 615 cm−1; HRMS (EI+) m/z calcd. for C15H22BrClO [M]+: 332.0543, found 332.0546.
4.6.2. Analysis of a natural sample of (+)-elatol (1)
A natural sample of (+)-elatol (1) provided by Prof. Mercedes Cueto was identical in all respects to the synthetic material except for its optical rotation: [α]25D +109.78° (c 0.045, CHCl3).
4.6.3. Characterization data for diastereomers from the initial ketone reduction of crude dibromide 59
cis-Bromohydrin 60

Rf 0.54 (30% EtOAc/hexanes eluent); 1H NMR (500 MHz, CDCl3) δ 6.14 (d, J = 3.9 Hz, 1H), 4.79 (d, J = 5.1 Hz, 1H), 4.22–4.27 (br m, 1H), 4.04–4.11 (m, 2H), 2.73–2.81 (br m, 1H), 2.50–2.59 (br m, 1H), 2.38 (d, J = 3.4 Hz, 1H), 2.10–2.18 (br m, 1H), 1.92–2.03 (br m, 1H), 1.76–1.85 (m, 4H), 1.53–1.60 (m, 1H), 1.14 (s, 3H), 1.07 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 143.3, 130.9, 129.6, 127.0, 68.6, 65.9, 47.7, 41.1, 36.9, 33.5, 30.2, 29.8, 24.3, 19.8, 19.0; IR (neat film, NaCl) ν 3541, 3455, 2976, 2932, 1678, 1438, 1219, 731 cm−1; HRMS (EI+) m/z calcd. for C15H21ClOBr81 Br [M]+: 411.9627, found 411.9620. The relative stereochemistry of the bromohydrin functionality was established by NOESY1D NMR spectroscopy and subsequent conversion to elatol (1).
trans-Bromohydrin 64

Rf 0.44 (30% EtOAc/hexanes eluent); 1H NMR (500 MHz, CDCl3) δ 6.00 (d, J = 3.4 Hz, 1H), 4.48 (d, J = 9.0 Hz, 1H), 4.35–4.41 (br m, 1H), 4.03–4.11 (m, 2H), 2.77–2.84 (br m, 1H), 2.54–2.63 (br m, 1H), 2.43 (d, J = 3.7 Hz, 1H), 2.10–2.19 (br m, 1H), 1.89–2.00 (br m, 1H), 1.87 (ddd, J = 12.2, 12.2, 5.0 Hz, 1H), 1.80–1.84 (br m, 3H), 1.62–1.69 (m, 1H), 1.16, (s, 3H), 1.05 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 143.6, 131.6, 129.6, 126.9, 73.3, 70.5, 47.4, 44.3, 36.3, 33.8, 31.4, 29.7, 24.6, 19.8, 17.6; IR (neat film, NaCl) ν 3383, 2977, 2932, 1677, 1438, 1039, 733 cm−1; HRMS (EI+) m/z calcd. for C15H21ClOBr81 Br [M]+: 411.9627, found 411.9621.
4.7. Procedures for the syntheses of enantioselective decarboxylative allylation products (+)-45, (+)-46, and (+)-24
4.7.1. Olefin (±)-66

A 250 mL round-bottomed flask equipped with a stir bar and a septum was flame dried under vacuum. After refilling with Ar, anhydrous THF (42 mL) and anhydrous diisopropylamine (1.6 mL, 11.4 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.57 M in hexanes, 4.1 mL, 10.5 mmol) was added dropwise via syringe, and the solution was stirred at 0 °C for 33 min. The resulting LDA solution was then cooled to −78 °C over 15 min. Vinylogous ester 11 (2.0 g, 10.2 mmol) in anhydrous THF (2 mL) was then added dropwise over 2 min via syringe, washing the original flask with anhydrous THF (2 × 2 mL). The reaction was then stirred for 1 h at −78 °C to provide a yellow solution. Methallyl bromide (65) (1.0 mL, 9.92 mmol) was added dropwise over 2 min via syringe. The cold bath was removed, and the reaction was allowed to warm to 23 °C as it was stirred for 26.6 h. Saturated NH4Cl (aq) (40 mL) was added, and the organic layer was separated. The aqueous layer was extracted with Et2O (2 × 150 mL), and the combined organic layers were washed with brine (2 × 200 mL). After drying over MgSO4, solvent was removed under reduced pressure. Flash chromatography over silica gel (15.5 × 5 cm, 15% Et2O/petroleum ether eluent) then provided olefin (±)-66 (2.135 g, 84%) as a slightly yellow liquid. Rf 0.10 (15% Et2O/petroleum ether eluent); 1H NMR (300 MHz, CDCl3) δ 5.24 (br s, 1H), 4.71–4.76 (br m, 1H), 4.64–4.69 (br m, 1H), 3.57 (d, J = 6.4 Hz, 2H), 2.12–2.40 (m, 5H), 2.02 (app. septet, J = 6.7 Hz, 1H), 1.78 (br s, 3H), 1.09 (s, 3H), 0.99 (s 3H), 0.97 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 201.4, 174.1, 144.6, 111.4, 100.5, 74.5, 41.6, 35.1, 34.7, 28.6, 27.7, 24.5, 22.3, 19.07, 19.06; IR (neat film, NaCl) ν 3074, 2962, 2933, 2875, 1661, 1615, 1471, 1425, 1404, 1382, 1364, 1315, 1299, 1278, 1222, 1192, 1178, 1152, 1131, 1008, 968, 954, 885, 834, 815, 686, 626 cm−1; HRMS (EI+) m/z calcd. for C16H26O2 [M]+: 250.1933, found 250.1945.
4.7.2. Enol carbonate 42

A 250 mL two-neck round-bottomed flask equipped with a 10 mL addition funnel, a stir bar, and two septa was flame dried under vacuum. After refilling with Ar, anhydrous THF (25 mL) and anhydrous diisopropylamine (0.67 mL, 4.78 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.57 M in hexanes, 1.7 mL, 4.37 mmol) was added dropwise via syringe, and the solution was stirred at 0 °C for 33 min. The resulting LDA solution was then cooled to −78 °C over 18 min. Olefin (±)-66 (1.0 g, 3.99 mmol) in anhydrous THF (5 mL) was then added dropwise over 8 min via the addition funnel, washing the funnel with THF (2.5 mL) upon complete addition. After stirring at −78 °C for 1 h, anhydrous N,N,N′,N′-tetramethylethylenediamine (0.72 mL, 4.80 mmol) was added dropwise over 2 min via syringe. After stirring at −78 °C for an additional 1 h, chloroformate 20 (0.68 g, 4.39 mmol) in anhydrous THF (5 mL) was then added dropwise over 19 min via the addition funnel, washing the funnel with THF (2.5 mL) upon complete addition. The reaction was stirred for an additional 3 h at −78 °C, whereupon saturated NaHCO3 (aq) (17 mL) and H2O (17 mL) were added. The mixture was allowed to warm to 23 °C with the aid of a 23 °C water bath, at which time the organic layer was immediately separated. The aqueous layer was extracted with Et2O (2 × 100 mL), and the combined organic layers were washed with brine (2 × 100 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over Florisil® (100–200 mesh, 13.5 × 5 cm, 1% Et2O/petroleum ether eluent until product begins to elute, then 2.5% Et2O/petroleum ether eluent) then provided enol carbonate 42 (973 mg, 66%) as a somewhat viscous colorless liquid containing residual Et2O (<1% by mass) and a minor unidentified impurity (4% by area) by 1H NMR spectroscopy. Rf 0.27 (2.5% Et2O/petroleum ether eluent); 1H NMR (500 MHz, CDCl3) δ 5.50–5.54 (m, 1H), 5.42–5.46 (m, 1H), 4.80 (s, 1H), 4.76–4.80 (br m, 1H), 4.72–4.76 (br m, 1H), 4.68–4.72 (m, 2H), 3.49 (d, J = 6.6 Hz, 2H), 2.76 (s, 2H), 2.19 (s, 2H), 1.98 (app. septet, J = 6.7 Hz, 1H), 1.71 (br s, 3H), 1.04 (s, 6H), 0.95 (d, J = 6.8 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 158.1, 152.7, 144.0, 142.6, 135.0, 120.5, 115.3, 111.3, 91.0, 73.8, 69.1, 43.4, 35.5, 33.4, 27.8, 25.6, 22.5, 19.2; IR (neat film, NaCl) ν 3077, 2960, 2932, 2913, 2874, 1763, 1663, 1648, 1638, 1620, 1470, 1465, 1444, 1421, 1382, 1367, 1363, 1331, 1276, 1242, 1217, 1174, 1150, 1099, 1040, 1023, 997, 969, 890, 782 cm−1; HRMS (EI+) m/z calcd. for C20H29ClO4 [M]+: 368.1754, found 368.1738.
4.7.3. α,ω-Diene (+)-45
To a flame-dried 100 mL round-bottomed flask equipped with a magnetic stir bar was added phosphinooxazoline ligand (−)-41 (54.4 mg, 0.0920 mmol) and bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (66.2 mg, 0.0812 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (22.5 mL, deoxygenated prior to use by sparging with N2 in a flame-dried flask for 2 h) was added via syringe, and the resulting solution was stirred for 30 min at 23 °C, during which time it turned dark yellow-brown. The solution was then cooled to 11 °C over 13 min. Concurrently, a flame-dried 1 dram screw-cap vial was charged with enol carbonate 42 (300.1 mg, 0.814 mmol). After sealing with a septum-bearing cap, the vial was placed under high vacuum and refilled with Ar. Deoxygenated anhydrous benzene (1 mL) was added via syringe, and the resulting solution was cooled to 11 °C. The enol carbonate solution was then transferred via cannula under positive N2 pressure to the catalyst solution, washing the original flask with deoxygenated anhydrous benzene (1 mL, allowed to cool briefly before transferring). The reaction, which turned green, was then stirred at 12 °C for 21.5 h. Solvent was removed under reduced pressure. The residue was suspended in Et2O and filtered through a plug of cotton to remove precipitated Pd. The filtrate was then concentrated under reduced pressure. The residue was suspended in 10% Et2O/pentane and applied to a silica gel column (11 × 2.5 cm). Flash chromatography (10% Et2O/pentane eluent) then provided α,ω-diene (+)-45 (242.9 mg, 92%) as an off-white solid. Observed 91% ee as determined by chiral HPLC analysis (Chiralpak® AD, 0.5% EtOH/hexanes, 1 mL/min, 254 nm, tR (major) = 10.4 min; tR (minor) = 12.6 min). Rf 0.16 (10% Et2O/petroleum ether eluent); [α]24D +7.15° (c 1.015, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.10–5.52 (br m, 1H), 5.28 (s, 1H), 5.27 (d, J = 0.97 Hz, 1H), 4.87 (br s, 1H), 4.85 (br s, 1H), 3.51–3.63 (m, 2H), 1.85–3.10 (br m, 6H), 2.02 (app. septet, J = 6.7 Hz, 1H), 1.72 (br s, 3H), 1.22 (br s, 3H), 1.16 (s, 3H), 0.97 (d, J = 6.8 Hz, 6H); 13C NMR (126 MHz, C6D6, overlapping of resonances was observed) δ 200.4 (br), 171.4, 142.6 (br), 117.5, 116.5 (br), 101.7, 74.2, 55.9, 43.6, 39.9 (br), 39.1, 27.9, 24.9 (br), 24.5 (br), 19.0; IR (neat film, NaCl) ν 3073, 2962, 2935, 2877, 1655, 1622, 1471, 1445, 1429, 1406, 1383, 1366, 1341, 1298, 1280, 1222, 1199, 1172, 1151, 1134, 1053, 1010, 968, 892, 856, 832, 789, 691, 653, 622 cm−1; HRMS (EI+) m/z calcd. for C19H29ClO2 [M]+: 324.1856, found 324.1865.
4.7.4. Olefin (±)-68

A 100 mL round-bottomed flask equipped with a stir bar and a septum was flame dried under vacuum. After refilling with Ar, anhydrous THF (26 mL) and anhydrous diisopropylamine (1.0 mL, 7.14 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.57 M in hexanes, 2.65 mL, 6.81 mmol) was added dropwise via syringe, and the solution was stirred at 0 °C for 30 min. The resulting LDA solution was then cooled to −78 °C over 20 min. Vinylogous ester 11 (1.29 g, 6.57 mmol) in anhydrous THF (1 mL) was then added dropwise over 1 min via syringe, washing the original flask with anhydrous THF (2 × 1 mL). The reaction was then stirred for 1 h at −78 °C to provide a yellow solution. Anhydrous hexamethylphosphoramide (1.25 mL, 7.18 mmol) was added in one portion via syringe over 1 min, and the reaction was stirred for 1 h at −78 °C. 5-Iodo-2-methyl-1-pentene (67)48 50 (1.67 g, 82.6% pure by mass [contained 15.8% pentane and 1.6% benzene by mass], 6.57 mmol) was added in one portion via syringe, washing the original flask with anhydrous THF (2 × 2 mL). The cold bath was removed, and the reaction was allowed to warm to 23 °C as it was stirred for 45 h. Saturated NH4Cl (aq) (40 mL) was added to give a colorless biphasic mixture, and the organic layer was separated. The aqueous layer was extracted with Et2O (2 × 150 mL), and the combined organic layers were washed with brine (2 × 200 mL). After drying over MgSO4, solvent was removed under reduced pressure. Flash chromatography over silica gel (15.5 × 5 cm silica, 10% Et2O/petroleum ether eluent until product begins to elute, then 20% Et2O/petroleum ether eluent) then provided olefin (±)-68 (1.261 g, 69%) as a very slightly yellow liquid. Rf 0.11 (10% Et2O/petroleum ether eluent); 1H NMR (500 MHz, CDCl3) δ 5.22 (s, 1H), 4.66–4.70 (m, 1H), 4.64–4.68 (m, 1H), 3.53–3.60 (m, 2H), 2.35 (d, J = 17.6 Hz, 1H), 2.16 (d, J = 17.8 Hz, 1H), 1.95–2.08 (m, 3H), 1.92 (dd, J = 6.5 Hz, 1H), 1.70 (s, 3H), 1.51–1.62 (m, 1H), 1.35–1.51 (m, 3H), 1.06 (s, 3H), 0.97 (s, 3H), 0.96 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 202.7, 174.4, 145.7, 109.9, 100.4, 74.5, 57.0, 41.2, 37.8, 34.9, 28.6, 27.7, 26.5, 25.8, 24.9, 22.3, 19.1; IR (neat film, NaCl) ν 3072, 2960, 2933, 2873, 1654, 1612, 1471, 1424, 1403, 1382, 1364, 1317, 1297, 1218, 1189, 1179, 1153, 1130, 1008, 968, 884, 842, 826 cm−1; HRMS (EI+) m/z calcd. for C18H30O2 [M]+: 278.2246, found 278.2250.
4.7.5. Enol carbonate 43

A 250 mL two-neck round-bottomed flask equipped with a 10 mL addition funnel, a stir bar, and two septa was flame dried under vacuum. After refilling with Ar, anhydrous THF (28 mL) and anhydrous diisopropylamine (0.72 mL, 5.14 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.57 M in hexanes, 1.85 mL, 4.75 mmol) was added dropwise via syringe, and the solution was stirred at 0 °C for 41 min. The resulting LDA solution was then cooled to −78 °C over 20 min. Olefin (±)-68 (1.20 g, 4.31 mmol) in anhydrous THF (5 mL) was then added dropwise over 6 min via the addition funnel, washing the funnel with THF (2.5 mL) upon complete addition. After stirring at −78 °C for 1 h, anhydrous N,N,N′,N′-tetramethylethylenediamine (0.78 mL, 5.20 mmol) was added dropwise over 1 min via syringe. After stirring at −78 °C for an additional 1 h, chloroformate 20 (737 mg, 4.76 mmol) in anhydrous THF (5 mL) was then added dropwise over 15 min via the addition funnel, washing the funnel with THF (2.5 mL) upon complete addition. The reaction was stirred for an additional 3 h at −78 °C, whereupon saturated NaHCO3 (aq) (17 mL) and H2O (17 mL) were added. The mixture was allowed to warm to 23 °C with the aid of a 23 °C water bath, at which time the organic layer was immediately separated. The aqueous layer was extracted with Et2O (2 × 100 mL), and the combined organic layers were washed with brine (2 × 100 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over Florisil® (100–200 mesh, 12 × 5 cm, 2.5% Et2O/petroleum ether eluent) then provided enol carbonate 43 (1.074 g, 63%) as a colorless liquid containing residual Et2O (<1% by mass) and a minor unidentified impurity (4% by area) by 1H NMR spectroscopy. Rf 0.21 (2.5% Et2 O/petroleum ether eluent); 1H NMR (500 MHz, CDCl3) δ 5.53–5.56 (m, 1H), 5.43–5.47 (m, 1H), 4.75 (s, 1H), 4.71–4.74 (m, 2H), 4.68–4.71 (br m, 1H), 4.65–4.68 (br m, 1H), 3.47 (d, J = 6.6 Hz, 2H), 2.18 (s, 2H), 1.92–2.05 (m, 5H), 1.71 (s, 3H), 1.47–1.55 (m, 2H), 1.07 (s, 6H), 0.94 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 157.6, 153.0, 145.6, 141.8, 135.1, 123.5, 115.4, 110.0, 91.0, 73.8, 69.1, 43.4, 38.0, 35.3, 27.8, 27.5, 26.0, 25.0, 22.3, 19.2; IR (neat film, NaCl) ν 3073, 2958, 2913, 2873, 2823, 1762, 1661, 1648, 1639, 1621, 1470, 1420, 1382, 1367, 1362, 1329, 1280, 1240, 1183, 1157, 1109, 1054, 1023, 1003, 965, 922, 887, 782 cm−1; HRMS (EI+) m/z calcd. for C22H33ClO4 [M]+: 396.2067, found 396.2083.
4.7.6. α,ω-Diene (−)-46
To a flame-dried 50 mL round-bottomed flask equipped with a magnetic stir bar was added phosphinooxazoline ligand (−)-41 (50.8 mg, 0.0859 mmol) and bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (61.5 mg, 0.0754 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (21 mL, deoxygenated prior to use by sparging with N2 in a flame-dried flask for >1 h) was added via syringe, and the resulting solution was stirred for 30 min at 23 °C, during which time it turned dark yellow-brown. The solution was then cooled to 10 °C over 15 min. Concurrently, a flame-dried 1 dram screw-cap vial with a magnetic stir bar was charged with enol carbonate 43 (299.6 mg, 0.755 mmol). After sealing with a septum-bearing cap, the vial was placed under high vacuum and refilled with Ar. Deoxygenated anhydrous benzene (1 mL) was added via syringe, and the resulting solution was cooled to 10 °C. The enol carbonate solution was then transferred via cannula under positive N2 pressure to the catalyst solution, washing the original flask with deoxygenated anhydrous benzene (1 mL, allowed to cool briefly before transferring). The reaction, which turned green, was then stirred at 14 °C for 23.3 h. Solvent was removed under reduced pressure. The residue was suspended in Et2O and filtered through a plug of cotton to remove precipitated Pd. The filtrate was then concentrated under reduced pressure. The residue was suspended in 10% Et2O/pentane and applied to a silica gel column (11 × 2.5 cm). Flash chromatography (10% Et2O/pentane eluent) then provided α,ω-diene (−)-46 (242.3 mg, 91%) as a viscous yellow liquid. Observed 90% ee as determined by chiral HPLC analysis (Chiralcel® OD-H, 0.5% EtOH/hexanes, 1 mL/min, 254 nm, tR (minor) = 6.8 min; tR (major) = 8.9 min). Rf 0.18 (10% Et2O/petroleum ether eluent); [α]24D −10.19° (c 0.515, benzene);1H NMR (500 MHz, C6D6) δ 5.31–5.34 (m, 1H), 5.19 (br s, 2H), 4.81 (br s, 1H), 4.79 (br s, 1H), 3.10–3.50 (br m, 1H), 3.01–3.12 (m, 2H), 2.67 (br d, J = 15.6 Hz, 1H), 2.30–2.70 (br m, 1H), 1.54–2.20 (br m, 6H), 1.67 (app. septet, J = 6.7 Hz, 1H), 1.64 (s, 3H), 1.38–1.54 (br m, 1H), 1.04 (br s, 3H), 0.95 (br s, 3H), 0.71 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, C6D6, overlapping of resonances was observed) δ 200.9, 171.7, 415.6, 142.5 (br), 116.9, 110.5, 100.8 (br), 74.2, 55.1, 43.2, 38.8, 38.2 (br), 30.1, 27.9, 24.8, 24.6, 23.6 (br), 22.4, 18.9; IR (neat film, NaCl) ν 3072, 2961, 2937, 2876, 1652, 1622, 1471, 1456, 1430, 1405, 1383, 1366, 1339, 1297, 1279, 1220, 1205, 1170, 1133, 1080, 1009, 968, 885, 847, 825, 736, 692, 655, 623 cm−1; HRMS (EI+) m/z calcd. for C21H33ClO2 [M]+: 352.2169, found 352.2175.
4.7.7. Olefin (±)-69

A 250 mL round-bottomed flask equipped with a stir bar and a septum was flame dried under vacuum. After refilling with Ar, anhydrous THF (42 mL) and anhydrous diisopropylamine (1.65 mL, 11.8 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.52 M in hexanes, 4.5 mL, 11.3 mmol) was added dropwise via syringe, and the solution was stirred at 0 °C for 47 min. The resulting LDA solution was then cooled to −78 °C over 52 min. 3-Isobutoxy-2-cyclohexen-1-one (19) (1.811 g, 10.8 mmol) in anhydrous THF (3 mL) was then added dropwise over 2 min via syringe, washing the original flask with anhydrous THF (3 mL). The reaction was then placed in an ice bath and stirred at 0 °C for 1.25 h. A yellow solution was obtained, which was subsequently cooled to −78 °C over 15 min. Anhydrous hexamethylphosphoramide (2.0 mL, 11.5 mmol) was added in one portion via syringe, and the reaction was stirred for 30 min at −78 °C. 4-Iodo-2-methyl-1-butene (18)17 (2.110 g, 10.8 mmol) in anhydrous THF (3 mL) was added via syringe, washing the original flask with anhydrous THF (3 mL). The cold bath was removed, and the reaction was allowed to warm to 23 °C as it was stirred for 24 h. Saturated NH4Cl (aq) (40 mL) was added, and the organic layer was separated. The aqueous layer was extracted with Et2O (2 × 150 mL), and the combined organic layers were washed with brine (2 × 200 mL). After drying over MgSO4, solvent was removed under reduced pressure. Flash chromatography over silica gel (10% Et2O/petroleum ether eluent until product begins to elute, then 25% Et2O/petroleum ether eluent) then provided olefin (±)-69 (751 mg, 30%) as a yellow liquid. Rf 0.04 (10% Et2O/petroleum ether eluent); 1H NMR (500 MHz, CDCl3) δ 5.30 (s, 1H), 4.69–4.73 (br m, 2H), 3.58 (dd, J = 9.5, 6.6 Hz, 1H), 3.56 (dd, J = 9.4, 6.5 Hz, 1H), 2.40–2.46 (m, 2H), 2.14–2.22 (m, 1H), 1.96–2.14 (m, 5H), 1.68–1.77 (m, 1H), 1.73 (br s, 3H), 1.39–1.49 (m, 1H), 0.967 (d, J = 6.59 Hz, 3H), 0.966 (d, J = 6.59 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 201.6, 176.9, 145.6, 110.1, 102.2, 74.7, 44.5, 35.1, 27.9, 27.7, 27.3, 26.1, 22.4, 19.07, 19.06; IR (neat film, NaCl) ν 3072, 2960, 2935, 2874, 1655, 1609, 1470, 1452, 1428, 1404, 1384, 1368, 1299, 1239, 1218, 1194, 1177, 1103, 1084, 1044, 990, 968, 952, 885, 833 cm−1; HRMS (EI+) m/z calcd. for C15H24O2 [M]+: 236.1776, found 236.1783.
4.7.8. Enol carbonate 44

A 100 mL round-bottomed flask equipped with a stir bar and a septum was flame dried under vacuum. After refilling with Ar, anhydrous THF (18 mL) and anhydrous diisopropylamine (0.51 mL, 3.64 mmol) were added via syringe, and the solution was cooled to 0 °C in an ice bath. n-BuLi (2.52 M in hexanes, 1.3 mL, 3.28 mmol) was added dropwise via syringe, and the solution was stirred for 30 min. The resulting LDA solution was cooled to −78 °C over 10 min. Olefin (±)-69 (714 mg, 3.02 mmol) in anhydrous THF (3 mL) was then added dropwise over 3 min via syringe, washing the original flask with anhydrous THF (3 mL). After stirring at −78 °C for 1 h, anhydrous N,N,N′,N′-tetramethylethylenediamine (0.54 mL, 3.6 mmol) was added over 1 min via syringe. After stirring at −78 °C for an additional 1.3 h, chloroformate 20 (517 mg, 3.34 mmol) in anhydrous THF (3 mL) was then added dropwise over 2 min via syringe, washing the original flask with anhydrous THF (3 mL). The reaction was stirred for an additional 2.7 h at −78 °C, at which time saturated NaHCO3 (aq) (13 mL) and H2O (13 mL) were added. The mixture was allowed to warm to 23 °C, at which time the mixture was extracted with Et2O (2 × 100 mL). The combined organic layers were washed with brine (2 × 100 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over Florisil® (100–200 mesh, 13 × 2.5 cm, 5% Et2O/petroleum ether eluent) then provided enol carbonate 44 (658 mg, 61%) as a colorless liquid containing a minor unidentified impurity (3% by area) by 1H NMR spectroscopy. Rf 0.15 (5% Et2O/petroleum ether eluent); 1H NMR (300 MHz, CDCl3) δ 5.52–5.56 (m, 1H), 5.43–5.47(m, 1H), 4.72–4.76 (m, 3H), 4.69–4.72 (m, 1H), 4.66–4.69 (m, 1H), 3.46 (d, J = 6.7 Hz, 2H), 2.23–2.40 (m, 4H), 2.13–2.23 (m, 2H), 2.02–2.13 (m, 2H), 1.96 (app. septet, J = 6.6 Hz, 1H), 1.70–1.74 (m, 3H), 0.94 (d, J = 6.7 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 158.5, 152.9, 145.4, 140.8, 135.1, 115.4, 115.1, 110.1, 91.4, 73.9, 69.2, 35.7, 27.9, 27.6, 27.1, 26.0, 22.3, 19.2; IR (neat film, NaCl) ν 3076, 2958, 2934, 2915, 2875, 2833, 1763, 1676, 1625, 1471, 1451, 1439, 1426, 1400, 1383, 1369, 1242, 1183, 1151, 1105, 1052, 1017, 1004, 988, 970, 950, 919, 888, 842, 781, 728, 712, 701, 644 cm−1; HRMS (EI+) m/z calcd. for C19H27ClO4 [M]+: 354.1598, found 354.1596.
4.7.9. α,ω-Diene (+)-24
To a flame-dried 50 mL round-bottomed flask equipped with a magnetic stir bar was added phosphinooxazoline ligand (−)-41 (56.2 mg, 0.0950 mmol) and bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (68.7 mg, 0.0843 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (24 mL, deoxygenated prior to use by sparging with N2 in a flame-dried flask for 1 h) was added via syringe, and the resulting solution was stirred for 32 min at 23 °C. The solution was then cooled to 12 °C over 15 min. Concurrently, a flame-dried 1 dram screw-cap vial was charged with enol carbonate 44 (299.5 mg, 0.844 mmol). After sealing with a septum-bearing cap, the vial was placed under high vacuum and refilled with Ar. Deoxygenated anhydrous benzene (1 mL) was added via syringe, and the resulting solution was cooled to 12 °C. The enol carbonate solution was then transferred via cannula to the catalyst solution at 12 °C, washing the original flask with deoxygenated anhydrous benzene (1 mL, allowed to cool briefly before transferring). The reaction was then stirred at 12 °C for 24.4 h, at which time it was filtered while still cold through a silica gel plug (4.5 × 1.5 cm), eluting with Et2O (200 mL). Solvent was removed under reduced pressure. The residue was suspended in Et2O and filtered through a plug of cotton to remove precipitated Pd. The filtrate was then concentrated under reduced pressure. The residue was suspended in 10% Et2O/pentane and applied to a silica gel column (12 × 2.5 cm). Flash chromatography (10% Et2O/pentane eluent) then provided α,ω-diene (+)-24 (210.7 mg, 80%) as a viscous colorless oil. Observed 94% ee as determined by chiral HPLC analysis (Chiralcel® OD-H, 1% EtOH/hexanes, 1 mL/min, 254 nm, tR (minor) = 6.6 min; tR (major) = 7.2 min). Rf 0.17 (10% Et2O/petroleum ether eluent); [α]25D +9.23° (c 1.04, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.28 (br s, 2H), 5.17 (br s, 1H), 4.64–4.71 (m, 2H), 3.58 (d, J = 6.4 Hz, 2H), 2.93 (dd, J = 14.5, 0.67 Hz, 1H), 2.38–2.60 (m, 3H), 1.82–2.18 (m, 5H), 1.58–1.82 (m, 2H), 1.71 (br s, 3H), 0.97 (d, J = 6.9 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 201.3, 175.9, 145.7, 139.0, 116.6, 109.7, 101.7, 74.7, 46.6, 43.2, 33.2, 32.0, 28.9, 27.7, 25.8, 22.7, 19.1; IR (neat film, NaCl) ν 3073, 2961, 2935, 2875, 1651, 1628, 1611, 1471, 1449, 1431, 1405, 1384, 1370, 1360, 1338, 1310, 1300, 1284, 1242, 1192, 1178, 1157, 1123, 1115, 1088, 1023, 993, 968, 952, 930, 886, 839, 800, 765, 752, 691, 644 cm−1; HRMS (EI+) m/z calcd. for C18H27ClO2 [M]+: 310.1700, found 310.1708.
4.8. Procedures for the synthesis of RCM product 54
4.8.1. α,ω-Diene 53

To an oven-dried 50 mL 2-neck round-bottomed flask equipped with a magnetic stir bar was added sodium hydride (100 mg, 4.17 mmol) and THF (10 mL) in a N2-filled glovebox. The flask was sealed with a septum and a hose adapter, and removed from the glovebox. Under a positive pressure of Ar, a reflux condenser was attached, and olefin 7049 51 (1.0 g, 4.38 mmol) was added via syringe. The suspension was stirred for 30 min, or until gas evolution ceased. 2,3-Dichloropropene (71) (0.40 mL, 4.34 mmol) was then added via syringe. The suspension was heated to 60 °C in an oil bath and stirred for 18 h. After cooling to 23 °C, H2O (2 mL) was added, and the mixture was diluted with Et2O (50 mL). The organic layer was separated, washed with brine (2 × 100 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over silica gel (10 × 2 cm, 10% EtOAc/hexane eluent) the provided α,ω-diene 53 (1.11 g, 84%) as a colorless liquid. Rf 0.47 (15% Et2O/pentane); 1H NMR (300 MHz, CDCl3) δ 5.29 (br s, 1H), 5.20 (br s, 1H), 4.70 (br s, 1H), 4.68 (br s, 1H), 4.19 (m, 4H), 3.06 (s, 2H), 2.12 (m, 2H), 1.88 (m, 2H), 1.71 (s, 3H), 1.25 (t, J = 4.2 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 170.7, 144.8, 137.6, 117.1, 110.7, 61.7, 56.7, 41.2, 32.4, 29.9, 22.6, 14.2; IR (neat film, NaCl) ν 2981, 1734, 1631, 1449, 1367, 1278, 1238, 1183, 1155, 1088, 1030, 890, 860 cm−1; HRMS (FAB+) m/z calcd. for C15H24ClO4 [M + H]+: 303.1363, found 302.1355.
4.8.2. Chloroalkene 54
To a flame-dried 10 mL 2-neck round-bottomed flask equipped with a magnetic stir bar and a condenser was added catalyst 49 (4.7 mg, 0.008 mmol). Anhydrous benzene (1.6 mL) was added via syringe. To the resulting solution was added α,ω-diene 53 (50 mg, 0.16 mmol) via syringe. The solution was placed in a 60 °C oil bath and stirred for 18 h. After cooling to 23 °C, solvent was removed under reduced pressure. Flash chromatography over silica gel (10 × 1 cm, 2.5 % Et2O/pentane eluent until product begins to elute, then 30% Et2O/pentane eluent) then provided chloroalkene 54 (11 mg, 24%) as a colorless oil. Rf 0.43 (15% Et2O/pentane); 1H NMR (300 MHz, CDCl3) δ 4.19 (q, J = 7.8 Hz, 4H), 2.84 (br s, 2H), 2.13 (app. s, 4H), 1.75 (app. t, J = 2.1 Hz, 3H), 1.24 (t, J = 7.5 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 170.5, 128.3, 122.7, 61.6, 54.6, 37.9, 28.9, 27.5, 19.6, 14.0; IR (neat film, NaCl) ν 2981, 1735, 1446, 1367, 1252, 1179, 1091, 1029 cm−1; HRMS (EI) m/z calcd. for C13H19Clo4 [M]+: 274.0972, found 274.0968.
4.9. Procedures for the synthesis of phosphinooxazoline ligand (+)-41
4.9.1. 2-Bromo-5-(trifluoromethyl)-benzoic acid (73)54

A 250 mL round-bottomed flask equipped with a stir bar and a condenser was charged with 2-bromo-5-(trifluoromethyl)-benzonitrile (72) (5.0 g, 20 mmol) and AcOH (22 mL, 384 mmol) to give a homogeneous solution. H2O (22 mL) was added and the benzonitrile oiled out to provide a biphasic mixture. Conc. H2SO4 (22 mL, 396 mmol) was added, and the mixture was stirred at 120 °C for 1 h and then at reflux for an additional 5 h. The mixture was cooled to 23 °C, and the pH was adjusted to 14 with KOH (aq) followed by NaOH (s). A significant exotherm was observed during this process, and an ice bath was employed to control temperature. H2O was added to dissolve all solids, and the aqueous solution was washed with EtOAc (3 × 200 mL). The pH of the aqueous solution was then adjusted to 1 with conc. HCl (aq), and the resulting biphasic mixture was extracted with EtOAc (3 × 200 mL). The combined organic layers were washed with brine (3 × 200 mL) and dried over MgSO4. Solvent was removed under reduced pressure, and the residue was azeotroped with heptane (4x) to remove any remaining AcOH. Residual solvent was then removed under high vacuum to provide 2-bromo-5-(trifluoromethyl)-benzoic acid (73)55 (4.42 g, 82%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 8.25 (d, J = 2.0 Hz, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.64 (dd, J = 8.1, 2.7 Hz, 1H);13C NMR (126 MHz, CDCl3) δ 169.6, 135.7, 131.0, 130.1 (q, JF = 33.9 Hz), 129.9 (q, JF = 3.5 Hz), 129.4 (q, JF = 3.8 Hz), 126.7 (q, JF = 1.4 Hz), 123.2 (q, JF = 272.5 Hz); 19F NMR (470 MHz, CDCl3) δ −63.3; IR (neat film, NaCl) ν 3454, 2957, 2925, 2872, 2853, 1664, 1618, 1586, 1513, 1464, 1444, 1419, 1376, 1365, 1342, 1301, 1248, 1220, 1171, 1138, 1092, 1072, 1039, 1018, 1010, 961, 908, 874, 861, 819, 745, 686, 666 cm−1; HRMS (EI+) m/z calcd. for C8H4F3BrO2 [M]+: 267.9347, found 267.9334.
4.9.2. 2-Bromo-5-(trifluoromethyl)-benzoyl chloride (74)

A 50 mL round-bottomed flask equipped with a stir bar was charged with 2-bromo-5-(trifluoromethyl)-benzoic acid (73) (1.719 g, 6.39 mmol), CH2Cl2 (13 mL), oxalyl chloride (1.1 mL, 13.0 mmol), and anhydrous DMF (40 μL, 0.517 mmol). A drying tube was attached to the flask, and the reaction was stirred for 12.7 h at 23 °C. Solvent was removed under reduced pressure, and the residue was suspended in hexanes and filtered through a plug of cotton to remove a precipitate. Solvent was removed under reduced pressure. The crude product was then distilled under high vacuum to provide 2-bromo-5-(trifluoromethyl)-benzoyl chloride (74) as a colorless liquid (1.787 g, 97%). 1H NMR (500 MHz, CDCl3) δ 8.27 (d, J = 2.0 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.65–7.70 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 165.1, 135.8, 135.7, 130.5 (q, JF = 3.4 Hz), 130.4 (q, JF = 34.1 Hz), 129.7 (q, JF = 3.8 Hz), 125.3 (q, JF = 1.2 Hz), 122.9 (q, JF = 272.8 Hz); 19F NMR (470 MHz, CDCl3) δ −63.4; IR (CHCl3, KBr) ν 1775, 1607, 1572, 1469, 1401, 1330 cm−1; HRMS (EI+) m/z calcd. for C8H3BrClF3O [M]+: 285.9008, found 285.9002.
4.9.3. Amide 7627 28

Salt 7550 52 (99% ee, 4.94 g, 12.3 mmol) was suspended in H2O (32 mL) and MeOH (16 mL). Conc. HCl (aq) (5.5 mL) was added, and the mixture was stirred for several minutes. The mixture was then filtered through a Büchner funnel into a round-bottomed flask equipped with a stir bar, washing with H2O (45 mL). The pH of the filtrate solution was adjusted to 10 with NaOH (s). K2CO3 (1.87 g, 13.5 mmol) and THF (30 mL) were then added. 2-Bromo-5-(trifluoromethyl)-benzoyl chloride (74) (3.991 g, 88.3% pure by mass [contained 11.7% benzene by mass], 12.3 mmol) was added, washing the original flask with THF (2 × 3 mL). The reaction mixture turned opaque, and an exotherm was observed. The mixture was stirred for 19 h at 23 °C, at which point it was extracted with Et2O (3 × 100 mL). The combined organic layers were washed with 10% HCl (aq) (100 mL) and brine (2 × 150 mL). After drying over MgSO4, solvent was removed under reduced pressure. The crude product was then recrystallized from EtOAc (17 mL)/hexanes (86.5 mL) at reflux to provide amide 76 (3.68 g, 82%) as fine white needles. Observed >99% ee as determined by chiral HPLC analysis (Chiralpak® AD, 5% i-PrOH/hexanes, 1 mL/min, 220 nm, tR (major) = 8.8 min; the minor enantiomer was not observed). A scalemic mixture afforded retention times of 6.6 and 8.9 min for the two enantiomers. [α]26D −11.78° (99.5% ee material, c 0.495, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 2.2 Hz, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.52–7.56 (m, 1H), 6.19 (br d, J = 8.6 Hz, 1H), 4.09 (ddd, J = 9.4, 7.3, 3.5 Hz, 1H), 3.98 (ddd, J = 11.2, 5.9, 3.5 Hz, 1H), 3.73 (ddd, J = 11.5, 7.3, 5.5 Hz, 1H), 1.99 (dd, J = 5.7, 5.7 Hz, 1H), 1.06 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 167.2, 138.7, 134.0, 130.3 (q, JF = 33.6 Hz), 127.8 (q, JF = 3.5 Hz), 126.7 (q, JF = 3.8 Hz), 123.0 (q, JF = 1.6 Hz), 123.3 (q, JF = 272.5 Hz), 62.7, 60.2, 33.9, 27.1; 19F NMR (470 MHz, CDCl3) δ −63.2; IR (neat film, NaCl) ν 3410, 3278, 3076, 2965, 2910, 2874, 1644, 1608, 1581, 1548, 1472, 1401, 1368, 1349, 1330, 1300, 1277, 1262, 1173, 1131, 1080, 1050, 1034, 1021, 999, 946, 908, 828, 796, 772, 733, 703, 668, 648 cm−1; HRMS (FAB+) m/z calcd. for C14H18BrNO2F3 [M + H]+: 368.0473, found 368.0487.
4.9.4. Oxazoline 7727 28

To a flame-dried 250 mL round-bottomed flask equipped with a magnetic stir bar and an oven-dried condenser was added amide 76 (3.67 g, 9.97 mmol). After sealing with a septum, the flask was placed under high vacuum and refilled with Ar. Anhydrous CH2Cl2 (40 mL) and anhydrous triethylamine (4.2 mL, 30.1 mmol) were added via syringe, and the reaction was cooled to 0 °C in an ice bath. Methanesulfonyl chloride (0.85 mL, 11.0 mmol) was added via syringe, and the reaction was stirred at 0 °C for 5 min. The reaction was then heated at reflux for 6 h, at which point additional CH2Cl2 (15 mL) was added to maintain the appropriate solvent volume. After stirring an additional 18 h at reflux, the reaction was cooled to 23 °C. The reaction was washed with H2O (50 mL), and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were washed with brine (150 mL) and dried over MgSO4. Solvent was removed under reduced pressure. Flash chromatography over silica gel (10 × 5 cm, 7.5% Et2O/petroleum ether eluent) then provided oxazoline 7727 28 (3.244 g, 93%) as a colorless viscous liquid. Rf 0.37 (7.5% Et2O/petroleum ether eluent); [α]24D +68.01° (c 1.035, CHCl3); lit.27 28 [α]25D −64.7° (c 1.26, CHCl3, S enantiomer); 1H NMR (500 MHz, CDCl3) δ 7.93 (d, J = 2.2 Hz, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.49–7.54 (m, 1H), 4.41 (dd, J = 10.3, 8.8 Hz, 1H), 4.28 (dd, J = 8.4, 8.4 Hz, 1H), 4.14 (dd, J = 10.3, 8.1 Hz, 1H), 1.01 (s, 9H).
4.9.5. Phosphinooxazoline ligand (+)-4127 28

To a flame-dried 250 mL round-bottomed Schlenck flask equipped with a magnetic stir bar and a septum was added copper(I) iodide (64.4 mg, 0.338 mmol). After sealing with a septum, the flask was wrapped in aluminum foil, placed under high vacuum, and refilled with Ar. Anhydrous toluene (11 mL), anhydrous N,N′-dimethylethylenediamine (79) (0.25 mL, 2.32 mmol), and phosphine 7851 53, 52 54 (1.35 g, 4.19 mmol) were added sequentially via syringe, and the reaction was stirred at 23 °C for 30 min. Oxazoline 77 (944 mg, 2.70 mmol) in anhydrous toluene (3 mL) was then added via syringe, washing the original flask with anhydrous toluene (2 × 4 mL). The flask was opened under positive Ar pressure, and Cs2CO3 (3.29 g, 10.1 mmol) was added. The flask was flushed with Ar and sealed with the stopcock, and the reaction was heated to 110 °C for 16.3 h. After cooling to 23 °C, the reaction was filtered through a pad of silica gel, eluting with CH2Cl2. Solvent was removed under reduced pressure. Flash chromatography over silica gel (18 × 2.5 cm, 1% Et2O/pentane eluent) then provided phosphinooxazoline (+)-4127 28 (1.17 g, 73%) as a white powder. Rf 0.087 (1% Et2O/petroleum ether eluent); [α]24D +15.04° (c 0.495, CHCl3); lit.27 28 [α]24D −16.0° (c 2.56, CHCl3, S enantiomer); 1H NMR (500 MHz, CDCl3) δ 8.23–8.27 (m, 1H), 7.54–7.62 (m, 5H), 7.27–7.37 (m, 4H), 6.95 (dd, J = 8.2, 3.3 Hz, 1H), 4.24 (dd, J = 10.3, 8.6 Hz, 1H), 4.09 (dd, J = 8.5, 8.5 Hz, 1H), 3.94 (dd, J = 10.1, 8.7 Hz, 1H), 0.68 (s, 9H).
4.10. Procedures for optimization studies for the enantioselective decarboxylative allylation and RCM
4.10.1. General procedure for enantioselective decarboxylative allylation screening (from Scheme 6, reaction b)7a
To a flame-dried 1 dram screw-cap vial equipped with a stir bar and a septum-bearing cap was added bis(3,5,3′,5′-dimethoxydibenzylideneacetone)palladium(0) (4.0 mg, 0.00491 mmol) and phosphinooxazoline ligand (−)-23 (2.5 mg, 0.00645 mmol). After sealing with the septum cap, the vial was placed under high vacuum and refilled with Ar. Anhydrous benzene (0.75 mL) was added via syringe, and the resulting solution was stirred for 29 min at 23 °C. Concurrently, a second flame-dried 1 dram screw-cap vial equipped with a septum-bearing cap was charged with enol carbonate 33 (17.5 mg, 99.5% pure by mass [contained 0.5% Et2O by mass], 0.050 mmol), and the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (0.75 mL) was added via syringe, and the resulting solution was transferred to the catalyst solution via syringe. The reaction was transferred to a 40 °C heating block and stirred for 68 min. After cooling to 23 °C, the reaction was filtered through a plug of silica gel in a pipette, eluting with Et2O. Solvent was removed under reduced pressure. 1H NMR spectroscopic analysis of the crude reaction mixture revealed >99% conversion and ≥99% α,ω-diene 34.
For runs in which the ee was determined, the crude product was subjected to purification by preparative thin-layer chromatography (0.25 mm thickness, 15% EtOAc/hexanes eluent) prior to chiral HPLC analysis.
For runs below 23 °C, the catalyst and substrate solutions were precooled for 5–7 min after metal-ligand complexation and prior to addition.
Scheme 4, reaction 26 → (+)-13 (24 °C conditions), Table 2, entries 2–3 and 5–6, and Table 3, entries 2 and 4 were conducted in an N2-filled glovebox.
For Table 3, entry 6, the anhydrous toluene was deoxygenated prior to use by freeze-pump-thawing under high vacuum in a flame-dried 1 dram screw-cap vial equipped with a septum-bearing cap for a total of four cycles.
4.10.2 Decarboxylative protonation of enol carbonate 26 (from Scheme 6, reaction a)25, 26
A flame-dried 1 dram screw-cap vial equipped with a stir bar and a septum-bearing cap was charged with powdered activated 4 Å molecular sieves (119 mg). The vial was placed under high vacuum, the sieves were flame-dried, and the vial was refilled with Ar. This was repeated for a total of three cycles. After cooling to 23 °C, Pd(OAc)2 (1.1 mg, 0.0049 mmol) and phosphinooxazoline ligand (−)-23 (2.4 mg, 0.0062 mmol) were added, and the flask was placed under high vacuum and refilled with Ar. Anhydrous benzene (0.75 mL) was added via syringe, and the reaction was placed in 40 °C heating block and stirred for 31 min. Concurrently, a second oven-dried 1 dram screw-cap vial equipped with a septum-bearing cap was charged with enol carbonate 26 (19.0 mg, 0.0496 mmol), and the vial was placed under high vacuum and refilled with Ar. Anhydrous benzene (0.75 mL) was added via syringe. To the catalyst solution at 40 °C was then added formic acid (19 μL, 0.504 mmol) followed by the enol carbonate 26 solution via syringe. The reaction was stirred at 40 °C for 3.1 h. After cooling to 23 °C, the reaction was filtered through a plug of silica gel in a pipette, eluting with Et2O. Solvent was removed under reduced pressure. 1H NMR spectroscopic analysis of the crude reaction mixture revealed >99% conversion and 99:1 olefin 27:α,ω-diene 13.
4.10.3. General procedure for RCM catalyst comparison (from Table 5, entry 3)8a
To a flame-dried 1 dram screw-cap vial equipped with a stir bar and a septum-bearing cap was added α,ω-diene (+)-13 (16.8 mg, 99.6% pure by mass [contained 0.4 % Et2O by mass], 0.0494 mmol) and 1,4-bis(trimethylsilyl)benzene (5.7 mg, 0.0256 mmol). The vial was brought into a N2-filled glovebox, and a solution of catalyst 49 in anhydrous C6D6 (0.00118 M, 1 mL, 0.00118 mmol) was added via syringe. The reaction solution was transferred to a screw-cap NMR tube equipped a septum-bearing cap, and the NMR tube was removed from the glovebox and placed in a 56–63 °C oil bath. Conversion was monitored by 1H NMR spectroscopy. The reaction was allowed to cool to 23 °C prior to 1H NMR analysis for each time point. The total time at 23 °C for each time point measurement was 45 min. Reaction times are given for the time the reaction spent at 60 °C only; time spent at 23 °C was not included.
Supplementary Material
Acknowledgments
This publication is based on work supported by Award No. KUS-11-006-02, made by King Abdullah University of Science and Technology (KAUST). Additionally, the authors wish to thank the NIH-NIGMS (R01 GM080269-01, postdoctoral fellowships to D.E.W. and I.C.S.), Abbott, Amgen, Bristol-Myers Squibb, Merck, and Caltech for generous funding; Materia, Inc. for their kind donation of catalyst 49 used in these studies; Professors Mercedes Cueto and Karen L. Erickson for their kind donation of natural samples of elatol (1); Professor Adusumilli Srikrishna for copies of 1H and 13C NMR spectra of synthetic (±)-laurencenone C ((±)-8) and (±)-α-chamigrene ((±)-5); and Professor Peter B. Dervan and David M. Chenoweth for use of their HPLC. Finally, B.M.S. thanks all of his current and former co-workers and colleagues who have made working at Caltech over the past 10 years such an enjoyable experience. As evidenced by the collaborative nature of this project, the spectacular environment at Caltech is one-of-a-kind and second-to-none.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data. Comparisons of published characterization data for natural and semisynthetic material with that of synthetic material for 1, 5, (+)-7, and 8; copies of 1H NMR, 13C NMR, and IR spectra for synthetic 1, 5, (+)-7, and 8; a copy of the 1H NMR spectrum for a natural sample of 1. Supplementary data associated with this article can be found in the online version, at doi:.
References
- 1.(a) Martín JD, Darias J. Algal Sesquiterpenoids. In: Scheuer PJ, editor. Marine Natural Products: Chemical and Biological Perspectives. Vol. I. Academic Press; New York: 1978. pp. 125–173. [Google Scholar]; (b) Erickson KL. Constituents of Laurencia. In: Scheuer PJ, editor. Marine Natural Products: Chemical and Biological Perspectives. Vol. V. Academic Press; New York: 1983. pp. 131–257. [Google Scholar]; (c) Dorta E, Díaz-Marrero AR, Cueto M, D'Croz L, Maté JL, Darias J. Tetrahedron Lett. 2004;45:7065–7068. doi: 10.1021/ol049287l. [DOI] [PubMed] [Google Scholar]
- 2.Elatol (1): Sims JJ, Lin GHY, Wing RM. Tetrahedron Lett. 1974;15:3487–3490.. Obtusol (2): González AG, Martín JD, Martín VS, Martínez-Ripoll M, Fayos J. Tetrahedron Lett. 1979;20:2717–2718.. Johnstonol (3): Sims JJ, Fenical W, Wing RM, Radlick P. Tetrahedron Lett. 1972;13:195–198.. Aplydactone (4): Fedorov SN, Radchenko OS, Shubina LK, Kalinovsky AI, Gerasimenko AV, Popov DY, Stonik VA. J. Am. Chem. Soc. 2001;123:504–505. doi: 10.1021/ja003254t.. α-Chamigrene (5): Ohta Y, Hirose Y. Tetrahedron Lett. 1968;9:2483–2485.. β-Chamigrene (6): Itô S, Endo K, Yoshida T, Yatagai M, Kodama M. Chem. Commun. 1967:186–188.. Proposed structure of Laurencenone B (7): Kennedy DJ, Selby IA, Thomson RH. Phytochemistry. 1988;27:1761–1766.. Neither the absolute configuration nor the optical rotation was specified. Laurencenone C (8): Asakawa Y, Tori M, Masuya T, Frahm J-P. Phytochemistry. 1990;29:1577–1584.. Also see ref. 2g. Majusculone (9): Suzuki M, Kurosawa E, Kurata K. Bull. Chem. Soc. Jpn. 1987;60:3795–3796..
- 3.(a) Granado I, Caballero P. Sci. Mar. 1995;59(Supl. 1):31–39. [Google Scholar]; (b) Vairappan CS, Daitoh M, Suzuki M, Abe T, Masuda M. Phytochemistry. 2001;58:291–297. doi: 10.1016/s0031-9422(01)00243-6. [DOI] [PubMed] [Google Scholar]; (c) Vairappan CS. Biomol. Eng. 2003;20:255–259. doi: 10.1016/s1389-0344(03)00067-4. [DOI] [PubMed] [Google Scholar]; (d) König GM, Wright AD. J. Nat. Prod. 1997;60:967–970. doi: 10.1021/np970181r. [DOI] [PubMed] [Google Scholar]
- 4.de Nys R, Leya T, Maximilien R, Afsar A, Nair PSR, Steinberg PD. Biofouling. 1996;10:213–224. doi: 10.1080/08927019609386281.. Martín JD, Pérez C, Ravelo JL. J. Am. Chem. Soc. 1986;108:7801–7811. doi: 10.1021/ja00284a052.. HeLa: IC50 = 4.1 μM (lag phase), 1.3 μM (log phase); Hep-2: IC50 = 2.4 μM (lag phase), 2.0 μM (log phase);Dias T, Brito I, Moujir L, Paiz N, Darias J, Cueto M. J. Nat. Prod. 2005;68:1677–1679. doi: 10.1021/np050240y..
- 5.For lead references on the total syntheses of chamigrene natural products, see: Hatsui T, Wang J-J, Takeshita H. Bull. Chem. Soc. Jpn. 1994;67:2507–2513.. Chen Y-J, Wang C-Y, Lin W-Y. Tetrahedron. 1996;52:13181–13188.. Srikrishna A, Lakshmi BV, Mathews M. Tetrahedron Lett. 2006;47:2103–2106.. Taber DF, Sikkander MI, Storck PH. J. Org. Chem. 2007;72:4098–4101. doi: 10.1021/jo070257g.. Srikrishna A, Babu RR. Tetrahedron. 2008;64:10501–10506.. Also see ref. 4b.
- 6.For our initial communication on the total syntheses of elatol (1) and the proposed structure of (+)-laurencenone B ((+)-7), see: White DE, Stewart IC, Grubbs RH, Stoltz BM. J. Am. Chem. Soc. 2008;130:810–811. doi: 10.1021/ja710294k..
- 7.(a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org. Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]; (b) Stewart IC, Benitez D, O'Leary DJ, Tkatchouk E, Day MW, Goddard WA, III, Grubbs RH. J. Am. Chem. Soc. 2009;131:1931–1938. doi: 10.1021/ja8078913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For the preparation of (+)-elatol (1) via the degradation of isoobtusol, see: González AG, Darias J, Díaz A, Fourneron JD, Martín JD, Pérez C. Tetrahedron Lett. 1976;17:3051–3054.. González AG, Martín JD, Martín VS, Norte M, Pérez R. Tetrahedron Lett. 1982;23:2395–2398.. Also see ref. 2b.
- 10.For the preparation of the proposed structure of (+)-laurencenone B ((+)-7) via the degradation of related chamigrene natural products including (+)-elatol (1), see: Brennan MR, Erickson K, Minott DA, Pascoe KO. Phytochemistry. 1987;26:1053–1057.. For the preparation of the proposed structure of laurencenone B (7) via the degradation of a related chamigrene natural product without specification of stereochemistry, see ref. 2g.
- 11.As the absolute configuration of the proposed structure of laurencenone B (7) was unknown, we chose to prepare the R-enantiomer for convenience.
- 12.For the preparation of trisubstituted chloroalkenes via RCM, see: Chao W, Weinreb SM. Org. Lett. 2003;5:2505–2507. doi: 10.1021/ol034775z.. Chao W, Meketa ML, Weinreb SM. Synthesis. 2004:2058–2061.. De Matteis V, van Delft FL, de Gelder R, Tiebes J, Rutjes FPJT. Tetrahedron Lett. 2004;45:959–963.. Dieltiens N, Claeys DD, Allaert B, Verpoort F, Stevens CV. Chem. Commun. 2005:4477–4478. doi: 10.1039/b508663a.. Yoshida K, Kawagoe F, Iwadate N, Takahashi H, Imamoto T. Chem. Asian J. 2006;1:611–613. doi: 10.1002/asia.200600133.. Chao W, Mahajan YR, Weinreb SM. Tetrahedron Lett. 2006;47:3815–3818.. Dieltiens N, Claeys DD, Zhdankin VV, Nemykin VN, Allaert B, Verpoot F, Stevens CV. Eur. J. Org. Chem. 2006:2649–2660.. Korboukh I, Kumar P, Weinreb SM. J. Am. Chem. Soc. 2007;129:10342–10343. doi: 10.1021/ja074108r..
- 13.For the preparation of a fully substituted vinyl fluoride via RCM, see: Marhold M, Buer A, Hiemstra H, van Maarseveen JH, Haufe G. Tetrahedron Lett. 2004;45:57–60..
- 14.For an example of a vinylogous ester in enantioselective decarboxylative allylation using a PHOX-Pd(0) catalyst, see: Levine SR, Krout MR, Stoltz BM. Org. Lett. 2009;11:289–292. doi: 10.1021/ol802409h.. For an example of a vinylogous thioester in enantioselective decarboxylative allylation using a PHOX-Pd(0) catalyst, see: Petrova KV, Mohr JT, Stoltz BM. Org. Lett. 2009;11:293–295. doi: 10.1021/ol802410t.. Also see ref. 14a.
- 15.Trost has recently reported the use of vinylogous ester and thioester derivatives in enantioselective decarboxylative allylation using a chiral bis(phosphine)-Pd(0) catalyst: Trost BM, Bream RN, Xu J. Angew. Chem. Int. Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421.. For use of this catalyst system in a formal total synthesis of platencin, see: Varseev GN, Maier ME. Angew. Chem. Int. Ed. 2009;48:3685–3688. doi: 10.1002/anie.200900447..
- 16.House HO, Fischer WF., Jr. J. Org. Chem. 1968;33:949–956. [Google Scholar]
- 17.Helmboldt H, Köhler D, Hiersemann M. Org. Lett. 2006;8:1573–1576. doi: 10.1021/ol060115t. [DOI] [PubMed] [Google Scholar]
- 18.(a) White DE. California Institute of Technology; Pasadena, CA: 2007. Unpublished work. [Google Scholar]; (b) Mohr JT. California Institute of Technology; Pasadena, CA: 2007. Unpublished work. [Google Scholar]
- 19.LDA, HMPA, THF, −78 °C, then 4-iodo-2-methyl-1-butene (18) −78 °C → 23 °C.
- 20.Takahashi K, Tanaka T, Suzuki T, Hirama M. Tetrahedron. 1994;50:1327–1340. [Google Scholar]
- 21.LDA, TMSCl, THF, −78 °C → 0 °C.
- 22.Use of Pd2(dba)3 in lieu of Pd(dmdba)2 led to significantly lower conversion.
- 23.10 mol% ligand (−)-23 was used here; 24 h reaction time.
- 24.(a) Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]; (b) Sherdan NH, Behenna DC, Virgil SC, Stoltz BM. Angew. Chem. Int. Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohr JT, Nishimata T, Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]
- 26.A separate experiment revealed no reactivity in the absence of a Pd(0) catalyst.
- 27.For preliminary results on the rate acceleration of enantioselective decarboxylative allylation of enol carbonates with electron-deficient PHOX ligands, see: Tani K, Behenna DC, McFadden RM, Stoltz BM. Org. Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s..
- 28.Et2O, −78 °C → 23 °C.
- 29.Imamoto T, Sugiura Y, Takiyama N. Tetrahedron Lett. 1984;25:4233–4236. [Google Scholar]
- 30.For similar challenges in the addition of methyl anion to analogous ketones in total syntheses of (±)-laurencenone C ((±)-8) and (±)-α-chamigrene ((±)-5), see ref. 5c.
- 31.[α]25D −42.01° (c 0.357, CHCl3, synthetic), [α]D +58.3° (c 0.36, CHCl3, semisynthetic, ref. 10).
- 32.See the supplementary data for a detailed comparison.
- 33.For additional total syntheses of (±)-α-chamigrene ((±)-5), see: Kanno S, Kato T, Kitahara Y. Chem. Commun. 1967:1257–1258.. Tanaka A, Uda H, Yoshikoshi A. Chem. Commun. 1968:56–57.. Kato T, Kanno S, Kitahara Y. Tetrahedron. 1970;26:4287–4292.. Kitagawa Y, Hashimoto S, Iemura S, Yamamoto H, Nozaki H. J. Am. Chem. Soc. 1976;98:5030–5031.. Fráter G. Chimia. 1977;31:63–64.. Fráter G. Helv. Chim. Acta. 1977;60:515–517.. Iwata C, Yamada M, Shinoo Y. Chem. Pharm. Bull. 1979;27:274–275.. White JD, Ruppert JF, Avery MA, Torii S, Nokami J. J. Am. Chem. Soc. 1981;103:1813–1821.. Iwata C, Miyashita K, Koga Y, Shinoo Y, Yamada M, Tanaka T. Chem. Pharm. Bull. 1983;31:2308–2312.. Plamondon J, Canonne P. Tetrahedron Lett. 1991;32:589–592.. For a formal total synthesis of (±)-α-chamigrene ((±)-5), see Subramanian T, Padmakumar R, Bhat SV. Synth. Commun. 1997;27:4067–4072..
- 34.For the preparation of laurencenone C (8) from laurencenone A, see ref. 2g. Neither the absolute configuration nor the optical rotation was specified for either compound. For the preparation of (−)-laurencenone C (8) from obtusol (2) and the preparation of (+)-laurencenone C ((+)-8) from isoobtusol, see ref. 2b and 9a.
- 35.For the preparation of (−)-α-chamigrene (5) from (−)-thujopsene, see Daeniker HU, Hochstetler AR, Kaiser K, Kitchens GC. J. Org. Chem. 1972;37:1–5.. Dauben WG, Aoyagi EI. J. Org. Chem. 1972;37:251–259.. The stereochemistry of α-chamigrene (5) was not determined. Kitchens GC, Hochstetler AR, Kaiser K. Acid Isomerization of Chamigrenes. 3,678,119. U.S. Patent. 1972 July 18;. The stereochemistry of α-chamigrene (5) was not determined. Kitchens GC, Hochstetler AR, Kaiser K. Acid Isomerization of Thujopsene and Novel Tricyclic Olerinic C15 H24 Hydrocarbons Formed Thereby. 3,681,470. U.S. Patent. 1972 August 1;. The stereochemistry of α-chamigrene (5) was not determined. Yarovaya OI, Polovinka MP, Korchagina DV, Gatilov Yu. V., Bagryanskaya I, Shcherbukhin VV, Shal'ko AA, Zenkovets GA, Barkhash VA. Russ. J. Org. Chem. 2001;37:362–374.; Zhurnal Organicheskoi Khimii. 2001;37:389–400.. For the preparation of (−)-α-chamigrene (5) from (−)-glanduliferol, see Suzuki M, Furusaki A, Kurosawa E. Tetrahedron. 1979;35:823–831.. For the preparation of (+)-α-chamigrene ((+)-5) from a (+)-bromo-α-chamigrene, see Guella G, Öztunç A, Mancini I, Pietra F. Tetrahedron Lett. 1997;38:8261–8264.. For the preparation of (+)-α-chamigrene ((+)-5) from a (−)-α-chamipinene, see Cool LG. Phytochemistry. 2005;66:249–260. doi: 10.1016/j.phytochem.2004.11.002..
- 36.Synthetic (−)-laurencenone C (8) was identical in all respects to the 1H NMR, 13C NMR, IR, and [α]D data reported for the natural product in ref. 2h except for a single 13C NMR resonance and the magnitude of its optical rotation: 13C NMR (126 MHz, CDCl3) δ 134.1 ppm (synthetic), 13C NMR (22.5 or 100 MHz, CDCl3) δ 131.4 ppm (natural); [α]26D −87.98° (c 1.00, CHCl3, synthetic), [α]D −43° (c 1.0, CHCl3, natural). We believe the reported 13C NMR peak to be a typographical error. Discrepancies between the published 1H NMR data for the natural product in ref. 2g and that of the synthetic material exist. No 13C NMR data was available for comparison from ref. 2g. No natural sample was available for comparison. Based on these observations, we conclude that the structure of the compound designated as laurencenone C (8) in ref. 2g has been incorrectly assigned. See the supplementary data for a detailed comparison.
- 37.Williams JR, Sarkisian GM. Synthesis. 1974:32–33. [Google Scholar]
- 38.Synthetic (−)-α-chamigrene (5) was identical in all respects to the 1H NMR, IR, and [α]D data reported for the natural product in ref. 2e except for the magnitude of its optical rotation: [α]26D −64.60° (c 0.21, CHCl3, synthetic), [α]D −14.5° (c 0.21, CHCl3, natural). No 13C NMR data was reported. No natural sample was available for comparison. See the supplementary data for a detailed comparison.
- 39.For an example of a sequential ketone reduction and directed reductive olefin transposition of a γ-halo-α,β-unsaturated ketone, see: Pattison VA. J. Org. Chem. 1970;35:2096–2099.. For an example of a sequential ester reduction and directed reductive olefin transposition of a γ-halo-α,β-unsaturated ester, see: Nakamura Y, Okada M, Horikawa H, Taguchi T. J. Fluorine Chem. 2002;117:143–148.. For an example of a directed reductive olefin transposition of a γ-benzyloxy-allylic alcohol, see: Hatakeyama S, Satoh K, Takano S. Tetrahedron Lett. 1993;34:7425–7428..
- 40.For examples of concomitant allylic and α'-bromination of enones, see Djerassi C, Rosenkranz G, Romo J, Kaufmann St., Pataki J. J. Am. Chem. Soc. 1950;72:4534–4540.. Brown FJ, Djerassi C. J. Org. Chem. 1981;46:954–963.. Kurzer F, Patel JN. Monatsh. Chem. 1987;118:793–812.. Oda T, Hashimoto H, Sato Y. Chem. Pharm. Bull. 1990;38:525–528. doi: 10.1248/cpb.38.525.. Shimizu T, Tobari A, Koyanagi J, Kawase M, Saito S. Chem. Pharm. Bull. 2001;49:23–28. doi: 10.1248/cpb.49.23.. Ma D, Tang G, Kozikowski AP. Org. Lett. 2002;4:2377–2380. doi: 10.1021/ol026125l.. Yick C-Y, Tsang T-K, Wong HNC. Tetrahedron. 2003;59:325–333..
- 41.Other electrophilic bromine sources examined consisted of CuBr2, pyridinium bromide perbromide, and N-bromosuccinimide.
- 42.Purification of α-bromoketone 59 was hampered by its poor stability to silica gel and reverse phase HPLC.
-
43.Determined by quenching the reaction at −78 °C in a separate run, resulting in the isolation a 3.9:1 mixture of alcohol diastereomers favoring 60.

- 44.Synthetic (+)-elatol (1) was identical in all respects to a natural sample provided by Prof. Karen L. Erickson except for the magnitude of its optical rotation: [α]23D +92.09° (c 0.22, CHCl3, synthetic), [α]25D +109.78° (c 0.045, CHCl3, natural). Based on 87% ee, the expected [α]D value for the synthetic material would be +95.5°, which differed from the observed value by 3.6%. The 1H NMR data also matched that reported for the natural product in ref. 2a. No 13C NMR or IR data was reported. See the supplementary data for a detailed comparison.
- 45.Et2O, −78 °C → 23 °C.
- 46.Toluene, −78 °C.
- 47.Takeda N, Imamoto T. Organic Syntheses. Vol. X. John Wiley & Sons, Inc.; Hoboken: 2004. pp. 200–204. Collect. [Google Scholar]
- 48.Boyer F-D, Ducrot P-H. Eur. J. Org. Chem. 1999:1201–1211. [Google Scholar]
- 49.Berlin JM, Campbell K, Ritter T, Funk TW, Chlenov A, Grubbs RH. Org. Lett. 2007;9:1339–1342. doi: 10.1021/ol070194o. [DOI] [PubMed] [Google Scholar]
- 50.Drauz K, Jahn W, Schwarm M. Chem. Eur. J. 1995;1:538–540. [Google Scholar]
- 51.van Doorn JA, Meijboom N. Recl. Trav. Chim. Pays-Bas. 1992;111:170–177. [Google Scholar]
- 52.Busacca CA, Lorenz JC, Grinberg N, Haddad N, Hrapchak M, Latli B, Lee H, Sabila P, Saha A, Sarvestani M, Shen S, Varsolona R, Wei X, Senanayake CH. Org. Lett. 2005;7:4277–4280. doi: 10.1021/ol0517832. [DOI] [PubMed] [Google Scholar]
- 53.Apparu M, Tiba YB, Léo P-M, Fagret D. Eur. J. Org. Chem. 2000:1007–1012. [Google Scholar]
- 54.Tagawa H, Kubo S. Chem. Pharm. Bull. 1984;32:3047–3052. doi: 10.1248/cpb.32.3047. [DOI] [PubMed] [Google Scholar]
- 55.Mongin F, Desponds O, Schlosser M. Tetrahedron Lett. 1996;37:2767–2770. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.