

Abstract
This report describes the scope and mechanism of the solvent-dependent, chemoselective oxidative coupling of 1-aryl-1,3-dicarbonyls with styrene using Ce(IV) reagents. Dihydrofuran derivatives are obtained when reactions are performed in methanol whereas nitrate esters can be selectively synthesized in acetonitrile and methylene chloride. Mechanistic studies are consistent with the rate of solvent-assisted deprotonation of a radical cation intermediate playing an integral role in the selective formation of products.
1. Introduction
In recent decades, the use of Ce(IV) reagents in single electron transfer (SET) reactions has steadily increased.1 Ceric ammonium nitrate (CAN) in particular has proven to be a cost-effective and synthetically versatile single electron oxidant. CAN is capable of generating radicals and radical cations that can further react generating carboncarbon and carbon-heteroatom bonds.2,3 Although traditionally restricted to aqueous or polar organic solvents, the replacement of the ammonium counterions of CAN with tetra-n-butylammonium yields ceric tetra-n-butylammonium nitrate (CTAN) which is more lipophilic resulting in increased solubility in less polar organic solvents.4 The single electron oxidative coupling of enolizable carbonyl and 1,3-dicarbonyl substrates to activated olefins has received a great deal of interest.5 Previous research from our group reported the solvent-dependent oxidative coupling of 1,3-dicarbonyl substrates to allyltrimethylsilane (Scheme 1).6 When reactions were performed in acetonitrile (MeCN), allylated products were obtained whereas dihydrofuran derivatives were obtained in methylene chloride (CH2Cl2). The basis for this solvent-selective chemoselectivity is a result of solvent-assisted desilylation of the β-silyl cation intermediate in more polar solvents such as MeCN leading to allylated products and inhibiting the cyclization pathway which results in dihydrofuran derivatives. A similar solvent-dependent chemoselectivity was observed for the oxidative addition of β-carbonyl imines to allyltrimethylsilane.7
Scheme 1.

Solvent-dependent synthesis of 2-allylated 1,3-dicarbonyl and dihydrofuran derivatives.
The oxidative coupling of 1,3-dicarbonyls to allyltrimethylsilane highlights the ability of solvent to have a significant impact on the reaction pathways of some carbon-carbon bond-forming events. Based on this precedent, the effect of solvent on the oxidative coupling of 1-aryl-1,3-dicarbonyl substrates to styrene was investigated. While previous research has examined similar synthetic systems, 1-aryl-1,3-dicarbonyl substrates were not used and reactions were performed only in polar solvents.8 The synthetic and mechanistic details for the Ce(IV)-mediated oxidative coupling of 1-aryl-1,3-dicarbonyl compounds to styrene are presented herein.
2. Results and discussion
2.1. Scope of reaction
In an initial study, when an equivalent of 1-phenyl-1,3-butanedione (1) was treated with 2 equivalents of CAN in MeCN in the presence of a slight excess of styrene, the nitrate ester derivative (1a) was formed as the major product in an isolated yield of 62 % (Table 1, entry 1). Interestingly, when the same reaction was performed in methanol (MeOH) with CAN, the dihydrofuran derivatives (1b) were produced in a combined 78% yield. To examine the scope of this solvent-dependent reaction, a variety of 1-aryl-1,3-dicarbonyl compounds were examined as substrates. As shown in entries 1–3 in Table 1, the synthesis worked well for a 1-aryl-1,3-diketone (1), a 1,3-diaryl-1,3-diketone (2), and 1-aryl-β-ketoesters (3–5). For the dihydrofuran syntheses using the 1-aryl-β-ketoesters, the products were obtained as single isomers. Nitrate esters 1a and 3a–5a were obtained as mixtures of diastereomers. These diastereomers proved to be inseparable by column chromatography but could be distinguished by the resonance for the benzylic proton.
Table 1.
Ce(IV)-mediated oxidative coupling of 1-aryl-1,3-dicarbonyls to styrene
| Entry | Substrate | Conditionsa | Yield (%)b | Ratio (a:b) | Productd |
|---|---|---|---|---|---|
| 1 |  |
A | 62 | 78:22 |  |
| B | 78 | 20:80 | |||
| 2 |  |
A | 50 | 60:40 |  |
| B | 84 | 10:90 | |||
| 3 | 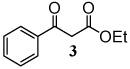 |
A | 61 | 77:23 |  |
| B | 78 | 16:84 | |||
| 4 |  |
A | 25 | 33:67 |  |
| B | 74 | 11:89 | |||
| 5 |  |
A | 67 | 100:trace |  |
| B | 25 | 70:30 |
Condition A: 2 equiv CAN in MeCN, r.t., 2 hrs; Condition B: 2 equiv CAN in MeOH, r.t., 2 hrs.
Isolated yield.
Ratios determined by 1H NMR by comparing the relative intensities of the proton signals from 5.6–6.0 ppm in the crude reaction mixture.
Nitrate esters are mixtures of diastereomers.
In addition to varying the types of 1-aryl-1,3-dicarbonyl compounds used, the effect of altering the electron density of the 1-aryl ring was examined. As shown in entry 4, when an electron-withdrawing fluorine was incorporated into the ring, the major product for the reaction performed in MeCN was dihydrofuran 4b instead of the expected nitrate ester 4a. Similarly, when the ring was activated by the addition of three methoxy substituents, the selectivity was significantly shifted towards the formation of nitrate ester 5a in MeOH, producing 5b in only a 25% yield. In addition, when the reaction was performed in MeCN, analysis of the crude reaction mixture by 1H NMR showed only trace amounts of dihydrofuran 5b. These results suggested a strong electronic effect with electron rich aryl rings favoring nitrate ester formation and electron poor aryl rings favoring the production of dihydrofurans.
The experiments described above show that both the solvent polarity and the electron density of the 1-aryl ring play a role in product distribution. Based on this observation, could the chemoselectivity be controlled by the use of an even less polar solvent? To examine this hypothesis, the oxidative addition of substrates 1–4 to styrene was performed in CH2Cl2 using CTAN as the oxidant (Table 2). For all four substrates, the selective formation of nitrate ester derivatives was improved when the reaction was performed in CH2Cl2 with CTAN when compared to MeCN and CAN. Whereas substrate 4 favored dihydrofuran formation in MeCN, the reaction in CH2Cl2 produced the desired nitrate ester 4a in a 51% yield.
Table 2.
Selective synthesis of nitrate ester derivatives in CH2Cl2a
| Entry | Substrate | Product | Ratio (a:b) | Yield (%)b |
|---|---|---|---|---|
| 1 | 1 | 1a | 88:12 | 66 |
| 2 | 2 | 2a | 85:15 | 66 |
| 3 | 3 | 3a | 82:18 | 66 |
| 4 | 4 | 4a | 66:34 | 51 |
2 equiv CTAN in CH2Cl2, r.t., 2 hrs.
Isolated yield.
With a simple procedure and mild reaction conditions, the oxidative addition of 1-aryl-1,3-dicarbonyls to styrene presented above provides an efficient approach to substituted nitrate esters and dihydrofurans selectively in moderate to very good yields. The ability to produce dihydrofuran derivatives is synthetically of interest since dihydrofuran moieties are present in a variety of natural products.9 Additionally, recent work from MacMillan et al. has highlighted the utility of benzylic nitrate esters in synthesis.10
2.2. Mechanistic studies
In order to fully elucidate the solvent-dependent chemoselectivity exhibited in this system, a thorough mechanistic analysis was performed. Preliminary studies were focused on the initial oxidation of the 1-aryl-1,3-diketone or β-ketoester in the absence of styrene to determine the impact of solvent on the mechanism of oxidation and the stability of the radical cation intermediate. Observed rate constants (kobs) for the oxidation of substrates 1 and 3 were obtained in all three solvents using either CAN or CTAN (k1 values, Table 3). These rate data were obtained by monitoring the decay of the Ce(IV) absorbance at 380 nm with a stopped-flow spectrophotometer. While the λmax of Ce(IV) is at 330 nm, the decay of Ce(IV) was monitored at 380 nm since the absorbance of the substrates overlapped at 330 nm. To assess the role of solvent, rate studies were performed first in the absence of styrene under pseudo-first order conditions keeping the substrate in excess with respect to the oxidant. Based on the previous studies of 1,3-dicarbonyls, it was postulated that the first step of the reaction involved the oxidation of the enol tautomer of the 1-aryl-1,3-dicarbonyl species by Ce(IV) to generate a radical cation.13,14 This supposition is also supported by the fact that many radical cations absorb in the range of 400 to 500 nm.15 To obtain a better understanding of the process, a time-resolved absorption spectrum was obtained for the oxidation of 3. As shown in the inset of Figure 1, a clear isosbestic point was observed at 420 nm. Since the substrates, Ce(IV) and Ce(III) do not absorb above 400 nm, the absorption at 460 nm was attributed to a radical cation intermediate. The rate of growth of the absorption at 460 nm for substrates 1 and 3 were recorded in each solvent and are included in Table 3. The growth of the radical cation absorption k2 was equal to the decay of Ce(IV) at 380 nm k2 within experimental error, a finding consistent with earlier studies on the Ce(IV)-mediated oxidation of 1-alkyl-1,3-diketones.13
Table 3.
Kinetic rate data for the Ce(IV)-mediated oxidation of 1-phenyl-1,3-butanedione and ethylbenzoylacetatea
| Entry | Substrate | Intermediate | Oxidant | Solvent | Rate constant of Ce(IV) decay at 380 nm k1 (sec− 1)b |
Rate constant of radical cation formation at 460 nm k2 (sec−1)b |
Rate constant of radical cation decay at 460 nm k3 (sec−1)b |
|---|---|---|---|---|---|---|---|
| 1 | 1 |  |
CAN | MeOH | 5.8 ± 0.6 × 102 | 6.0 ± 0.2 × 102 | 4.1 ± 0.1 × 10−2 |
| MeCN | 8.3 ± 0.2 | 8.7 ± 0.1 | 5.8 ± 0.2 × 10−3 | ||||
| CTAN | MeCN | 6.0 ± 0.3 | 6.2 ± 0.1 | 5.1 ± 0.5 × 10−3 | |||
| CH2Cl2 | 3.4 ± 0.3 | 3.4 ± 0.1 | 1.7 ± 0.1 × 10−3 | ||||
| 2 | 3 |  |
CAN | MeOH | 3.5 ± 0.3 × 102 | 3.7 ± 0.2 × 102 | 3.2 ± 0.1 × 10−1 |
| MeCN | 6.2 ± 0.1 | 6.3 ± 0.1 | 9.0 ± 0.3 × 10−2 | ||||
| CTAN | MeCN | 3.8 ± 0.4 | 3.9 ± 0.1 | 8.8 ± 0.2 × 10−2 | |||
| CH2Cl2 | 1.4 ± 0.1 | 1.6 ± 0.1 | 1.5 ± 0.1 × 10−2 |
[Ce(IV)]=1 mM, [substrate]=20 mM at 25°C.
Average of at least two runs.
Figure 1.

Time-resolved absorption spectrum observed from CTAN and ethylbenzoylacetate (3) in CH2Cl2 ([3] = 50 mM, [CTAN] = 1 mM) from 400–500 nm at 25°C. Spectrum was obtained by taking 10 scans every 5 nm over a period of 50 msec.
The rate data obtained indicated a clear trend based on the polarity of the solvent. The rate of decay of Ce(IV) increased with solvent polarity being fastest in MeOH and slowest in CH2Cl2. Furthermore, the rate of oxidation of diketone 1 and β-ketoester 3 is roughly 2 orders of magnitude faster in MeOH than in MeCN. The impact of solvent polarity and the relative rate differences among the solvents examined are consistent with earlier studies on 1-alkyl-1,3-diketones and related silyl enol ethers.13
Next, the impact of solvent on the lifetime of the radical cation was examined by monitoring its decay at 460 nm. Examination of the observed rate constants of radical cation decay (k3) contained in Table 3 shows a clear dependence on solvent polarity in the order of MeOH > MeCN > CH2Cl2. The k3 value is 4–7 times greater in MeOH than in MeCN whereas k3 is 3–6 times greater in MeCN than in CH2Cl2. The general trend for the stability of radical cations in the solvents examined is the same as in previous studies of 1,3-diketones and β-silyl enol ethers.13 However, the difference in the rates of radical cation decay of 1 and 3 among the solvents examined is less. Previous studies on radical cations derived from 1-alkyl-1,3-diketones showed a large difference among the solvents with decays in MeOH on the order of 15 to 100 times faster than in MeCN.13 It is likely that the presence of the 1-phenyl group stabilizes the radical cation intermediate thereby tempering the impact of solvent.
To further probe the role of solvent, 2,2-dideuterio-1-phenyl-1,3-butanedione was prepared and the rate of decay of its radical cation was measured under conditions identical to those described previously. The data are displayed in Table 4. The kH/kD values for both MeCN and CH2Cl2 were greater than 2 (entries 2 and 3, Table 4), a finding consistent with studies reported by Schmittel for the deprotonation of the anisyldimesitylethenol radical cation.16 The kH/kD value for MeOH (entry 1, Table 4) was 1.5. The lower value in MeOH is likely due to exchange between solvent and deuterium in the substrate.
Table 4.
Observed rate constants for the decay of the radical cation of 2,2-dideuterio-1-phenyl-1,3-butanedione in MeOH, MeCN, and CH2Cl2a
| Entry | Oxidant/ Solvent |
kD (sec− 1)b | kH/kD |
|---|---|---|---|
| 1 | CAN/ MeOH |
2.7 ± 0.1 × 10−2 | 1.5 ± 0.1 |
| 2 | CAN/ MeCN |
2.7 ± 0.1 × 10−3 | 2.2 ± 0.1 |
| 3 | CTAN/ CH2Cl2 |
7.3 ± 0.1 × 10−4 | 2.4 ± 0.1 |
[Ce(IV)]=1 mM, [substrate]=20 mM at 25°C.
Average of at least two runs.
Both the observation that the radical cations of 1-aryl-1,3-diketones and 1-aryl-β-ketoesters decay faster in more polar solvents and the results from the deuterium isotope study agree with the known solvent-assisted mechanism of O-H bond cleavage17,18 and are consistent with previous mechanistic studies on the role of solvent in the decay of radical cations derived from 1-alkyl-1,3-diketones.13
The studies described to this point support solvent playing an important role in the oxidation of substrate and in the stability of the initial radical cation intermediate. In the absence of styrene, the decay of the radical cation was a result of deprotonation. Next, a series of experiments was performed to determine the mechanistic role of styrene in the reaction. In these studies, the decay of the radical cation of 1-phenyl-1,3-butanedione (1) was monitored in the presence of increasing concentrations of styrene in all three solvents under pseudo-first order conditions with respect to the oxidant. The data for these experiments are contained in Table 5.
Table 5.
Rate order of styrene for decay of radical cation at 460 nm.
| Entry | Oxidant | Solvent | Styrene Rate Ordera,b |
|---|---|---|---|
| 1 | CAN | MeOH | 0.28 ± 0.01 |
| 2 | CAN | MeCN | 0.97 ± 0.05 |
| 3 | CTAN | CH2Cl2 | 1.02 ± 0.06 |
Average of at least 2 runs.
Determined from the slope for the plot of lnkobs vs. ln[styrene].
These experiments clearly showed that the rate order of styrene was 1 in MeCN and CH2Cl2 whereas it was significantly less than unity (0.28) in MeOH. These results indicated that reaction of radical cation with styrene was the rate-limiting step of the reaction in MeCN and CH2Cl2. Previous studies from our group have shown that radical cations derived from 1,3-diketones and related silyl enol ethers are deprotonated by MeOH through solvent-assisted deprotonation whereas in CH2Cl2 and MeCN, the intermediates are deprotonated through a unimolecular mechanism.13 Based on this precedent, the rate order of styrene in MeOH was interpreted as being consistent with deprotonation of the radical cation by solvent prior to addition to styrene.
Taken together, these studies indicated several key details about the mechanism of the Ce(IV)-mediated oxidative coupling of 1-aryl-1,3-dicarbonyls to styrene. First, the rates of oxidation of substrates by Ce(IV), the rates of radical cation formation and the rates of decay of the radical cations were solvent-dependent (MeOH > MeCN > CH2Cl2). Second, styrene was first order in both MeCN and CH2Cl2. Finally, a fractional rate order of styrene for the decay of the radical cation in MeOH indicated solventassisted deprotonation of the radical cation to a radical species prior to addition to styrene.
From the experimental results and points described above, the mechanism provided in Scheme 2 is proposed to explain the solvent-dependent chemoselectivity of the oxidative coupling of 1-aryl-1,3-dicarbonyls to styrene. Initial oxidation of the enol tautomer (6’) by Ce(IV) produces radical cation 7. In MeOH, solvent-assisted deprotonation of the radical cation yields radical intermediate 8. After the addition to styrene to form 9, rotation around one of the carbonyl-CH bonds and another single electron oxidation by Ce(IV) produces cation 10. Cyclization and deprotonation of 10 result in dihydrofuran derivative 11. Conversely in less polar solvents such as MeCN and CH2Cl2, radical cation 7 adds directly to styrene producing intermediate 12. This intermediate has restricted rotation since the proton is shared by the two carbonyl groups. Oxidation to cation 13 followed by internal nitrate ligand transfer from cerium yields nitrate ester 14.
Scheme 2.

Proposed mechanism for the solvent-dependent chemoselective oxidative addition of 1-aryl-1,3-dicarbonyls to styrene.
Observations from reactions involving substrates 4 and 5 indicate an electronic effect consistent with the proposed mechanism. Electron-donating groups on the aryl ring stabilize radical cation 7. This enhanced stability assists in direct radical cation addition to styrene prior to deprotonation. Conversely, electron-withdrawing groups on the aryl ring would destabilize radical cation 7 and as a result, deprotonation to radical 8 would become the favored pathway.
The key feature of the above mechanism is that direct addition of the radical cation to styrene before deprotonation provides a conformationally restricted intermediate inhibiting the intramolecular C-O bond formation that produces dihydrofurans. If this hypothesis is correct, addition of a basic solvent to the reaction performed in CH2Cl2 (or MeCN) should provide the dihydrofuran as the major product. To test this supposition, the reaction of substrate 2 with styrene employing CTAN as the oxidant was conducted in CH2Cl2 containing 5 equiv of MeOH. Dihydrofuran 2b was obtained in 80% yield.
3. Conclusions
A solvent-dependent chemoselective method for the Ce(IV)-mediated oxidative coupling of 1-aryl-1,3-dicarbonyls to styrene producing substituted dihydrofuran and nitrate ester derivatives has been developed. Reactions performed in MeOH yielded predominantly dihydrofuran derivatives whereas reactions in MeCN or CH2Cl2 favored the formation of nitrate esters. The reaction is general, working for a variety of 1-aryl-1,3-dicarbonyls and generating the desired products in good to very good yields. The reaction conditions are straightforward with short reaction times at room temperature. A thorough mechanistic analysis was consistent with the rate of solvent-assisted deprotonation of an initial radical cation intermediate playing an integral role in the selective formation of products. To the best of our knowledge, this approach is the first reported in which the reaction pathway is controlled by the lifetime of a radical cation intermediate. Further studies of other solvent-dependent reaction systems involving radical cations are currently underway.
4. Experimental
4.1. Instrumentation
Mechanistic rate data and time-resolved spectra were obtained using a computer-controlled stopped-flow reaction spectrophotometer from Applied Photophysics Limited. Column chromatography was performed using the automated CombiFlash® Rf system from Teledyne Isco, Inc. 1H, 13C and NOESY NMR spectra were recorded on a Bruker 500 MHz spectrometer. Mass spectra were obtained using a HP 5890 series GC-MS instrument. A Satellite FTIR from Thermo-Mattson was used to obtain IR spectra. LC-HRMS data were recorded at the Mass Spectrometry Facility at Notre Dame University.
4.2. Materials and methods
Acetonitrile (MeCN) and methylene chloride (CH2Cl2) were purified with a Pure Solv solvent purification system from Innovative Technology, Inc. Methanol (MeOH) was dried with activated 3Å molecular sieves prior to use. All 1-aryl-1,3-dicarbonyl substrates were purchased commercially and used without further purification. Styrene was filtered through a plug of neutral alumina to remove stabilizers. CAN was purchased commercially and used without further purification. CTAN was synthesized using a previously reported procedure.4 Products were separated using prepacked silica gel columns with a gradient elution of either ethyl acetate:hexanes or ether:hexanes.
For the mechanistic studies the Ce(IV) oxidants and substrates were prepared separately in the appropriate solvent in a glovebox, transported in airtight syringes, and injected into the stopped-flow spectrophotometer. The cellblock and the drive syringes of the stopped-flow spectrophotometer were flushed at least three times with dry, degassed solvent to make the system anaerobic. Temperature in the stopped-flow spectrophotometer was maintained at 25°C using a NESLAB RTE-111.
4.3. General procedures
4.3.1. Synthesis of nitrate ester derivatives
All glassware was flame-dried before use. The 1-aryl-1,3-dicarbonyl substrate (1 mmol) was dissolved in 15 mL of either MeCN or CH2Cl2 respectively. Styrene (1.1 mmol) was added dropwise and the reaction was purged with N2 gas. CAN or CTAN (2.1 mmol) was dissolved in 5 mL MeCN or CH2Cl2 respectively and added to the reaction via syringe with stirring. After stirring at room temperature for 2 hrs, solvent was removed via rotary evaporation. Water was added and then extracted three times with ether. The organic layers were combined, dried with MgSO4, filtered and concentrated. The nitrate ester products 1a–5a were purified via automated flash chromatography. Compounds 1a–5a were characterized by 1H NMR, 13C NMR, 1H NMR, 13C NMR, GC-MS, IR and LC-HRMS. NMR spectra are included in the Supplementary Information.
4.3.2. Synthesis of dihydrofuran derivatives
All glassware was flame-dried before use. The 1-aryl-1,3-dicarbonyl substrate (1 mmol) was dissolved in 15 mL of MeOH. Styrene (1.1 mmol) was added dropwise and the reaction was purged with N2 gas. CAN (2.1 mmol) was dissolved in 5 mL MeOH and added to the reaction via syringe with stirring. After stirring at room temperature for 2 hrs, solvent was removed via rotary evaporation. Water was added and then extracted three times with ether. The organic layers were combined, dried with MgSO4, filtered and concentrated. The dihydrofuran products 1b–5b were purified via automated flash chromatography. Compounds 1b–5b were characterized by 1H NMR, 13C NMR, GC-MS, IR and LC-HRMS. NMR spectra are included in the Supplementary Information.
4.4. Characterization of products
4.4.1. 3-Benzoyl-4-oxo-1-phenylpentyl-1-nitrate (1a): mixture of diastereomers
Light yellow oil. 1H NMR (CDCl3, 500MHz) – δ 7.96–7.88 (m, 4H), 7.67–7.61 (m, 2H), 7.55–7.47 (m, 4H), 7.44–7.30 (m, 8H), 5.83 (t, 1H, J=7.2 Hz), 5.75 (dd, 1H, J=2.3 Hz, 6.0 Hz), 4.61–4.56 (m, 2H), 2.58–2.52 (m, 4H), 2.17–2.13 (s, 6H). 13C NMR (CDCl3, 125MHz) – δ 202.2, 195.4, 137.2, 134.3, 134.2, 129.3, 129.1, 129.0, 128.8, 128.7, 126.3, 83.2, 82.8, 58.7, 58.4, 33.4, 33.3, 28.8, 28.5. MS [m/z (rel int)] 264 (M+, 1), 221 (65), 203 (18), 173 (15), 105 (98), 77 (90), 51 (30). IR (KBr) v (cm−1) 3508, 3448, 3062, 3035, 2962, 2930, 1720, 1635, 1555, 1447, 1358, 1274, 1071, 964, 855, 758, 698. LC-HRMS calcd. for C18H17NNaO5 [M+Na] 350.0999, found 350.1006; calc. for C18H17O2 [M-ONO2] 265.1223, found 265.1225.
4.4.2. 3-Benzoyl-4,5-dihydro-2-methyl-5-phenyl-furan (1b): major isomer
Yellow oil. 1H NMR (CDCl3, 500MHz) – δ 7.62–7.58 (m, 2H), 7.51–7.34 (m, 8H), 5.66 (dd, 1H, J=1.1 Hz, 9.0 Hz), 3.51 (ddd, 1H, J=1.4 Hz, 4.1 Hz, 10.5 Hz), 3.17 (ddd, 1H, J=1.4 Hz, 5.9 Hz, 8.8 Hz), 1.95 (br. t, 3H, J=1.4 Hz). 13C NMR (CDCl3, 125MHz) – δ 168.5, 141.0, 131.1, 128.7, 128.3, 127.8, 125.8, 83.4, 39.5, 15.5. MS [m/z (rel int)] 264 (M+, 36), 221 (15), 203 (8), 171 (9), 105 (94), 91 (17), 77 (100), 51 (35). IR (KBr) v (cm−1) 3448, 3417, 3385, 3355, 3060, 3033, 2927, 1714, 1639, 1563, 1448, 1352, 1274, 1225, 970, 895, 852, 700. LC-HRMS calcd. for C18H17O2 [M+H] 265.1223, found 265.1223.
4.4.3. 3-Benzoyl-4-oxo-1,4-diphenylbutyl-1-nitrate (2a)
Yellow oil. 1H NMR (CDCl3, 500MHz) – δ 7.97-7.92 (m, 2H), 7.87-7.83 (m, 2H), 7.64-7.56 (m, 2H), 7.52-7.42 (m, 4H), 7.41-7.38 (m, 4H), 5.92 (dd, 1H, J=3.7 Hz, 5.2 Hz), 5.38 (dd, 1H, J=2.2 Hz, 5.5 Hz), 2.72-2.59 (m, 2H). 13C NMR (CDCl3, 125MHz) – δ 195.0, 194.9, 137.4, 135.4, 135.2, 134.0, 129.3, 129.1, 129.0 (2), 128.6, 128.5, 126.2, 83.3, 52.4, 33.9. MS [m/z (rel int)] 326 (M+, 1), 239 (67), 222 (7), 161 (9), 105 (100), 77 (58), 51 (11). IR (KBr) v (cm−1) 3701, 3619, 3063, 2934, 1693, 1637, 1600, 1569, 1449, 1270, 1189, 1104, 995, 853, 853, 699. LC-HRMS calcd. for C23H20NO5 [M+H] 390.1336, found 390.1324; calc. for C23H19NNaO5 [M+Na] 412.1155, found 412.1143; C23H19O2 [M-ONO2] 327.1380, found 327.1391.
4.4.4. 3-Benzoyl-4,5-dihydro-2,5-diphenyl-furan (2b)
Colorless oil. 1H NMR (CDCl3, 500MHz) – δ 7.51–7.18 (m, 11H), 7.13–7.06 (m, 4H), 5.85 (t, 1H, J=9.8 Hz), 3.72 (dd, 1H, J=4.8 Hz, 10.3 Hz), 3.40 (dd, 1H, J=6.2 Hz, 8.9Hz). 13C NMR (CDCl3, 125MHz) – δ 193.4, 165.4, 141.1, 139.0, 131.2, 130.1, 129.5, 128.9, 128.8, 128.3, 127.7 (2), 125.9, 111.8, 83.2, 41.2. MS [m/z (rel int)] 326 (M+, 10), 223 (13), 134 (51), 121 (100), 105 (70), 91 (15), 77 (80), 51 (16). IR (KBr) v (cm−1) 3698, 3598, 3057, 2955, 2866, 1601, 1491, 1447, 1354, 1231, 1112, 1016, 918, 879, 695. LC-HRMS calcd. for C23H19O2 [M+H] 327.1380, found 327.1389.
4.4.5. Ethyl 3-benzoyl-1-phenylbutan-4-oate-1-nitrate (3a): mixture of diastereomers
Light yellow oil. 1H NMR (CDCl3, 500MHz) – δ 7.98-7.91 (m, 4H), 7.50-7.33 (m, 15H), 5.93 (dd, 1H, J=3.2 Hz, 5.6 Hz), 5.83 (dd, 1H, J=2.8 hz, 5.8 Hz), 4.50-4.42 (m, 2H), 4.20-4.12 (m, 4H), 2.66-2.54 (m, 4H), 1.21-1.15 (m, 6H). 13C NMR (CDCl3, 125MHz) – δ 193.9, 193.8, 168.9 (2), 137.2, 137.1, 135.6, 135.5, 134.0, 133.8, 129.2 (2), 129.0 (2), 128.9 (2), 128.8, 128.7, 128.6, 126.4, 126.3, 83.0 (2), 61.9 (2), 50.3, 50.2, 33.5, 13.9. MS [m/z (rel int)] 294 (M+, 1), 238 (33), 133 (35), 105 (100), 77 (60), 51 (20). IR (KBr) v (cm−1) 3457, 3357, 3063, 3035, 2983, 2904, 1736, 1688, 1636, 1450, 1274, 1196, 1095, 1021, 855, 755, 696, 592. LC-HRMS calcd. for C19H20NO6 [M+H] 358.1285, found 358.1288; calc. for C19H19NNaO6 [M+Na] 380.1105, found 380.1099; C19H19O3 [M-ONO2] 295.1329, found 295.1346.
4.4.6. Ethyl-4,5-dihydro-2,5-diphenyl-3-furancarboxylate (3b)
Light yellow oil. 1H NMR (CDCl3, 500MHz) – δ 7.88–7.85 (m, 2H), 7.46–7.33 (m, 8H), 5.73 (dd, 1H, J=2.1 Hz, 8.7 Hz), 4.18–4.12 (m, 2H), 3.58 (dd, 1H, J=4.5 Hz, 10.8 Hz), 3.16 (dd, 1H, 6.6 Hz, 8.6 Hz), 1.21 (t, 3H, J=7.2 Hz). 13C NMR (CDCl3, 125MHz) – δ 198.9, 165.2, 164.8, 141.7, 130.4, 129.4, 128.7, 128.1, 127.6, 125.7, 102.1, 82.5, 59.8, 39.9, 14.2. MS [m/z (rel int)] 294 (M+, 13), 247 (26), 220 (4), 115 (25), 105 (100), 77 (43). IR (KBr) v (cm−1) 3658, 3514, 3061, 3033, 2979, 1693, 1628, 1493, 1452, 1373, 1334, 1241, 1156, 1082, 1029, 928, 828, 758, 694. LC-HRMS calcd. for C19H19O3 [M+H] 295.1329, found 295.1349.
4.4.7. Methyl 3-(4-fluorobenzoyl)-1-phenylbutan-4-oate-1-nitrate (4a): mixture of diastereomers
Colorless oil. 1H NMR (CDCl3, 500MHz) – δ 8.00–7.94 (m, 4H), 7.43–7.32 (m, 8H), 7.19–7.13 (m, 4H), 5.91 (dd, 1H, J=3.6 Hz, 5.4 Hz), 5.81 (dd, 1H, J=3.2 Hz, 5.6 Hz), 4.47–4.41 (m, 2H), 3.72-3.69 (s, 6H). 13C NMR (CDCl3, 125MHz) – δ 192.1 (2), 169.2, 167.2, 165.2 (2), 137.1, 137.0, 131.5, 131.5 (2), 131.4, 129.3 (2), 129.0 (2), 126.3 (2), 116.2, 116.0, 82.9 (2), 53.0, 50.0, 49.8, 33.6, 33.5. MS [m/z (rel int)] 384 (M+, 10), 281 (11), 257 (13), 207 (100), 195 (92), 115 (19), 105 (27), 77 (16). IR (KBr) v (cm−1) 3472, 3361, 3069, 3033, 2955, 2902, 1741, 1687, 1638, 1600, 1506, 1442, 1273, 1240, 1161, 1097, 1009, 852, 744, 700. LC-HRMS calcd. for C18H17FNO6 [M+H] 362.1034, found 362.1034; calc. for C18H16FNNaO6 [M+Na] 384.0854, found 384.0843; C18H16FO3 [M-ONO2] 299.1078, found 299.1092.
4.4.8. Methyl-4,5-dihydro-2-(4-fluoro-phenyl)-5-phenyl-3-furancarboxylate (4b)
Colorless oil. 1H NMR (CDCl3, 500MHz) – δ 7.97-7.91 (m, 2H), 7.44-7.38 (m, 4H), 7.38-7.32 (m, 1H), 7.13-7.06 (m, 2H), 5.73 (dd, 1H, J=2.2 Hz, 8.6 Hz), 3.70 (s, 3H), 3.58 (dd, 1H, J=4.6 Hz, 10.8 Hz), 3.16 (dd, 1H, J=6.7 Hz, 8.6 Hz). 13C NMR (CDCl3, 125MHz) – δ 165.5, 164.9, 163.9, 162.9, 141.4, 131.7, 131.6, 128.8, 128.2, 125.7, 114.9, 114.7, 101.6, 82.5, 51.1, 39.8. MS [m/z (rel int)] 384 (M+, 6), 370 (13), 281 (12), 207 (96), 195 (100), 105 (42), 77 (39). IR (KBr) v (cm−1) 3067, 3031, 2949, 2871, 1701, 1621, 1507, 1442, 1341, 1240, 1156, 1085, 913, 841, 759, 700. LC-HRMS calcd. for C18H16FO3 [M+H] 299.1078, found 299.1097.
4.4.9. Ethyl 3-(3,4,5-trimethoxybenzoyl)-1-phenylbutan-4-oate-1-nitrate (5a): mixture of diastereomers
Colorless oil. 1H NMR (CDCl3, 500MHz) – δ 7.41-7.34 (m, 8H), 7.25, 7.20 (m, 4H), 5.89, (dd, 1H, J=5.0 Hz, 4.8 Hz), 5.84 (dd, 1H, J=2.3 Hz, 6.0 Hz), 4.46 (dd, 1H, J=3.9 Hz, 5.0 Hz) 4.41 (t, 1H, J=7.1 Hz), 4.24-4.15 (m, 4H), 3.96-3.88 (m, 18H), 2.64-2.49 (m, 4H), 1.26-1.19 (m, 6H). 13C NMR (CDCl3, 125MHz) – δ 192.6 (2), 169.0 (2), 153.2 (2), 137.3, 137.2, 130.7, 130.6, 129.3, 129.2, 129.0 (2), 126.3, 126.2, 106.2, 106.1, 83.2, 83.1, 62.0, 61.0, 56.3 (2), 50.3, 50.2, 33.8, 14.0 (2). MS [m/z (rel int)] 298 (M+, 8), 265 (6), 170 (16), 123 (100), 95 (25). IR (KBr) v (cm−1) 3644, 3553, 3337, 2978, 2944, 2839, 2255, 1734, 1681, 1636, 1584, 1502, 1456, 1416, 1332, 1272, 1237, 1125, 1004, 914, 854, 735, 704. LC-HRMS calcd. for C22H26NO9 [M+H] 448.1602, found 448.1596; C22H25O6 [M-ONO2] 385.1646, found 385.1648.
4.4.10. Ethyl-4,5-dihydro-5-phenyl-2-(3,4,5-trimethoxyphenyl)-3-furancarboxylate (5b)
Colorless oil. 1H NMR (CDCl3, 500MHz) – δ 7.44–7.33 (m, 5H), 7.33–7.30 (s, 2H), 5.71 (dd, 1H, J=1.8 Hz, 8.9 Hz), 4.20–4.13 (m, 2H), 3.90 (s, 9H), 3.58 (dd, 1H, J=4.5 Hz, 10.8 Hz), 3.16 (dd, 1H, J=6.6 Hz, 8.8 Hz), 1.25 (t, 3H, J=7.1 Hz). 13C NMR (CDCl3, 125MHz) – δ 165.2, 156.0, 152.4, 141.6, 128.7, 128.2, 125.7, 124.8, 107.1, 101.8, 82.3, 60.9, 59.8, 56.2, 40.4, 14.4. MS [m/z (rel int)] 298 (M+, 1), 105 (100), 77 (60), 51 (25). IR (KBr) v (cm−1) 3632, 3502, 2944, 2839, 1692, 1635, 1583, 1501, 1459, 1417, 1348, 1293, 1241, 1125, 1094, 1007, 914, 851, 734. LC-HRMS calcd. for C22H25O6 [M+H] 385.1646, found 385.1651.
Supplementary Material
Acknowledgements
RAF is grateful to the National Institutes of Health (1R15GM075960-01) for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Nair V, Deepthi A. Chem. Rev. 2007;107:1862. doi: 10.1021/cr068408n. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Balagopal L, Rajan R, Mathew J. Acc. Chem. Res. 2004;37:21. doi: 10.1021/ar030002z. [DOI] [PubMed] [Google Scholar]; (c) Molander GA. Chem. Rev. 1992;92:29. [Google Scholar]
- 2.(a) Paolobelli AB, Ceccherelli P, Pizzo F. J. Org. Chem. 1995;60:4954. [Google Scholar]; (b) Nair V, Mathew J, Prabhakaran J. Chem. Soc. Rev. 1997;26:127. [Google Scholar]
- 3.(a) Nair V, Nair LG, George TG, Augustine A. Tetrahedron. 2000;56:7607. [Google Scholar]; (b) Jiao J, Nguyen LX, Patterson DR, Flowers RA., II Org. Lett. 2007;9:1323. doi: 10.1021/ol070159h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muathen HA. Ind. J. Chem. 1991;30B:522. [Google Scholar]
- 5.(a) Roy SC, Mandal PK. Tetrahedron. 1996;52:2193. [Google Scholar]; (b) Hwu JR, Chen CN, Shiao SS. J. Org. Chem. 1995;60:856. [Google Scholar]; (c) Baciocchi E, Ruzziconi R. J. Org. Chem. 1986;51:1645. [Google Scholar]; (d) Citterio A, Sebastiano R, Marion A, Santi R. J. Org. Chem. 1991;56:5328. [Google Scholar]; (e) Lee YR, Kim BS, Kim DH. Tetrahedron. 2000;56:8845. [Google Scholar]; (f) Lee YR, Kim BS. Tetrahedron Letters. 1997;38:2095. [Google Scholar]
- 6.Zhang Y, Raines AJ, Flowers RA., II Org. Lett. 2003;5:2003. doi: 10.1021/ol034763d. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Raines AJ, Flowers RA., II J. Org. Chem. 2004;69:6267. doi: 10.1021/jo048955d. [DOI] [PubMed] [Google Scholar]
- 8.(a) Baciocchi E, Ruzziconi R. J. Org. Chem. 1991;56:4772. [Google Scholar]; (b) Baciocchi E, Giese B, Farshchi H, Ruzziconi R. J. Org. Chem. 1990;55:5688. [Google Scholar]; (c) Heiba EAI, Dessau RM. J. Org. Chem. 1974;39:3456. [Google Scholar]
- 9.(a) Krmzgl S, Gren N, Yang SW, Cordell GA, Bozok-Johansson C. J. Nat. Prod. 1997;60:378. doi: 10.1021/np9605652. [DOI] [PubMed] [Google Scholar]; (b) Weng JR, Tsao LT, Wang JP, Wu RR, Lin CN. J. Nat. Prod. 2004;67:1796. doi: 10.1021/np049811x. [DOI] [PubMed] [Google Scholar]
- 10.Graham TH, Jones CM, Jui NT, MacMillan DWC. J. Amer. Chem. Soc. 2008;130:16494. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Forsey SP, Rajapaksa D, Taylor NJ, Rodrigo R. J. Org. Chem. 1989;54:4280. [Google Scholar]; (b) Garzino F, Méou A, Brun P. Eur. J. Org. Chem. 2003:1410. [Google Scholar]
- 12.Yang FZ, Trost MK, Fristad WE. Tetrahedron Letters. 1987;28:1493. [Google Scholar]
- 13.Jiao J, Zhang Y, Devery JJ, II, Xu L, Deng J, Flowers RA., II J. Org. Chem. 2007;72:5486. doi: 10.1021/jo0625406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmittel M, Burghart A. Angew. Chem. Int. Ed. Engl. 1997;36:2550. [Google Scholar]
- 15.(a) Courtneidge JL, Davies AG. Acc. Chem. Res. 1987;20:90. [Google Scholar]; (b) Schepp NP. J. Org. Chem. 2004;69:4931. doi: 10.1021/jo049603+. [DOI] [PubMed] [Google Scholar]; (c) Dombrowski G. J. Org. Chem. 2005;70:3791. doi: 10.1021/jo047813g. [DOI] [PubMed] [Google Scholar]; (d) Bietti M, Capone A. J. Org. Chem. 2006;71:5260. doi: 10.1021/jo060678i. [DOI] [PubMed] [Google Scholar]
- 16.Schmittel M, Gescheidt G, Rock M. Angew. Chem. Int. Ed. Engl. 1994;33:1961. [Google Scholar]
- 17.Bockman TM, Kochi JK. J. Chem. Soc. Perkin Trans. 1996;2:1633. [Google Scholar]
- 18.Schmittel M, Heinze J, Trenkle H. J. Org. Chem. 1995;60:2726. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.