

Abstract
Short- and medium-chain acyl coenzyme A (acyl-CoA) synthetases catalyze the formation of acyl-CoA from an acyl substrate, ATP, and CoA. These enzymes catalyze mechanistically similar two-step reactions that proceed through an enzyme-bound acyl-AMP intermediate. Here we describe the characterization of a member of this enzyme family from the methane-producing archaeon Methanosarcina acetivorans. This enzyme, a medium-chain acyl-CoA synthetase designated MacsMa, utilizes 2-methylbutyrate as its preferred substrate for acyl-CoA synthesis but cannot utilize acetate and thus cannot catalyze the first step of acetoclastic methanogenesis in M. acetivorans. When propionate or other less favorable acyl substrates, such as butyrate, 2-methylpropionate, or 2-methylvalerate, were utilized, the acyl-CoA was not produced or was produced at reduced levels. Instead, acyl-AMP and PPi were released in the absence of CoA, whereas in the presence of CoA, the intermediate was broken down into AMP and the acyl substrate, which were released along with PPi. These results suggest that although acyl-CoA synthetases may have the ability to utilize a broad range of substrates for the acyl-adenylate-forming first step of the reaction, the intermediate may not be suitable for the thioester-forming second step. The MacsMa structure has revealed the putative acyl substrate- and CoA-binding pockets. Six residues proposed to form the acyl substrate-binding pocket, Lys256, Cys298, Gly351, Trp259, Trp237, and Trp254, were targeted for alteration. Characterization of the enzyme variants indicates that these six residues are critical in acyl substrate binding and catalysis, and even conservative alterations significantly reduced the catalytic ability of the enzyme.
AMP-forming acetyl coenzyme A (acetyl-CoA) synthetase (Acs; acetate:CoA ligase [AMP forming], EC 6.2.1.1), which catalyzes the activation of acetate to acetyl-CoA, is a member of the acyl-adenylate-forming enzyme superfamily (8), which consists of acyl- and aryl-CoA ligases, nonribosomal peptide synthetases that mediate the synthesis of peptide and polyketide secondary metabolites, such as gramicidin and tyrocidine, and the enzymes firefly luciferase and α-aminoadipate reductase. Although these enzymes share the property of forming an acyl-adenylate intermediate and are structurally related, they share limited sequence homology and catalyze unrelated reactions in which the intermediate serves different functions for different members of this enzyme family.
A two-step mechanism for Acs (equations 1 and 2) in which the reaction proceeds through an acetyl-AMP intermediate has been proposed based on evidence including detection of an enzyme-bound acetyl-AMP (2-4, 38):
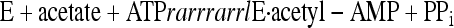 |
(1) |
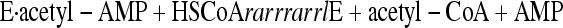 |
(2) |
In the CoA-dependent first step of the reaction, an enzyme-bound acetyl-AMP intermediate is formed from acetate and ATP, and inorganic pyrophosphate (PPi) is released. In the second step, the acetyl group is transferred to the sulfhydryl group of CoA and AMP is released. Other short- (Sacs) and medium-chain acyl-CoA synthetases (Macs) follow a similar reaction mechanism using acyl substrates other than acetate (8, 15).
In the 2.3-Å structure of trimeric Saccharomyces cerevisiae Acs1 in a binary complex with AMP (19), the C-terminal domain is positioned away from the N-terminal domain in a conformation for catalysis of the first step of the reaction (equation 1). The 1.75-Å structure of the monomeric Salmonella enterica Acs (AcsSe) (13) in complex with both CoA and adenosine-5′-propylphosphate, an inhibitor of the related propionyl-CoA synthetase (12, 15), which mimics the acetyl-adenylate intermediate, reveals that the C-terminal domain of Acs is rotated approximately 140° toward the N-terminal domain to form the complete active site for catalysis of the second half-reaction (equation 2). In this orientation, the CoA thiol is properly positioned for nucleophilic attack on the acetyl group. In structure/function studies of 4-chlorobenzoate:CoA ligase (CBAL), a distant member of the acyl- and aryl-CoA synthetase subfamily of the acyl-adenylate-forming enzyme superfamily, Wu et al. (39) and Reger et al. (28) provide evidence that PPi produced in the first step of the reaction dissociates from the enzyme before the switch from the first conformation to the second conformation required for CoA binding and catalysis of the second step of the reaction.
Acs and Sacs/Macs are widespread in all three domains of life and play a key role in archaea, as suggested by the finding that several thermophilic archaea have multiple open reading frames (ORFs) (up to seven) that encode putative Sacs or Macs (33). The chemolithoautotrophic methanoarchaeon Methanothermobacter thermautotrophicus has two ORFs with high identity to Acs and a third ORF that is likely to encode a Macs. M. thermautotrophicus Acs1 (Acs1Mt) has been biochemically and kinetically characterized, has been shown to have a strong preference for acetate as the acyl substrate, and can also utilize propionate but not butyrate (16, 17).
Methanosarcina and Methanosaeta are the only two methanoarchaea isolated that are able to utilize acetate as substrate for methane production. Unlike Methanosaeta species, which utilize Acs for catalyzing the first step of methanogenesis (18, 34), Methanosarcina species employ the acetate kinase-phosphotransacetylase pathway for activation of acetate to acetyl-CoA, and an Acs activity has not been observed in Methanosarcina (1, 23, 30, 32). Surprisingly, an Acs-related sequence was identified in the Methanosarcina acetivorans genome. Here we describe the kinetic characterization this enzyme, designated MacsMa, and show that it utilizes longer acyl substrates than Acs. The preferred acyl substrate was shown to be 2-methylbutyrate, and 2-methylbutyryl-CoA, AMP, and PPi were the products of the reaction, as expected. However, when propionate was used as the acyl substrate, propionyl-CoA was not produced. Instead, in the absence of CoA, propionyl-AMP and PPi were released, whereas in the presence of CoA, the propionyl-AMP intermediate was broken down into AMP and propionate and released along with PPi. Intermediate results were obtained with other acyl substrates, with both acyl-CoA and acyl-AMP production observed.
The 2.1-Å crystal structure of MacsMa (31), determined in the absence of any substrate, revealed the enzyme to be in a conformation similar to that of the S. enterica Acs (13) with respect to the position of the C-terminal domain. Through inspection of the MacsMa structure and alignment of Acs, Sacs, and Macs sequences, we identified six residues that form the putative acyl substrate-binding pocket. Individual alterations at these residues dramatically diminished enzyme activity and indicate that the acyl substrate-binding pocket of MacsMa has a very precise architecture that cannot be perturbed.
MATERIALS AND METHODS
Cloning and sequencing the M. acetivorans macs gene.
The M. acetivorans macs gene (GenBank accession no. NP_617808.1, GI:20091733) was PCR amplified from genomic DNA using the primer pair 5′-CGCACCCATATGACTTCCTTGCTGAGCCAATTTGTTTCC-3′ and 5′-CTAAACCTCGAGTCATTGGCTCTGGTCCTTGTCACGG-3′. The PCR product was cloned into the pET19b expression vector (Novagen, Inc.) using the NdeI and XhoI sites introduced during PCR amplification. The sequence of the cloned macs gene was confirmed by Sanger-style sequencing at the Clemson University Genomics Institute.
Site-directed mutagenesis.
Site-directed mutagenesis of the gene encoding MacsMa was performed using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Mutagenic primers were 30 to 40 nucleotides in length, with the altered base(s) located at the center of the primer. The alterations were confirmed by Sanger-style sequencing at the Clemson University Genomics Institute.
Heterologous enzyme production in Escherichia coli.
MacsMa was heterologously produced in E. coli RosettaBlue(DE3)placI (Novagen, Inc.). Cultures were grown at 37°C in LB medium containing 50 μg/ml ampicillin and 34 μg/ml chloramphenicol to an A600 of ∼0.6, and recombinant protein production was induced by the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a 0.5 mM final concentration. Cells were grown overnight at ambient temperature and harvested by centrifugation.
Purification of MacsMa.
Cells with heterologously produced MacsMa were suspended in ice-cold buffer A (25 mM Tris, 150 mM NaCl, 10% glycerol, 20 mM imidazole [pH 7.4]) and disrupted by two passages through a French pressure cell at 138 MPa, and the cell lysate was clarified by ultracentrifugation at 100,000 × g for 1 h. The supernatant was applied to a 5-ml His-Trap HP column (GE Healthcare) equilibrated with buffer A. After the column was washed with buffer A to remove unbound and weakly bound proteins, a linear gradient from 0 mM to 500 mM imidazole in buffer A was applied to elute MacsMa. The electrophoretically pure protein was dialyzed against buffer containing 25 mM Tris and 10% glycerol [pH 7.4], aliquoted, and stored at −20°C. Activity was found to be stable for greater than 6 months at this temperature. Protein concentrations were determined by the Bradford method (5) using bovine serum albumin as the standard.
Hydroxamate assay for acyl-CoA and acyl-adenylate production.
Production of acyl-CoA and acyl-adenylate was determined by the hydroxamate reaction (22, 29), in which activated acyl groups are converted to a ferric hydroxamate complex that can be detected spectrophotometrically at 540 nm. Reaction mixtures contained 100 mM Tris and 600 mM hydroxylamine-HCl [pH 7.5] with varied concentrations of the acyl substrate and Mg-ATP in the presence or absence of varied concentrations of CoA. Reactions (0.3-ml mixtures) were terminated by the addition of 2 volumes of stop solution (1 N HCl, 5% trichloroacetic acid, 1.25% FeCl3), and the change in absorbance at 540 nm was determined spectrophotometrically. Product formation was determined by comparison to a standard curve generated with known concentrations of acetyl phosphate. Reaction times were empirically determined such that the rate of the reaction remained linear over the time period. The standard reaction temperature of 55°C was based on the optimal temperature determined for MacsMa. In cases in which the acyl substrate was soluble in ethanol but not water, the final concentration of ethanol in the reaction was kept constant at 2%. This concentration was determined to have minimal effect on MacsMa enzymatic activity. All reactions were performed in triplicate.
Molybdate assay for inorganic pyrophosphate production.
Production of pyrophosphate was determined by direct reaction of PPi with molybdate reagent to produce a PPi-molybdate complex which can be measured at 580 nm (27). Reaction mixtures (0.3 ml) contained 50 mM Tris-HCl [pH 7.5], 4 mM MgCl2, 10 mM dithiothreitol, and varied concentrations of the acyl substrate, ATP, and CoA. Reaction mixtures were preincubated at 55°C for 5 min, and reactions were initiated by the addition of enzyme. After 10 min, the reactions were terminated by the addition of 80 μl H2O, 50 μl 2.5% molybdate reagent (2.5% ammonium molybdate in 5 N H2SO4), 50 μl 0.5 M 2-mercaptoethanol, and 20 μl Eikonogen reagent (25 mM sodium sulfite, 13 mM 1-amino-2-naphthol-4-sulfonic acid, 963 mM sodium meta-bisulfite). After color development at ambient temperature for 10 min, the absorbance at 580 nm was measured and PPi concentration was determined by comparison to a PPi standard curve. All reactions were performed in triplicate.
Spectroscopic assay for acyl-CoA thioester bond formation.
Acyl-CoA formation was measured using a spectrophotometric assay that detects thioester bond formation (21). The reaction mixture contained 100 mM Tris-HCl (pH 7.5) and various concentrations of the acyl substrate and Mg-ATP in the presence or absence of 0.5 mM CoA. Reactions (0.5-ml mixtures) were initiated by the addition of enzyme, and the change in absorbance at 233 nm was monitored continuously using a diode array spectrophotometer. Acs1Mt and Archaeoglobus fulgidus Acs2 (Acs2Af) (16) were used as control enzymes for detection of propionyl-CoA formation by this assay. All reactions were performed in triplicate.
Coupled-enzyme assay for AMP production.
AMP production was detected via a coupled-enzyme assay in which myokinase (MK; AMP + ATP → 2 ADP), pyruvate kinase (PK; phosphoenolpyruvate + ADP → pyruvate + ATP), and lactate dehydrogenase (LDH; pyruvate + NADH → lactate + NAD+) couple AMP production to NADH oxidation (15). Reaction mixtures (0.5 ml) contained 50 mM HEPES (pH 7.5), 1 mM MgCl2, 3 mM phosphoenolpyruvate, 2 mM glutathione, 1 unit PK, 1.5 units LDH, and 0.2 mM NADH, with varied concentrations of acyl substrate and CoA. As 55°C is the optimal temperature for MacsMa, the stability and activities of the coupling enzymes were tested at this temperature. For the length of the reaction, PK and LDH were determined to be sufficiently stable and active at 55°C to not be rate limiting. To determine kinetic parameters for CoA, both ATP and the acyl substrate were held constant at saturating concentrations and the concentration of CoA was varied from 0.01 to 2 mM. The reaction was initiated by the addition of enzyme, and the change in absorbance at 340 nm was monitored. An extinction coefficient of 6220 M−1 min−1 for oxidation of NADH to NAD+ was used to calculate the reaction rate. All reactions were performed in triplicate.
Determination of kinetic parameters.
For determination of apparent kinetic parameters, the concentration of one substrate was varied and the other(s) held constant at a saturating level (generally 5 to 10 times the Km value for that substrate). Concentrations for the varied substrate generally ranged from 0.2 to 5 to 10 times the Km value, and 8 to 12 concentrations were used for each determination. The apparent steady-state kinetic parameters kcat and kcat/Km and their standard errors were determined using nonlinear regression to fit the data to the Michaelis-Menten equation using Kaleidagraph (Synergy Software). MacsMa followed Michaelis-Menten-like kinetics for all substrates.
Molecular modeling.
MacsMa variants were modeled on the wild-type MacsMa structure (Protein Database accession no. 3ETC) via DS Modeler (Accelrys) using the standard parameters. The structures were visualized with DS Visualizer (Accelrys) and DS Viewer Pro 5.0 (Accelrys).
Sequence analysis.
Sacs and Macs amino acid sequences were aligned by Clustal X (36) using a Gonnet PAM 250 weight matrix with a gap opening penalty of 10.0 and a gap extension penalty of 0.05.
RESULTS
Although M. acetivorans utilizes the acetate kinase-phosphotransacetylase pathway for the activation of acetate to acetyl-CoA in the first step of acetoclastic methane production and an Acs activity has not previously been detected in Methanosarcina species (1, 23), a BLAST search of the M. acetivorans genome (GenBank accession no. NC_003552, GI 20088899) revealed a sequence (NP_617808.1, GI 20091733) that shows identity and similarity to Acs. The M. acetivorans enzyme was heterologously produced in E. coli as a soluble, active protein and purified for biochemical and kinetic characterization to determine whether it has activity toward acetate and might thus play a role in acetate activation in M. acetivorans.
The preferred acyl substrate for acyl-CoA synthesis is 2-methylbutyrate.
The acyl-CoA synthetase activity of this enzyme was determined using the hydroxamate assay with straight- and branched-chain acyl substrates. The enzyme displayed little activity with acetate but substantial activity with a number of longer straight- and branched-chain acyl substrates. This enzyme thus appears to be a medium-chain acyl-CoA synthetase and has been designated MacsMa. Interestingly, significant enzymatic activity was observed for many of these substrates in the absence of CoA. There was little difference in activity in the presence or absence of CoA with acetate, propionate, hexanoate, heptanoate, octanoate, and 3-methyl or 4-methyl branched-chain acyl compounds or with 2-methyl branched-chain acyl compounds longer than 2-methylvalerate (data not shown). The level of activity observed in the presence of 1 mM CoA with butyrate, 2-methylpropionate, or 2-methylvalerate as the acyl substrate was slightly higher than that observed in the absence of CoA, but the difference was less than 2-fold. As the hydroxamate assay utilized for these initial experiments can detect activated acyl substrates, including acyl-CoA and acyl-AMP, the activity observed in the absence of CoA was likely due to production of an acyl-AMP (see below).
The only acyl substrate for which substantial activity was observed in the presence but not the absence of CoA was 2-methylbutyrate. Production of 2-methylbutyryl-CoA, AMP, and PPi by MacsMa was confirmed to verify acyl-CoA synthetase activity. Acyl-CoA thioester bond formation measured using the spectroscopic assay was absolutely CoA dependent, as expected. PPi production with 2-methylbutyrate as the acyl substrate in the presence or absence of CoA was measured using the molybdate assay. The requirement of CoA for PPi production (or release) is consistent with results observed with Acs1Mt and Acs2Af (Table 1), proven acetyl-CoA synthetases from the archaea Methanothermobacter thermautotrophicus and Archaeoglobus fulgidus (16), respectively. AMP production with 2-methylbutyrate, measured using the coupled-enzyme assay, likewise was CoA dependent (Table 1). These results suggest that the M. acetivorans enzyme catalyzes acyl-CoA synthesis, with 2-methylbutyrate as the acyl substrate following the two step acyl-CoA synthetase reaction mechanism. As 2-methylbutyrate appears to be the only acyl substrate for which acyl-CoA synthesis is the primary activity supported, kinetic parameters for the acyl-CoA synthetase activity of MacsMa were determined only with 2-methylbutyrate (Table 2).
TABLE 1.
Product formation in the absence versus the presence of CoA
| Product measured (assay) | Enzyme | Substrate | % activity in the absence vs the presence of CoA |
|---|---|---|---|
| PPi (molybdate assay) | MacsMa | 2-Methylbutyrate | 3.1 ± 0.1 |
| MacsMa | Propionate | 48.1 ± 0.7 | |
| Acs1Mt | Acetate | 0.1 ± 0.01 | |
| Acs2Af | Acetate | 0.33 ± 0.02 | |
| AMP (coupled-enzyme assay) | MacsMa | Propionate | 2.0 ± 0.3 |
| MacsMa | 2-Methylbutyrate | 0.6 ± 0.03 | |
| Acyl-AMP + acyl-CoA (hydroxamate assay) | MacsMa | Propionate | 91.7 ± 0.8 |
| Acs1Mt | Propionate | 0.7 ± 0.09 |
TABLE 2.
Kinetic parameters for acyl-CoA formation in the presence of 2-methylbutyrate
| Substrate | Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) |
|---|---|---|---|
| CoA | 4.09 ± 0.45 | 2.15 ± 0.10 | 0.53 ± 0.030 |
| 2-Methylbutyrate | 8.92 ± 0.51 | 1.58 ± 0.07 | 0.18 ± 0.002 |
| ATP | 4.21 ± 0.11 | 1.94 ± 0.02 | 0.46 ± 0.006 |
MacsMa releases propionyl-adenylate rather than propionyl-CoA as a product.
A possible explanation for the enzymatic activity observed in the absence of CoA with most acyl substrates is that MacsMa has difficulty retaining the acyl-adenylate intermediate when suboptimal acyl substrates are utilized and instead releases some proportion of it as a product, precluding the second step of the reaction. As the enzyme displayed nearly identical activities in the presence and absence of 1 mM CoA with propionate as the acyl substrate in the hydroxamate assay (Table 1), release of the intermediate was examined further using propionate as the substrate. To verify that the hydroxamate assay can detect free but not enzyme-bound acyl-adenylate, Acs1Mt enzymatic activity with propionate was assessed using the hydroxamate assay in the absence and presence of saturating concentrations of CoA. No activity was detected for Acs1Mt in the absence of CoA (Table 1), suggesting that only free activated acyl groups are detected and not the enzyme-bound acyl-adenylate intermediate produced in the first step of the acyl-CoA synthetase reaction.
As further confirmation, enzymatic reactions were performed in the presence or absence of CoA and analyzed by centrifugal ultrafiltration in which molecules smaller than 10,000 Da flowed through the filter and the enzyme was retained. The presence of activated acyl groups was detected in the flowthrough fraction by addition of hydroxylamine and conversion to the ferric hydroxamate complex. For Acs1Mt, activated acyl groups were detected in the flowthrough fraction in reactions performed in the presence but not in the absence of CoA, consistent with retention of the acetyl-adenylate intermediate after the first step of the reaction and release of the acetyl-CoA product after the second step. For MacsMa, activated acyl groups were detected in the flowthrough fraction in reactions performed in the absence of CoA, indicating that the propionyl-adenylate was released.
PPi production in the absence of CoA was determined for MacsMa, Acs1Mt, and Acs2Af. PPi production, measured using the molybdate assay, was detected only in the presence, not in the absence, of CoA for Acs1Mt and Acs2Af using acetate as the acyl substrate (Table 1). Likewise, for MacsMa, PPi production was detected in the presence but not the absence of CoA with 2-methylbutyrate as the acyl substrate. However, substantial PPi production was detected in the absence of CoA for MacsMa when propionate was the acyl substrate (Table 1). AMP, a product of the second step of the acyl-CoA synthetase reaction, was detected only in the presence, not in the absence, of CoA for MacsMa with either propionate or 2-methybutyrate as the acyl substrate, using the coupled-enzyme assay for AMP production (Table 1).
To determine whether MacsMa can catalyze the synthesis of propionyl-CoA, acyl-CoA thioester bond formation was measured using the spectroscopic assay with propionate and ATP at saturating concentrations. No thioester bond-forming activity was detected in the absence of CoA or with increasing CoA concentrations from 0.1 mM to 0.5 mM. In control reactions conducted using Acs1Mt and Acs2Af, with acetate or propionate as the acyl substrate, thioester bond formation was detected in the presence but not the absence of CoA.
Overall, the results suggest that MacsMa behaves as an acyl-CoA synthetase in the presence of 2-methylbutyrate but as an acyl-adenylate synthetase which releases acyl-AMP as a product when propionate is the acyl substrate (equation 3), precluding the second step of the acyl-CoA synthetase reaction (equation 2).
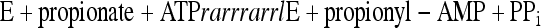 |
(3) |
This activity has also been shown for MacsMa homologs from Methanothermobacter thermautotrophicus and Methanococcus maripaludis (C. Ingram-Smith, C. Goodman, J. Neuffer, and K. S. Smith, data not shown) and is not an anomaly of the M. acetivorans enzyme.
CoA displays noncompetitive inhibition of the formation of a free propionyl-adenylate and initiates the release of AMP.
To determine whether MacsMa retains the ability to bind CoA when functioning as an acyl-adenylate synthetase, we examined the effect of CoA on propionyl-adenylate production. The propionyl-adenylate release activity of MacsMa, measured using the hydroxamate assay, was found to decrease almost 60% at 20 mM CoA (Fig. 1A), and the Ki of CoA was determined to be ∼14 mM (Fig. 1B). To investigate the nature of CoA inhibition, propionyl-adenylate production was measured using the hydroxamate assay in the presence of increasing levels of CoA, with the concentration of either propionate or ATP varied and the other held constant. CoA displayed noncompetitive inhibition versus both propionate and ATP (Fig. 1C and D). In a further examination of CoA inhibition of the MacsMa propionyl-adenylate synthetase activity, AMP production in the presence and absence of CoA with propionate as the acyl substrate was determined using the coupled-enzyme assay. Activity increased ∼50-fold in the presence of 1 mM CoA versus in the absence of CoA (Table 1). These results combined with our other results suggest that CoA affects retention of propionyl-adenylate such that the second step of the acyl-CoA synthetase reaction can be initiated but cannot be completed to form propionyl-CoA.
FIG. 1.

CoA inhibition of MacsMa acyl-adenylate synthetase activity with propionate was measured using the hydroxamate assay. (A) Plot of MacsMa activity in the presence of increasing CoA concentrations. Activities are reported as percentages of the activity observed in the absence of CoA (100%). (B) Inverse plot of MacsMa activity in the presence of increasing CoA concentrations. (C) Reciprocal plot of 1/v values (where v is velocity) versus 1/[propionate] values at different CoA concentrations. (D) Reciprocal plot of 1/v versus 1/[ATP] at different CoA concentrations. The amount of enzyme in each reaction mixture was 53 μg.
Kinetic characterization of the acyl-adenylate synthetase activity of MacsMa.
The acyl substrate range and kinetic parameters for acyl-adenylate production and release in the absence of CoA have been determined for MacsMa using the hydroxamate assay (Table 3). The highest catalytic efficiency (kcat/Km) and turnover number (kcat) were observed with propionate, which also has the highest Km. Although the Km values for propionate and butyrate are similar, the kcat value observed with butyrate was over 2-fold reduced versus that with propionate and is reflected in the approximately 2-fold reduction in catalytic efficiency. The enzyme had 4- to 10-fold-reduced Km values for 2-methylpropionate, 2-methylbutyrate, and 2-methylvalerate compared to values for propionate, and the turnover numbers for each of these substrates were also reduced. MacsMa was unable to utilize valerate, hexanoate, 3-methylbutyrate, 3-methylvalerate, or 4-methylvalerate for its acyl-adenylate synthetase activity.
TABLE 3.
Kinetic parameters for acyl substrate utilization in acyl-adenylate formationa
| Substrate | Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) |
|---|---|---|---|
| Acetate | — | — | — |
| Propionate | 29.1 ± 0.30 | 4.78 ± 0.04 | 0.164 ± 0.001 |
| Butyrate | 27.9 ± 0.77 | 2.01 ± 0.02 | 0.073 ± 0.002 |
| 2-Methylpropionate | 6.3 ± 0.06 | 0.54 ± 0.007 | 0.085 ± 0.001 |
| 2-Methylbutyrate | 2.7 ± 0.16 | 0.21 ± 0.003 | 0.075 ± 0.003 |
| 2-Methylvalerate | 5.3 ± 0.17 | 0.33 ± 0.003 | 0.062 ± 0.001 |
—, activity was too low for determination of kinetic parameters.
Using propionate as the acyl substrate, ATP, CTP, GTP, TTP, UTP, ITP, and ADP were tested for their ability to serve as the nucleotide substrate for acyl-adenylate synthesis. Activity, as measured by the hydroxamate assay, was observed only with ATP, and thus kinetic parameters for ATP were determined with each acyl substrate (Table 4). The kcat and Km values determined for ATP varied in a pattern similar to those observed for the partner acyl substrate. The lowest Km value for ATP was observed with 2-methylbutyrate as the acyl substrate, with an 18-fold-reduced Km compared to that observed with propionate; in turn, the turnover rate decreased approximately 67-fold. A corresponding decrease in both the kcat and Km values for ATP was observed as those values for acyl substrates decreased. Thus, the lower saturating concentration of acyl substrate for the enzyme results in the lower turnover rate for release of acyl-adenylate as a product.
TABLE 4.
Kinetic parameters for ATP utilization in acyl-adenylate synthesis
| Acyl substrate | Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) |
|---|---|---|---|
| Acetate | — | — | — |
| Propionate | 6.90 ± 0.75 | 10.67 ± 1.34 | 1.54 ± 0.07 |
| Butyrate | 6.72 ± 0.51 | 4.98 ± 0.09 | 0.74 ± 0.04 |
| 2-Methylpropionate | 1.66 ± 0.05 | 1.09 ± 0.004 | 0.66 ± 0.02 |
| 2-Methylbutyrate | 0.38 ± 0.01 | 0.16 ± 0.005 | 0.43 ± 0.01 |
| 2-Methylvalerate | 3.51 ± 0.10 | 0.39 ± 0.002 | 0.11 ± 0.002 |
—, activity was too low for determination of kinetic parameters.
An Acs1Mt Trp416Gly variant releases a free acyl-adenylate with propionate and butyrate in the absence of CoA.
To determine if other acyl-CoA synthetases have the ability to catalyze the formation and release of acyl-adenylate, an Acs1Mt Trp416Gly variant was utilized to examine the acyl-adenylate synthetase activity with propionate and butyrate. This variant is Macs-like in that replacement of Trp416 by Gly expanded the acyl substrate-binding pocket such that the enzyme was able to accommodate substrates that were as long as octanoate as well as branched-chain acyl substrates for acyl-CoA synthesis (17). Using the hydroxamate assay with the preferred acyl substrate 4-methylvalerate, the Acs1Mt Trp416Gly variant behaved as an acyl-CoA synthetase, with only 6.4% ± 0.1% activity observed in the absence versus the presence of CoA. However, substantial acyl-adenylate synthetase activity was observed with propionate or butyrate as the substrate, with 64.1% ± 1.0% and 67.4% ± 0.9% activity, respectively, in the absence versus the presence of CoA. Thus, enzymatic activity increased only ∼1.5- to 1.6-fold in the presence of CoA with propionate and butyrate, versus a 15.6-fold increase in the presence of CoA with 4-methylbutyrate as the substrate.
The results from the hydroxamate assay were confirmed by measuring the production of PPi by the molybdate assay. Again, only 6.4% ± 0.1% activity was observed in the absence versus the presence of CoA with 4-methylvalerate as the substrate, whereas 36.6% ± 0.7% and 67.3% ± 2.5% more activity was observed with propionate and butyrate, respectively, in the absence versus the presence of CoA. The 1.5- to 2.7-fold increase in activity in the presence of CoA with propionate and butyrate as the acyl substrates versus a 15.6-fold increase with 4-methylvalerate is similar to the results observed in the hydroxamate assay. These results suggest that Acs1Mt Trp416Gly behaves similarly to MacsMa, catalyzing acyl-adenylate synthesis and release with short acyl substrates but acyl-CoA synthesis with its preferred branched-chain substrate 4-methylvalerate.
Investigation of acyl-binding pocket residues.
Shah et al. (31) have solved the structure of MacsMa and proposed a putative acyl-binding pocket composed at least in part of Trp259, Gly351, Trp237, Trp254, Lys256, and Cys298. Trp259 is proposed to play a role in truncating the acyl substrate-binding pocket by forming a “floor” to prevent the enzyme from accommodating larger acyl substrates (31). Alignment of Acs and Macs sequences (Fig. 2) demonstrates that Gly351 of MacsMa is replaced by Trp416 in Acs1Mt, which has been shown to limit substrate range in Acs (17). Trp254 is highly conserved among Macs and Acs sequences but is replaced by His207 in 4-chlorobenzoate:CoA ligase (CBAL), which is also a member of the adenylate-forming enzyme superfamily and catalyzes the synthesis of 4-chlorobenzoyl:CoA in a two-step reaction. Lys256 in MacsMa is replaced in Acs1Mt by Thr313, which has also been shown to play a very important role in the acetate-binding pocket (17). Interestingly, the structure of MacsMa revealed an interaction between Lys256 and Cys298, which pulls the Lys256 side chain away from the acyl-binding pocket (31).
FIG. 2.

Partial sequence alignment of Macs and Sacs sequences. Sequences were aligned using Clustal X (36). Residues proposed to be important in the acyl substrate-binding pocket of MacsMa or Acs1Mt are in boldface. MacsMa, M. acetivorans Macs (GI 20091733); MacsMm, Methanococcus maripaludis Macs (GI 159905782); Acs1Mt, M. thermautotrophicus Acs1 (GI 82541817); AcsSe, S. enterica Acs (GI 16767525); Acs1Sc, S. cerevisiae Acs1 (GI 6319264); MacsMt, Methanothermobacter thermautotrophicus Macs (GI 110613493).
MacsMa variants altered in the acyl substrate-binding pocket were created as follows: Trp259→Phe, Tyr, Arg, and Ala; Gly351→Trp; Trp237→Tyr and Ala; Trp254→Tyr, His and Ala; Lys256→Thr and Leu; and Cys298→Tyr, Ser, and Ala. The variant enzymes were all soluble, with the exception of the Trp237Ala and Trp254Ala variants. Enzymatic activity was examined using the hydroxamate assay in the presence or absence of CoA, with a wide range of acyl substrates, including both straight-chain (acetate to octanoate) and branched-chain (2-methylpropionate to 4-methylvalerate) acyl compounds, but little-to-no acyl-adenylate synthetase or acyl-CoA synthetase activity was detected, except with the Lys256Leu variant. In confirmation of this result, PPi production was examined using the molybdate assay and found to be barely detectable with propionate and 2-methylbutyrate as the acyl substrates for testing acyl-adenylate synthetase in the absence of CoA and acyl-CoA synthetase activity in the presence of CoA, respectively.
The Lys256Leu variant had significantly reduced acyl-adenylate synthetase activity with a dramatically decreased turnover number (kcat) compared to that of the wild-type enzyme (0.22 ± 0.01 s−1 versus 4.78 ± 0.04 s−1) and an increased Km for propionate (101.1 ± 9.6 mM versus 29.1 ± 0.3 mM for the wild-type enzyme), as measured by the hydroxamate assay. As for other variants altered in the putative acyl pocket, the acyl-CoA synthetase activity with 2-methylbutyrate was significantly diminished for the Lys256Leu variant, such that kinetic parameters could not be determined. Interestingly, the optimal temperature for this variant decreased by 10°C relative to that observed for the wild-type enzyme, and the variant also displayed lower thermostability than the wild-type enzyme (data not shown), suggesting that Lys256 plays an important role in the structural stability of the enzyme.
DISCUSSION
MacsMa is a medium-chain acyl-CoA synthetase with 2-methylbutyrate as the preferred acyl substrate.
The characterized members of the short- and medium-chain acyl-CoA synthetase family of enzymes are the acetyl- and propionyl-CoA synthetases, which utilize the short-chain acyl substrates acetate and propionate (6, 7, 9, 15-18), and Sa, MACS1, LAE, and FadK, which utilize a range of slightly longer straight- and branched-chain acyl substrates (10, 11, 25). Consistent with its being phylogenetically more closely positioned with the Sa, MACS1, and LAE sequences rather than Acs sequences, MacsMa was found to utilize 2-methylbutyrate as its preferred acyl substrate for acyl-CoA synthesis. The production of 2-methylbutyryl-CoA, AMP, and PPi were confirmed, indicating that MacsMa catalyzes the two-step acyl-CoA synthetase reaction with an acyl-adenylate intermediate.
Surprisingly, MacsMa was discovered to catalyze the first step of the acyl-CoA synthetase reaction and to release the acyl-adenylate intermediate as a free product when short-chain acyl substrates, such as propionate, were used. Four lines of evidence support this conclusion: (i) activity was observed in the absence of CoA using the hydroxamate reaction, which can detect activated acyl groups, including both acyl-CoA and acyl-AMP; (ii) the enzyme-bound acyl-adenylate produced in the first step of the acyl-CoA synthetase reaction of Acs1Mt could not be detected by this assay, but a free acyl-adenylate was detected for MacsMa; (iii) PPi was found to be the other product of the reaction catalyzed by MacsMa in the absence of CoA; and (iv) little-to-no AMP production was observed in the absence of CoA for MacsMa, indicating that the propionyl adenylate was not degraded into propionate and AMP. However, CoA was found to inhibit the release of the propionyl-adenylate from the enzyme and instead initiated the release of AMP.
Similar results were obtained for a Trp416Gly variant of Acs1Mt. This variant is Macs-like in that it has a broader acyl substrate range and prefers longer acyl substrates than wild-type Acs1Mt. With short-chain acyl substrates, the variant catalyzed the first step of the acyl-CoA synthetase reaction and released the acyl-adenylate in the absence of CoA. The results observed with MacsMa and the Macs-like Trp416Gly Acs1Mt variant suggest that although the medium-chain acyl-CoA synthetases may have the ability to utilize a broad range of substrates for the acyl-adenylate-forming first step of the reaction, the intermediate may not be suitable for the thioester-forming second step, in which case the enzyme has devised a mechanism for breaking down and releasing the intermediate.
MacsMa is not unique in its release of the acyl-adenylate intermediate.
The duality of the MacsMa enzymatic activity is not unprecedented. LAE, a Macs which catalyzes the first step of lipoylation in mammals, has been shown to catalyze the formation of either an acyl-CoA or an acyl-GMP, depending on the nucleotide triphosphate substrate utilized. This enzyme utilizes GTP for acyl-guanidylate synthesis but utilizes ATP for acyl-CoA synthesis (11). Lipoyl-GMP appears to be easily released from LAE, whereas lipoyl-AMP is retained in the active site as an intermediate for use in the second step of acyl-CoA synthesis. Purified LAE from bovine liver mitochondria exhibited the highest acyl-GMP synthetase activity with octanoate, decanoate, and lipoate but had the highest acyl-CoA synthetase activity with hexanoate and moderate activity with butyrate or octanoate (11). The lipoyl-GMP product is subsequently utilized by lipoyltransferase for lipoylation of other proteins.
The bovine LAE characterized by Fujiwara et al. (11) shows 75% identity and 84% similarity to the human MACS1 characterized by Fujino et al. (10), suggesting that these are homologs. MACS1 and Sa were shown to require both ATP and CoA for activity (10); however, the spectrophotometric and isotopic enzymatic assays used in those studies measure the formation of acyl-CoA and would not detect the formation of a free acyl-adenylate. Thus, it is possible that these enzymes may also have acyl-activating activity in the presence of GTP, as observed for LAE. MacsMa was tested for both acyl-CoA synthetase and acyl-activating activity with ATP and GTP but was unable to utilize GTP for either enzymatic activity.
Trivedi et al. (37) identified a class of FadD enzymes from Mycobacterium tuberculosis that activate long-chain fatty acids for utilization by polyketide synthases. These enzymes, designated FAAL for fatty acyl-AMP ligase, activate long-chain fatty acids as acyl-adenylates rather than acyl-CoA thioesters (37). Interestingly, these enzymes catalyzed fatty acyl-AMP production even in the presence of CoA. Whether these FAALs can catalyze acyl-CoA thioester formation in the presence of a nucleotide substrate other than ATP was not determined. The FadDs are distantly related to the short- and medium-chain acyl-CoA synthetases, including MacsMa, MACS1, and LAE, and there is low sequence identity (∼20%) between these groups even though they are part of the acyl-adenylate-forming enzyme superfamily.
Comparison of the acyl-binding pockets in Acs and MacsMa.
The acyl substrate-binding pocket of Acs1 from M. thermautotrophicus has been investigated previously (17). Four important amino acid residues, Ile312, Thr313, Val388, and Trp416, are well conserved in Acs and have been shown to be critical in acyl substrate binding and catalysis (17). Comparison of the structures of the acyl-binding pockets in MacsMa (31) and Salmonella enterica Acs (AcsSe) (13) revealed that MacsMa has a much shallower acyl-binding pocket than AcsSe and that residue Trp259 in MacsMa forms a “floor” to prevent longer acyl substrates from binding and thus has a function similar to that of Trp414 in AcsSe. Alteration of the Trp416 residue in Acs1Mt equivalent to Gly expanded the acyl substrate-binding pocket so that 4-methylvalerate was the preferred acyl substrate (17).
The role of Trp259 in determining acyl substrate utilization by MacsMa was examined by analyzing variants altered at this position. The five purified variants displayed little-to-no acyl-adenylate or acyl-CoA synthetase activity, which suggests an essential role for this residue in acyl substrate binding and catalysis. Although Tyr and Phe are considered to be conservative amino acid replacements for Trp, modeled structures for these variants indicate that the benzoyl ring side chains of Phe and Tyr are twisted 62° and 32°, respectively, in relation to the side chain of Trp259, which may be responsible for the severe reduction in the acyl-adenylate and acyl-CoA synthetase activities.
As seen by sequence alignment (Fig. 2), the acetate-binding pocket residue Thr313 of Acs1Mt is replaced by Lys256 in MacsMa. Alteration of Thr313 to Lys in Acs1Mt was shown to cause the enzyme to release acyl-adenylate as a free product before the thioester-forming step of the reaction (17). This Acs variant had only weak activity with acetate and could no longer utilize propionate. Sa also has Lys in the position equivalent to Thr313 of Acs1Mt yet catalyzes the two-step reaction to produce acyl-CoA (10, 11, 25). Whether Sa releases the acyl-adenylate with other, smaller acyl substrates is not known.
The structure of MacsMa revealed an uncommon interaction between Lys256 and Cys298 (31). Therefore, the effects on enzymatic activity of amino acid substitutions at both of these residues were analyzed. With the exception of the Lys256Leu variant, no detectable acyl-adenylate synthetase or acyl-CoA synthetase activity was observed for variants altered at either Lys256 or Cys298. For the Lys256Leu variant, the kcat for the propionyl-adenylate synthetase activity was greatly diminished and no acyl-CoA synthetase activity was evident. The decreased temperature optimum for this variant suggests that Lys256 may have both a catalytic and a structural function in MacsMa. Modeled structures for both the Lys256Thr and Lys256Leu variants revealed that the position of Cys298 is altered such that there is no longer an interaction with Thr or Leu at position 256. These results suggest that the interaction between the ɛ-amino group of Lys256 and Cys298 may be crucial for both structural stability and acyl-adenylate synthetase activity.
Finally, Trp237 and Trp254 in the acyl substrate-binding pocket were targeted for kinetic analysis. Ala substitution at either residue resulted in insoluble recombinant enzyme. Although other variants altered at Trp237 or Trp254 were soluble, the purified enzymes showed little of either enzymatic activity, as observed for other MacsMa variants altered in the acyl substrate-binding pocket. Thus, residues Trp259, Lys256, Cys298, Trp254, and Trp237 appear to play essential functions in acyl substrate binding and catalysis, and even conservative changes in these residues resulted in a loss of nearly all the acyl-adenylate synthetase/acyl-CoA synthetase activity. Overall, these results suggest that the acyl substrate-binding pocket of MacsMa has a very specific architecture and cannot withstand even minor changes.
A possible physiological role for MacsMa.
BLAST searches of the nonredundant protein sequence database at the National Center for Biotechnology Information and the Integrated Microbial Genomes system available through the DOE Joint Genome Institute (http://img.jgi.doe.gov) combined with phylogenetic analysis revealed that an ORF encoding a putative MacsMa ortholog is present only in the genomes of a limited number of bacteria or archaea. Within the archaea, MacsMa orthologs are found only in the strictly anaerobic methanogenic archaea, and of the 31 finished or draft methanoarchaeal genome sequences available through the Integrated Microbial Genomes system, only six do not have an ORF encoding Macs and five have two putative macs genes. Within the bacteria, MacsMa orthologs have been identified only in select strict anaerobes.
Inspection of the region surrounding the macs genes in the methanoarchaeal genomes revealed gene conservation that may suggest a role for these Macs enzymes. The vorCBA operon encoding the three subunits of ketoisovalerate oxidoreductase (Vor) (35) is found adjacent to the macs gene in 17 of the 25 methanoarchaeal genomes with macs. Of the eight genomes that have macs but lack vorCBA, all but two instead have the kor operon, encoding 2-ketoglutarate ferredoxin oxidoreductase. The genomes that lack macs also lack vorCBA.
Fusion of macs and vorC in Methanosaeta thermophila and the presence of the vorCBA operon downstream of the majority of the methanoarchaeal macs genes suggest that Macs and Vor may interact and have integrated roles. The most straightforward explanation is that branched-chain acyl-CoA produced by Macs serves as a substrate for Vor. Vor, originally discovered in the peptide-utilizing hyperthermophilic archaea Pyrococcus and Thermococcus (14, 20), is responsible for CoA-dependent oxidation of branched-chain 2-ketoacids derived from valine, leucine, isoleucine, and methionine (14, 20). The acyl-CoA derivatives are then converted to fatty acids and CoA in an ATP-generating reaction catalyzed by the ADP-forming acetyl-CoA synthetase (24). M. thermautotrophicus has been shown to assimilate acetate into alanine, propionate into isoleucine, succinate into glutamate, and phenylacetate into phenylalanine (35), indicating that Vor and the related enzymes Por (pyruvate oxidoreductase), Kor (2-ketoglutarate oxidoreductase), and Ior (indolepyruvate oxidoreductase) have an anabolic function in the biosynthesis of amino acids (35).
Proteomic and microarray data on expression of macs are incomplete for the methanoarchaeal species. However, microarray data from M. maripaludis indicate that macs is expressed in both defined rich medium and minimal medium (40). Both microarray and proteomics studies suggest that vorCBA and macs are coordinately regulated, as was expected if the protein products form a pathway (26, 40).
Conclusions.
The results presented here suggest that although acyl-CoA synthetases may have the ability in vitro to utilize a broad range of acyl substrates for the acyl-adenylate-forming first step of the reaction, the intermediate may not be suitable for the thioester-forming second step. Although it is unlikely that this acyl-adenylate synthetase activity plays a physiological role as a mechanism for the enzyme to release an unfavorable acyl substrate after the first step of the reaction, this activity provides additional insight into the mechanism of acyl-CoA synthetases and suggests that the role of active-site architecture in substrate selection for the first step of the reaction has implications for the second step of the reaction as well.
Acknowledgments
Genomic DNA from M. acetivorans was kindly provided by Kevin Hill and James G. Ferry (Pennsylvania State University).
This work was supported by NIH award GM69374-01A1 to K.S.S., the South Carolina Experiment Station (project SC-1700198 grant to K.S.S.), and Clemson University. L.L.C. was supported by National Science Foundation award 0333210 to K.S.S.
This paper is Technical Contribution 5861 of the Clemson University Experiment Station.
Footnotes
Published ahead of print on 17 September 2010.
REFERENCES
- 1.Aceti, D. J., and J. G. Ferry. 1988. Purification and characterization of acetate kinase from acetate-grown Methanosarcina thermophila. Evidence for regulation of synthesis. J. Biol. Chem. 263:15444-15448. [PubMed] [Google Scholar]
- 2.Anke, H., and L. B. Spector. 1975. Evidence for an acetyl-enzyme intermediate in the action of acetyl-CoA synthetase. Biochem. Biophys. Res. Commun. 67:767-773. [DOI] [PubMed] [Google Scholar]
- 3.Berg, P. 1956. Acyl adenylates: an enzymatic mechanism of acetate activation. J. Biol. Chem. 222:991-1013. [PubMed] [Google Scholar]
- 4.Berg, P. 1956. Adenylates: the synthesis and properties of adenyl acetate. J. Biol. Chem. 222:1015-1034. [PubMed] [Google Scholar]
- 5.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 6.Brasen, C., and P. Schonheit. 2005. AMP-forming acetyl-CoA synthetase from the extremely halophilic archaeon Haloarcula marismortui: purification, identification and expression of the encoding gene, and phylogenetic affiliation. Extremophiles 9:355-365. [DOI] [PubMed] [Google Scholar]
- 7.Brasen, C., C. Urbanke, and P. Schonheit. 2005. A novel octameric AMP-forming acetyl-CoA synthetase from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. FEBS Lett. 579:477-482. [DOI] [PubMed] [Google Scholar]
- 8.Chang, K. H., H. Xiang, and D. Dunaway-Mariano. 1997. Acyl-adenylate motif of the acyl-adenylate/thioester-forming enzyme superfamily: a site-directed mutagenesis study with the Pseudomonas sp. strain CBS3 4-chlorobenzoate:coenzyme A ligase. Biochemistry 36:15650-15659. [DOI] [PubMed] [Google Scholar]
- 9.Eggen, R. I., A. C. Geerling, A. B. Boshoven, and W. M. de Vos. 1991. Cloning, sequence analysis, and functional expression of the acetyl coenzyme A synthetase gene from Methanothrix soehngenii in Escherichia coli. J. Bacteriol. 173:6383-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujino, T., Y. A. Takei, H. Sone, R. X. Ioka, A. Kamataki, K. Magoori, S. Takahashi, J. Sakai, and T. T. Yamamoto. 2001. Molecular identification and characterization of two medium-chain acyl-CoA synthetases, MACS1 and the Sa gene product. J. Biol. Chem. 276:35961-35966. [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara, K., S. Takeuchi, K. Okamura-Ikeda, and Y. Motokawa. 2001. Purification, characterization, and cDNA cloning of lipoate-activating enzyme from bovine liver. J. Biol. Chem. 276:28819-28823. [DOI] [PubMed] [Google Scholar]
- 12.Grayson, N. A., and R. B. Westkaemper. 1988. Stable analogs of acyl adenylates. Inhibition of acetyl- and acyl-CoA synthetase by adenosine 5′-alkylphosphates. Life Sci. 43:437-444. [DOI] [PubMed] [Google Scholar]
- 13.Gulick, A. M., V. J. Starai, A. R. Horswill, K. M. Homick, and J. C. Escalante-Semerena. 2003. The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42:2866-2873. [DOI] [PubMed] [Google Scholar]
- 14.Heider, J., X. Mai, and M. W. Adams. 1996. Characterization of 2-ketoisovalerate ferredoxin oxidoreductase, a new and reversible coenzyme A-dependent enzyme involved in peptide fermentation by hyperthermophilic archaea. J. Bacteriol. 178:780-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horswill, A. R., and J. C. Escalante-Semerena. 2002. Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: residue Lys592 is required for propionyl-AMP synthesis. Biochemistry 41:2379-2387. [DOI] [PubMed] [Google Scholar]
- 16.Ingram-Smith, C., and K. S. Smith. 2007. AMP-forming acetyl-CoA synthetases in Archaea show unexpected diversity in substrate utilization. Archaea 2:95-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingram-Smith, C., B. I. Woods, and K. S. Smith. 2006. Characterization of the acyl substrate binding pocket of acetyl-CoA synthetase. Biochemistry 45:11482-11490. [DOI] [PubMed] [Google Scholar]
- 18.Jetten, M. S., A. J. Stams, and A. J. Zehnder. 1989. Isolation and characterization of acetyl-coenzyme A synthetase from Methanothrix soehngenii. J. Bacteriol. 171:5430-5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jogl, G., and L. Tong. 2004. Crystal structure of yeast acetyl-coenzyme A synthetase in complex with AMP. Biochemistry 43:1425-1431. [DOI] [PubMed] [Google Scholar]
- 20.Kletzin, A., and M. W. Adams. 1996. Molecular and phylogenetic characterization of pyruvate and 2-ketoisovalerate ferredoxin oxidoreductases from Pyrococcus furiosus and pyruvate ferredoxin oxidoreductase from Thermotoga maritima. J. Bacteriol. 178:248-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, H. Y., K. B. Na, H. M. Koo, and Y. S. Kim. 2001. Identification of active site residues in Bradyrhizobium japonicum acetyl-CoA synthetase. J. Biochem. 130:807-813. [DOI] [PubMed] [Google Scholar]
- 22.Lipmann, F., and L. C. Tuttle. 1945. A specific micromethod for determination of acyl phosphates. J. Biol. Chem. 159:21-28. [Google Scholar]
- 23.Lundie, L. L., Jr., and J. G. Ferry. 1989. Activation of acetate by Methanosarcina thermophila. Purification and characterization of phosphotransacetylase. J. Biol. Chem. 264:18392-18396. [PubMed] [Google Scholar]
- 24.Mai, X., and M. W. Adams. 1996. Purification and characterization of two reversible and ADP-dependent acetyl coenzyme A synthetases from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 178:5897-5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan-Kiss, R. M., and J. E. Cronan. 2004. The Escherichia coli fadK (ydiD) gene encodes an anaerobically regulated short chain acyl-CoA synthetase. J. Biol. Chem. 279:37324-37333. [DOI] [PubMed] [Google Scholar]
- 26.Porat, I., W. Kim, E. L. Hendrickson, Q. Xia, Y. Zhang, T. Wang, F. Taub, B. C. Moore, I. J. Anderson, M. Hackett, J. A. Leigh, and W. B. Whitman. 2006. Disruption of the operon encoding Ehb hydrogenase limits anabolic CO2 assimilation in the archaeon Methanococcus maripaludis. J. Bacteriol. 188:1373-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Putinis, R. F., and E. W. Yamada. 1975. Colorimetric determination of inorganic pyrophosphate by a manual or automated method. Anal. Biochem. 68:185-195. [DOI] [PubMed] [Google Scholar]
- 28.Reger, A. S., R. Wu, D. Dunaway-Mariano, and A. M. Gulick. 2008. Structural characterization of a 140° domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47:8016-8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose, I. A., M. Grunberg-Manago, S. F. Korey, and S. Ochoa. 1954. Enzymatic phosphorylation of acetate. J. Biol. Chem. 211:737-756. [PubMed] [Google Scholar]
- 30.Rother, M., and W. W. Metcalf. 2004. Anaerobic growth of Methanosarcina acetivorans C2A on carbon monoxide: an unusual way of life for a methanogenic archaeon. Proc. Natl. Acad. Sci. U. S. A. 101:16929-16934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah, M., C. Ingram-Smith, L. Cooper, K. Smith, and A. M. Gulick. 2009. The 2.1Å crystal structure of an acyl-CoA synthetase from Methanosarcina acetivorans reveals an alternate acyl binding pocket for small branched acyl substrates. Proteins 77:685-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh-Wissmann, K., and J. G. Ferry. 1995. Transcriptional regulation of the phosphotransacetylase-encoding and acetate kinase-encoding genes (pta and ack) from Methanosarcina thermophila. J. Bacteriol. 177:1699-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith, K. S., and C. Ingram-Smith. 2007. Methanosaeta, the forgotten methanogen. Trends Microbiol. 15:150-155. [DOI] [PubMed] [Google Scholar]
- 34.Teh, Y. L., and S. H. Zinder. 1992. Acetyl-coenzyme A synthetase in the thermophilic, acetate-utilizing methanogen Methanothrix sp strain CALS-1. FEMS Microbiol. Lett. 98:1-8. [Google Scholar]
- 35.Tersteegen, A., D. Linder, R. K. Thauer, and R. Hedderich. 1997. Structures and functions of four anabolic 2-oxoacid oxidoreductases in Methanobacterium thermoautotrophicum. Eur. J. Biochem. 244:862-868. [DOI] [PubMed] [Google Scholar]
- 36.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. J. Higgins. 1997. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trivedi, O. A., P. Arora, V. Sridharan, R. Tickoo, D. Mohanty, and R. S. Gokhale. 2004. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 428:441-445. [DOI] [PubMed] [Google Scholar]
- 38.Webster, L. T., Jr. 1963. Studies of the acetyl coenzyme A synthetase reaction. I. Isolation and characterization of enzyme-bound acetyl adenylate. J. Biol. Chem. 238:4010-4015. [PubMed] [Google Scholar]
- 39.Wu, R., J. Cao, X. F. Lu, A. S. Reger, A. M. Gulick, and D. Dunaway-Mariano. 2008. Mechanism of 4-chlorobenzoate:coenzyme A ligase catalysis. Biochemistry 47:8026-8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia, Q., E. L. Hendrickson, Y. Zhang, T. Wang, F. Taub, B. C. Moore, I. Porat, W. B. Whitman, M. Hackett, and J. A. Leigh. 2006. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time PCR. Mol. Cell Proteomics 5:868-881. [DOI] [PMC free article] [PubMed] [Google Scholar]