

Abstract
Initial rate studies have revealed dramatic acceleration in aerobic Pd(II)-catalyzed C–H olefination reactions of phenylacetic acids when mono-N-protected amino acids are used as ligands. In light of these findings, systematic ligand tuning was undertaken, which has resulted in drastic improvements in substrate scope, reaction rate, and catalyst turnover. We present evidence from intermolecular competition studies and kinetic isotope effect experiments that implies that the observed rate increases are a result of acceleration in the C–H cleavage step. Furthermore, these studies suggest that the origin of this phenomenon is a change in the mechanism of C–H cleavage from electrophilic palladation to proton abstraction.
1. Introduction
Pd(0)-catalyzed reactions of aryl and alkyl halides (R–X) have revolutionized the field of synthetic organic chemistry since the early 1970s, when the first reports describing the catalytic coupling of aryl halides and olefins were independently disclosed by Mizoroki and Heck.1 Subsequent to that work, an array of carbon–carbon2–8 and carbon–heteroatom8–12 bond–forming reactions have been developed based on Pd(0)/Pd(II) redox catalysis, with increasing levels of efficiency, practicality, and reliability. The power of this class of catalytic reactions stems from the diverse reactivity of the [Pd(II)–R] intermediates, which are commonly generated from oxidative addition of the R–X group to Pd(0) (Scheme 1).
Scheme 1.
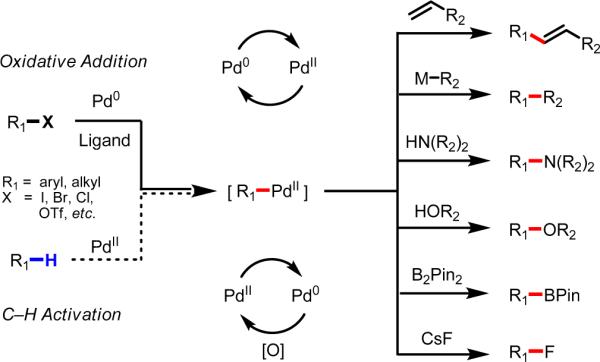
The versatile reactivity of [Pd(II)–R] intermediates.
Fascinated and inspired by this diverse reactivity, our research group has focused on mimicking this catalysis using Pd(II)-mediated C–H activation as an entry point to form the [Pd(II)–R] species.13–14 At the outset we recognized that the precise molecular structure of [Pd(II)–R] intermediates generated in this manner, which could exist as monomeric, dimeric, or trimeric species, would likely be more complex than that of those made from oxidative addition of an aryl halide to Pd(0), which are predominantly monomeric. Nevertheless, our group15–19 and others20–29 have demonstrated several catalytic reactions in which [Pd(II)–R] intermediates generated by C–H activation react with strong oxidants to induce reductive elimination from high-energy Pd(III)30,31 or Pd(IV)32 species. Despite our early success in this direction, the bulk of our efforts have centered on developing novel carbon–carbon and carbon–heteroatom bond–forming reactions along a Pd(II)/Pd(0) catalytic cycle. This strategy has proven to be a remarkably fruitful research area, as many of the reactions depicted in Scheme 1 have already been demonstrated.33–40
In comparison to the state of the art in traditional Pd(0) coupling chemistry, C–H activation reactions using Pd(II)/Pd(0) catalysis remain underdeveloped. The principal driving force that has propelled Pd(0)-catalyzed coupling chemistry during the past several decades has been the development of bulky phosphine and N-heterocyclic carbene (NHC) ligands that promote both the oxidative addition and reductive elimination steps, thereby improving the reaction rate, lowering the requisite catalyst loading, and expanding the substrate scope.41 The advancements in ligand design have resulted in truly practical and operationally simple reactions that have found a myriad of applications, including numerous examples in natural product total synthesis.42
In sharp contrast, though several notable achievements have been made in the area of Pd(II)-catalyzed C–H activation,14 a pervasive limitation that has hampered efforts in this field is the dearth of suitable ligand scaffolds that can accelerate C–H cleavage. (Throughout the text, we use the term “C–H cleavage” to refer to the general process of carbon–hydrogen bond breaking along any of a variety of mechanistic pathways.) In this respect, several challenges must be overcome to design suitable ligands. One problem is that many of the common phosphine and NHC ligands are too strong of σ-donors and outcompete the substrate molecule for binding to Pd(II) or render the metal-center overly electron-rich for C–H activation. A second related problem is devising reaction conditions that facilitate controlled assembly of a pre-transition state Pd(II) complex with one molecule of substrate and one molecule of ligand because the two different molecules must have well matched coordinative affinity for Pd(II) (Scheme 2).
Scheme 2.

Depiction of three scenarios for the pre-transition state coordination structures prior to Pd(II)-mediated C–H cleavage: (I) The substrate contains a strong directing group (DG) and is dominantly bound to Pd(II), precluding ligand (L) coordination. The reaction may take place, but the transition state energy for C–H cleavage will be unaffected by the ligand. (II) The substrate and ligand possess matched coordinative affinities for Pd(II), allowing one molecule of each to bind. The reaction may take place, and the transition state energy for C–H cleavage will be affected by ligand binding. (III) The ligand is a strong σ-donor and outcompetes substrate molecules for coordination to Pd(II). The reaction will not take place.
To engineer a reaction system in which the ligand plays a dominant role in influencing the reactivity and/or selectivity, utilizing a functional group capable of directing C–H cleavage via weak coordination is highly advantageous. Our group has pursued this strategy by developing reactions that are compatible with substrates containing commonly occurring Lewis basic moieties that form low-energy interactions with Pd(II)14i (e.g., carboxylic acids, triflimides, and alcohols). In these cases, C–H cleavage does not occur as easily as with many of the other commonly utilized directing groups (e.g., pyridines, oxazolines and oximes), which has given us a unique opportunity to probe how external ligands change the activation energy of the C–H cleavage step. Recently, our group has reported successful examples of using chiral mono-N-protected amino acid ligands to control the enantio-43,44 and positional45 selectivity of C–H cleavage, which serves as evidence that the ligands are coordinated to Pd(II) during the C–H cleavage event and are influencing the corresponding transition state energies. Moreover, as part of our group's interest in Pd(II)-catalyzed C–H olefination,35,44–56 a research field that was initiated by the seminal contributions of Fujiwara and Moritani in the late 1960s,33–34 we have found success in using mono-N-protected amino acid ligands to promote C–H activation of generally unreactive substrates that are strongly electron-deficient or those that contain remote, weakly coordinating directing groups.45,49g,49i Given the need for new ligands to accelerate C–H activation and enable the diverse reactivity depicted in Scheme 1, at this stage it is critical to elucidate more fully whether the improved reactivity observed using amino acid ligands is a result of ligand-induced acceleration (i.e., a decrease in the activation energy) and if so, whether the amino acid core structure can be further tuned to offer improved reactivity, reduced reaction times, and lower catalyst loading.57
Herein we report the results of our efforts to investigate these questions. Initial rate studies have revealed a dramatic rate increase in the presence of amino acid ligands, which, taken in the context of other data, is suggestive of acceleration in the C–H cleavage step. The results from these initial rate studies prompted us to undertake further ligand optimization, which has led to drastically improved reaction conditions. Finally, we present evidence from intermolecular competition experiments and kinetic isotope (KIE) experiments that suggest that coordination of amino acid ligands to Pd(II) leads to a change in the reaction mechanism.
Results and Discussion
2.1. Preliminary Initial Rate Studies
We have previously reported a Pd(II)-catalyzed mono-selective C–H olefination reaction of phenylacetic acid substrates.45 In that report, we demonstrated preliminary results utilizing mono-N-protected amino acid ligands to promote C–H olefination with unreactive substrates, including hydrocinnamic acids (which are challenging because of the remoteness of the carboxylate directing group) and phenylacetic acid substrates bearing electron-withdrawing groups (e.g., CF3 and NO2).
We began our investigation by revisiting the data from our initial report. In particular we were interested in determining whether the improved yield after 48 h stemmed from improved catalyst lifetime or increased initial reaction rate (Scheme 3). Using the C–H olefination of 1 to 1a as a model study, we ran parallel reactions, quenching them at regular intervals and determining the conversion by 1H NMR of the crude reaction mixtures. We then plotted the conversion versus time under four different reaction conditions: (1) without Boc-Val-OH, without 1,4-benzoquinone (BQ), (2) without Boc-Val-OH, with BQ, (3) with Boc-Val-OH, without BQ, (4) with Boc-Val-OH, with BQ. The results from these studies are shown in Figure 1.
Scheme 3.

Preliminary results for ligand-promoted C–H olefination of 1.45
Figure 1.

Initial rate studies of C–H olefination of o-trifluoromethylphenylacetic acid (1). BQ (5 mol%) and Boc-Val-OH (10 mol%). Each data point represents the average of three trials, with the exception of the Boc-Val-OH trials from 60 min to 120 min, which represent single trials. See Supporting Information for experimental details.
In the absence of BQ, we observed a 36-fold rate increase for reactions with Boc-Val-OH compared to those without this ligand (from 1.3 × 10−4 M/min to 4.7 × 10−3 M/min). To our surprise, we observed that BQ substantially decreased the reaction rate both with and without Boc-Val-OH. Previously, during our investigation to develop a highly mono-selective C–H olefination reaction with phenylacetic acid substrates, BQ was found to be beneficial for improving the overall yield after 48 h and controlling the mono/di selectivity with more reactive substrates. The observation that BQ leads to a decrease in the initial reaction rate, is consistent with it being found to improve the mono/di selectivity with more reactive substrates. Additionally, though BQ lowers the initial reaction rate, it also seems to be capable of improving the overall turnover number given a sufficiently long reaction time (particularly in the case of reactions run in the absence of amino acid ligands).
In light of these results, we wondered whether this trend was unique to electron-deficient substrates. Thus, we performed an analogous set of experiments with a more electron-rich substrate, 2, and found a 15-fold rate increase for reactions using with Boc-Val-OH compared to those without it (from 3.5 × 10−4 M/min to 5.2 × 10−3 M/min) (Figure 2).
Figure 2.

Kinetic studies of C–H olefination of o-tolylacetic acid (2). BQ (5 mol%) and Boc-Val-OH (10 mol%). Each data point represents the average of three trials, with the exception of the Boc-Val-OH trials from 60 min to 120 min, which represent single trials. See Supporting Information for experimental details.
With this information in hand, we next sought to determine optimal conditions for Pd(II)-catalyzed C–H olefination of 1 in an effort to improve the reaction rate, catalytic turnover, and substrate scope, which we viewed as central to advancing the versatility and practicality of the transformation.14i
2.2. Ligand Optimization
Our efforts to develop an improved reaction protocol for our aerobic Pd(II)-catalyzed C–H olefination reaction centered on the identification of an optimal ligand in terms of reaction rate and overall yield. To begin, we sought to identify the most active ligand backbones by examining a set of commercially N-Boc-protected amino acids (Table 1). For our screening studies, we selected a highly abridged reaction time of 20 minutes in order to see the comparative kinetic behavior of the different ligands. Notably, in the absence of ligand, the reaction was found to give less than 5% conversion (Entry 1). Prior to our investigation, we hypothesized that the bite angle between the two coordination sites on the amino acid ligand could have a dramatic impact on the catalytic activity of Pd(II), as it would presumably effect the geometry of the coordination assembly in the pre-transition state. We probed Boc-Gly-OH (Entry 9) (with no substitution at the alpha position, and hence a large bite angle) and a quaternary substituted amino acids with a smaller bite angles (Entry 14), and found that both of these ligand structures resulted in slower reactions than mono-α-substituted ligands. Among this group, Boc-Val-OH was the best, providing 1a in 46% conversion after 20 minutes (Entry 3). As a control experiment, we also examined other commonly used organic acids, representative examples of which are shown in Table 1 (Entries 17 and 18; see Supporting Information for additional results). These other acids uniformly gave low conversions, suggesting that coordination of both the carboxylate group and the protected amino group with Pd(II) is crucial for rate acceleration.
Table 1.
Optimization of the ligand backbone using Boc-protected amino acids.

| Entry | Ligand | % Conv. |
|---|---|---|
| 1 | --- | <5 |
| 2 | BQ | <5 |
| 3 | Boc-Val-OH | 46b |
| 4 | Boc-lle-OH | 37 |
| 5 | Boc-Leu-OH | 37 |
| 6 | Boc-t-Leu-OH | 38 |
| 7 | Boc-Nva-OH | 30 |
| 8 | Boc-Ala-OH | 28 |
| 9 | Boc-Gly-OH | 17 |
| 10 | Boc-Phe-OH | 33 |
| 11 | Boc-Ser-OH | 8 |
| 12 | Boc-β-Ala-OH | 8 |
| 13 |

|
6 |
| 14 |

|
21 |
| 15 |

|
5 |
| 16 |

|
16 |
| 17 | PivOH | <5 |
| 18 | p-TsOH·H2O | 0 |
aThe conversion was determined by 1H NMR analysis of the crude reaction mixture.
Average of three trials.
Following identification of valine as a highly reactive amino acid backbone, we sought to optimize the N-protecting group (Table 2). In an initial control experiment, we found that N-protection was required, as the reaction did not proceed with unprotected H-Val-OH (Entry 1). With this information in hand, we examined several commercially available N-protected valine amino acid ligands (Entries 1–3, 6, 7, and 12) and subsequently prepared several new ligands (Entries 4, 5, and 8–11). Because Boc protecting groups were found to be highly reactive, we examined other carbamate protecting groups with different steric properties (Entries 7–11 and 13), but did not observe any improvement. We moved on to amide protecting groups (Entries 3–5 and 12). Among the amide protecting groups examined, ligands bearing less sterically demanding groups (Entries 3 and 6) were found to give better conversions than those with sterically bulky protecting groups (Entries 4, 5, and 12), with Ac-Val-OH giving the best conversion (57%) (Entry 3).
Table 2.
Optimization of the N-protecting group on valine.

| Entry | Ligand | % Conv. |
|---|---|---|
| 1 | H-Val-OH | 0 |
| 2 | Boc-Val-OH | 46b |
| 3 | Ac-Val-OH | 57 |
| 4 | Ada-Val-OHc | 4 |
| 5 | Piv-Val-OH | 2 |
| 6 | Formyl-Val-OH | 31 |
| 7 | Fmoc-Val-OH | 19 |
| 8 | MeO2C-Val-OH | 21 |
| 9 | EtO2C-Val-OH | 26 |
| 10 | i-BuO2-C-Val-OH | 27 |
| 11 | Cbz-Val-OH | 18 |
| 12 | Bz-Val-OH | 3 |
| 13 | Men-Val-OHd | 39 |
aThe conversion was determined by 1H NMR analysis of the crude reaction mixture.
Average of three trials.
Ada = Adamantyl(OC).
Men = (−)-Menthyl(O2C).
Having found that the acetyl protecting group was highly reactive when used with valine, we wondered whether additional fine-tuning of the ligand backbone of N-acetyl-protected amino acids might result in further improvement in the catalyst activity (Table 3). To our delight, when we examined the N-acetyl-protected versions of a selected set of ligands from Table 1, we found improved yields for nearly every ligand that we probed relative to its Boc-protected counterpart. Ac-Ile-OH and Ac-Ala-OH were found to be the most reactive, giving 1a in 72% and 71% conversion, respectively. Quantitative conversion of 1 to 1a could be achieved by extending the reaction time to 2 hours (Entry 5).
Table 3.
Reexamination of alternative ligand backbones using Ac as the protecting group.a

| Entry | Ligand | % Conv. |
|---|---|---|
| 1 | Ac-Val-OH | 57 |
| 2 | Ac-Leu-OH | 55 |
| 3 | Ac-Ala-OH | 71 |
| 4 | Ac-Gly-OH | 51 |
| 5 | Ac-Ile-OH | 72b[>99]c |
| 6 | Ac-Ile-OH | 93d |
| 7 | Ac-Phe-OH | 60 |
| 8 |

|
5 |
| 9 |

|
31 |
The conversion was determined by 1H NMR analysis of the crude reaction mixture.
Average of three trials.
The bracketed value represents the conversion after 2 h.
110 °C.
We also probed the effect of amino acid ligand loading and found that when the reaction was run with anywhere between 5–15 mol% Ac-Ile-OH, the conversions were similar after 20 min. 2.5 mol% Ac-Ile-OH led to lower conversion (see Supporting Information). Similarly, we found that between 1 and 2 equivalents of olefin led to similar reaction rates and overall yields; with 3 equivalents of olefin, the reaction rate decreased. Because the reagents used in these experiments were readily available, we elected to use 2 equiv. Ac-Ile-OH (relative to Pd(OAc)2) and 2 equiv. olefin (relative to substrate) as the conditions for the investigations below (Sections 2.3–2.5). The reaction could be run effectively at temperatures between 50–110 °C (see Supporting Information), with lower temperatures giving decreased reaction rates. For example, at 50 °C, the reaction took 48 hours to reach >90% conversion. Running the reaction at 110 °C, >90% conversion could be achieved after 20 minutes (Entry 6, Table 3). The use of higher temperatures, such as 130 °C, reduced the reaction rate, presumably due to catalyst decomposition.
2.3 Low Catalyst Loadings/Air (1 atm) as the Reoxidant
Having identified Ac-Ile-OH as a superior ligand for reaction rate (i.e., turnover frequency, TOF) in the Pd(II)-catalyzed ortho-C–H olefination of 1, we became interested in examining the efficiency of the catalytic turnover under these conditions. We began by reducing the catalyst loading to 1 mol% Pd(OAc)2 and running parallel reactions under 1 atm O2 (Table 4). Gratifyingly, we found that efficient catalysis could still be achieved under these conditions, such that 1a could be prepared in 96% conversion (94% yield) after 4 h.
Table 4.
C–H olefination of 1 using 1 mol% Pd(OAc)2 and 1 atm O2.a

| Entry | Time (h) | % Conv. |
|---|---|---|
| 1 | 0 | 0 |
| 2 | 1 | 34 |
| 3 | 2 | 67 |
| 4 | 3 | 93 |
| 5 | 4 | 98 (94) |
The conversion was determined by 1H NMR analysis of the crude reaction mixture. Isolated yield is given in parentheses.
This result encouraged us to further lower the catalyst loading to 0.2 mol% Pd(OAc)2 (Table 5). Again, we found that highly efficient catalysis could be maintained under these conditions, and we were able to synthesize 1a with 91% conversion (83% yield) after an extended reaction time of 48 h. Notably, this result represents a TON of 455, which is among the highest reported for an aerobic C–H activation reaction.46,47 We further demonstrated the scalability of this chemistry by using the conditions described in Table 5 to prepare over 1 gram of 1a from 1 using only 2.2 mg of Pd(OAc)2. Following an aqueous quench and extraction with organic solvent, 1a could be obtained from the crude reaction mixture in 76% yield after recrystallization (Scheme 4).
Table 5.
C–H olefination of 1 using 0.2 mol% Pd(OAc)2 and 1 atm O2.a

| Entry | Time (h) | % Conv. |
|---|---|---|
| 1 | 0 | 0 |
| 2 | 4 | 15 |
| 3 | 20 | 52 |
| 4 | 48 | 91 (83) |
The conversion was determined by 1H NMR analysis of the crude reaction mixture.
Scheme 4.

Gram-scale synthesis of 1a using 0.2 mol% Pd(OAc)2 and 1 atm O2.
Though 1 atm O2 is a highly convenient oxidant, in terms of operational simplicity, being able to use 1 atm air (which is 21% O2) is highly desirable because it obviates the need to store compressed O2 in the laboratory. To examine whether O2 could be replaced with air, we used 1 mol% Pd(OAc)2 and ran parallel reactions in sealed tubes under air (Table 6). (Having a sealed vessel is necessary due to the volatility of ethyl acrylate.) Under these conditions, 1a could be synthesized in 87% conversion (78% yield) after an extended reaction time of 48 h.
Table 6.
C–H olefination of 1 using 1 mol% Pd(OAc)2 and 1 atm air.a

| Entry | Time (h) | % Conv. |
|---|---|---|
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 6 |
| 4 | 12 | 29 |
| 5 | 24 | 68 |
| 6 | 48 | 87 (78) |
The conversion was determined by 1H NMR analysis of the crude reaction mixture. Isolated yield is given in parentheses.
Given the low catalyst loading, relatively mild reaction conditions, and the fact that the only byproduct in the catalytic cycle is water, this chemistry is highly atom-economical58 and falls in line with goals of green chemistry.59
2.4. Substrate Scope for Accelerated C–H Olefination
Under the optimized conditions, a wide variety of phenylacetic acid substrates were highly reactive, generally giving the desired C–H olefinated products in quantitative yields (Table 7). The substrate scope was found to be remarkably broad, as the reaction tolerated electron-donating methoxy (5a and 10a) and alkyl groups (2a and 4a), as well as fluorides (6a) and chlorides (7a), the latter offering a unique opportunity for subsequent Pd(0)-mediated coupling chemistry. Bromides and iodides were found to be unreactive in C–H olefination. Arenes bearing electron-withdrawing substituents, including trifluoromethyl (1a and 3a), nitro (8a), and ketone (9a) groups, gave good to excellent yields. In an effort to demonstrate the potential applicability of this chemistry in drug diversification, we targeted ketoprofen (9) and naproxen (10), two commercially available non-steroidal anti-inflammatory drugs (NSAIDs), and observed that they could be directly ortho-olefinated to give 9a and 10a in nearly quantitative isolated yields.
Table 7.

| Product | Ligand | % Conv. |
|---|---|---|

|
--- | 7c |
| BQ | 3c | |
| Ac-Ile-OH | >99 (96) | |

|
--- | 25c |
| BQ | 7c | |
| Ac-Ile-OH | 98 (97) | |

|
--- | 2 |
| BQ | 1 | |
| Ac-Ile-OH | >99 (98) | |

|
--- | 64 |
| BQ | 9 | |
| Ac-Ile-OH | 88 (83)d | |

|
--- | 93 (92) |
| BQ | 12 | |
| Ac-Ile-OH | >99 (95) | |

|
--- | 6 |
| BQ | 2 | |
| Ac-Ile-OH | >99 (99) | |

|
--- | 8e |
| BQ | 3e | |
| Ac-Ile-OH | >99 (99)e | |

|
--- | 0 |
| BQ | 0 | |
| Ac-Ile-OH | 72 (70)f | |

|
--- | 10 |
| BQ | 5 | |
| Ac-Ile-OH | >99 (98) | |

|
--- | 44g |
| BQ | 23 | |
| Ac-Ile-OH | >99 (94) |
5 mol% BQ (when used), 10 mol% Ac-Ile-OH (when used).
The conversion was determined by 1H NMR analysis of the crude reaction mixture. Isolated yield is given in parentheses.
Average of three trials.
An additional 11% of the di-ortho-olefinated product was observed by 1H NMR.
6 h.
An additional 6% of the decarboxylated product was formed.
An additional 4% of a positional isomer was formed.
For each substrate, we ran two control experiments in the absence of Ac-Ile-OH: with BQ (our original mono-selective procedure) and without. In all cases, the presence of Ac-Ile-OH led to higher conversions, but the improvement was most pronounced with electron-poor substrates. Interestingly, for electron-rich arenes (2a, 4a, 5a, and 10a), relatively high conversions could be obtained after 2 h without Ac-Ile-OH if BQ was removed. As discussed above, in the absence of amino acid ligands, BQ allows for control of mono/di-selectivity and leads to higher TONs in many instances; however, in the case of these particular substrates (i.e., electron-rich arenes containing an ortho or meta-blocking group) BQ does not offer a clear benefit and only serves to reduce the reaction rate. In general, electron-deficient substrates were found to exhibit low reactivity in the absence of ligand (1a and 3a). The presence of a nitro group rendered the arene totally unreactive (8a) without ligand.
In accordance with our earlier observations,45 8a was found to decarboxylate under the reaction conditions, leading to exclusive formation of the corresponding decarboxylated product 8a' after 48 h. This reaction is triggered by the installation of the electron-withdrawing olefin, which further stabilizes anionic charge at the benzylic position through resonance effects. It is important to note here, that prior to our identification of Ac-Ile-OH as a ligand for accelerated C–H olefination, 8a could not be obtained in appreciable yields because using other ligands, it was found to decarboxylate to 8a' faster than it could be generated from C–H olefination of 8. In contrast, under these new conditions, 8a could be isolated in 70% yield after 2 h. This reaction was monitored over time using 1H NMR, and the results are depicted in Table 8.
Table 8.
Accelerated Pd(II)-catalyzed C–H olefination of 8.a

| Entry | Time (h) | %8 | %8a | % 8a' |
|---|---|---|---|---|
| 1 | 0 | 100 | 0 | 0 |
| 2 | 2 | 19 | 72 (70) | 6 (6) |
| 3 | 6 | 15 | 43 | 29 |
| 4 | 12 | 6 | 39 | 36 |
| 5 | 24 | 5 | 15 | 59 |
| 6 | 48 | 4 | 0 | 75 (71) |
The % composition was determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard. Isolated yield is given in parentheses. Less than 5% of 2-nitrotoluene (from direct decarboxylation of 8) was observed by 1H NMR in entries 2–6.
The phenylacetic acid substrates shown in Table 7 all contain a sterically bulky blocking substituent at the ortho- or meta- positions. For those that are not of this type, the ligand-accelerated C–H olefination conditions that we report here will lead to significant formation of the di-ortho-olefinated byproduct over time. Indeed, concurrent to this work, we developed a robust method for direct 2,6-diolefination of phenylacetic and hydrocinnamic acids (Scheme 5).49i For 2,6-diolefination, Ac-Ile-OH, which we use throughout this paper, gave irreproducible results, and Ac-Val-OH was found to be superior. Current investigations are underway in our laboratory to design a ligand that will offer enhanced reactivity but will stop after a single olefination event, thereby obviating the need for a proximate blocking group.
Scheme 5.

A representative example of our previously reported 2,6-diolefination reaction of phenylacetic acids using Ac-Val-OH.49i
We next sought to examine the scope of olefin coupling partners that could be used under our accelerated reaction conditions (Table 9). A variety of acrylates (1a–1c), styrenes (1d and 1e), and vinyl ketones (1g and 1h) were found to be highly reactive, generally offering quantitative yields after 2 h (10 h in the case of 1e and 1h).
Table 9.

| Product | Additive/Ligand | % Conv. |
|---|---|---|

|
--- | 7c |
| BQ | 3c | |
| Ac-Ile-OH | >99 (96) | |

|
--- | 6 |
| BQ | 2 | |
| Ac-Ile-OH | >99 (99) | |

|
--- | 8 |
| BQ | 3 | |
| Ac-Ile-OH | >99 (90) | |

|
--- | 7 |
| BQ | 2 | |
| Ac-Ile-OH | >99 (99) | |

|
--- | 13d |
| BQ | 4d | |
| Ac-Ile-OH | 70 (65)d | |

|
--- | 0d |
| BQ | 0d | |
| Ac-Ile-OH | 82 (62)d | |

|
--- | 0 |
| BQ | 0 | |
| Ac-Ile-OH | >99 (94) | |

|
--- | 0d |
| BQ | 0d | |
| Ac-Ile-OH | >99 (97>d |
5 mol% BQ (when used), 10 mol% Ac-Ile-OH (when used).
The conversion was determined by 1H NMR analysis of the crude reaction mixture. Isolated yield is given in parentheses.
Average of three trials.
10 h.
Consistent with our earlier observation,45,49i linear alkenes were also compatible, fashioning the non-conjugated product 1f. We hypothesize that this product is formed as a consequence of the mechanistic scenario depicted in Scheme 6, wherein coordination of the carboxylate directing group restricts bond rotation, which makes β-hydride elimination away from the aromatic ring more kinetically favorable. Notably, this result represents a formal C–H allylation, and as such, provides access to a novel class of non-conjugated products. Efforts to carry out C–H olefination with vinyl sulfones, sulfonates, phosphonates, and nitriles were unsuccessful. Moreover, internal alkenes, such as trans-methyl crotonate and trans-methyl cinnamate, were found to be unreactive with 1 under these conditions.
Scheme 6.

Mechanistic hypothesis to explain the preferential formation of 1f, rather than the thermodynamically favored conjugated product.
It is worth noting here that the phenylacetic acid substrate scope and olefin scope studies were run using 5 mol% Pd(OAc)2 under 1 atm O2, but using lower catalyst loadings (0.2–1 mol%) and/or air as the oxidant would likely give similar yields after extended reaction times (4–48 h), as demonstrated above.
2.5. Mechanistic Considerations
To elucidate fully the origin of the observed ligand-promoted acceleration, insights from a number of different techniques are needed, including isolation and characterization of the putative intermediates, kinetic determination of the rate law, and computational modeling of possible reaction pathways. In collaboration with other research groups, our laboratory is currently pursuing these investigations. In the meantime, we have performed two key sets of experiments, measuring the intermolecular KIE and observing the relationship between initial rate and the electronic properties of the substrate through competition experiments. The resulting data provide evidence that the overall rate increase stems from acceleration in the C–H cleavage step and that the mechanism of C–H cleavage changes from electrophilic palladation to proton abstraction when amino acid ligands are coordinated to Pd(II).
Historically speaking, the mechanism of C(sp2)–H cleavage by Pd(II) has attracted a great deal of attention.60 During the past few decades, three leading transition state proposals have emerged, with the operative mechanism thought to depend on the individual system in question (Scheme 7). One mechanism is electrophilic palladation via an arenium (Wheland) intermediate, originally proposed by Ryabov and coworkers in 1985 (A).60a In this case, Pd(II) coordinates to the π-system of the arene, and the resulting Wheland species transfers a proton to a bound acetate to generate the cyclopalladated intermediate. In electrophilic palladation, the efficacy of C–H activation is highly dependent on the electronic properties of the arene, with electron-rich substrates giving better reactivity. A second mechanism, oxidative addition, was proposed by Canty and van Koten in 1995, in which the C–H bond oxidatively adds to Pd(II) to generate a short-lived Pd(IV) species that can reductively eliminate HX to generate the Pd(II)-cyclopalladated intermediate (B).60b Lastly a third mechanism is proton abstraction, first put forward by Martinez in 1997 (C).60c,d Subsequent computational work by Davies and Macgregor in 2005 supported a similar mechanism (D).60f In proton abstraction, C–H cleavage proceeds via the concerted transfer of the hydrogen atom to an intramolecular base, without substantial build up of positive charge on the arene. Martinez suggested that this process proceeds via a four-membered transition state,60c but later evidence from Davies and Macgregor pointed to a six-membered transition state.60f Proton abstraction is thought to operate via an agostic interaction, rather than through a Wheland intermediate;60f as such, the rate dependence of the electronic properties of substituents is less pronounced.
Scheme 7.

Mechanistic models for the C–H cleavage transition state with Pd(II).
It is worth mentioning that proton abstraction with Pd(II) is conceptually related to the mechanism for C–H activation in Pd(0)/ArX chemistry (E–G, Scheme 8).61 However, the latter belongs to a different reactivity paradigm, in which the Pd(II) species is attached to a phosphine ligand and an aryl fragment and is thus rendered nucleophilic. Though the mechanism for Pd(II)-mediated C–H cleavage has been studied for a longer period of time, arguably the mechanism at play in Pd(0)/ArX chemistry is better understood due to pioneering studies by Eschavarren and Fagnou.61b–f
Scheme 8.

Conceptually relevant proton abstraction mechanistic proposals for Pd(0)/ArX catalytic systems.
Regarding our studies of Pd(II)-mediated C–H cleavage with phenylacetic acids, qualitatively speaking, the data that we obtained using our original Pd(II)-catalyzed mono-selective C–H olefination reaction45 are consistent with an electrophilic palladation mechanism. The reaction was found to be high yielding when substrates with electron-donating groups (e.g., methoxy and methyl) were used, and to be low yielding or unreactive for substrates with electron-withdawing groups (e.g., CF3 and NO2). In particular, without amino acid ligands, we found substrate 8, which contains an ortho-nitro group, to be totally unreactive after 48 h, with only starting material recovered (and <5% decarboxylation) (Scheme 9). In sharp contrast, in the presence of Ac-Ile-OH gave 70% yield of 8a after only 2 h. Viewed in conjunction with data in Figures 1 and 2, this data suggests two points: (1) the amino acid ligands are not merely enhancing the TON, but are generating a more reactive catalyst, (2) electrophilic palladation no longer seems to be the operative mechanism.
Scheme 9.

Qualitative evidence for a change in the mechanism of C–H cleavage: C–H activation of highly electron-deficient arenes in the presence of Ac-Ile-OH.
To investigate whether a departure from the electrophilic palladation pathway or a drastic increase in the electrophilic property of the Pd(II) catalyst is responsible for the reactivity of 8, we carried out intermolecular competition experiments between electron-rich and electron-poor substrates (Table 10). We selected 1 and 2 for analysis because they are roughly isosteric to one another and do not contain strongly chelating functional groups on the aromatic ring. We began by submitting a one-to-one mixture of 1 and 2 to the reaction conditions in the absence of amino acid ligand and found that 2 reacted preferentially, consistent with our hypothesis for electrophilic palladation. We then repeated these experiments in the presence of two ligands: Boc-Val-OH, which we found to be an active ligand in our early studies,45 and Ac-Ile-OH, which proved to be optimal throughout this investigation. When Boc-Val-OH was used, the relative reaction rate of 1 to 2 increased to the point where the two substrates were reacting almost at the same speed (i.e., k1/k2 changed from 0.22 to 0.58). Strikingly, when we examined Ac-Ile-OH, we found a reversal in relative reactivity, such that electron-poor substrate 1 now reacted almost twice as fast as electron-rich substrate 2 (k1/k2 = 1.87). This same trend held for individually measured rates for the single-component reactions of 1 and 2 (Table 11).
Table 10.
One-pot intermolecular competition experiment data.a

| Entry | Ligand | Time (min) | % Conv. 1a | % Conv. 2a | k1/k2 |
|---|---|---|---|---|---|
| 1 | --- | 120 | 3 | 14 | 0.22 |
| 2 | Boc-Val-OH | 10 | 18 | 30 | 0.58 |
| 3 | Ac-Ile-OH | 10 | 40 | 21 | 1.87 |
The conversion was determined by 1H NMR analysis of the crude reaction mixture. See Supporting Information for experimental details.
Table 11.
Comparison of initial rates for single-component reactions of 1 and 2 under the different conditions shown.a

| Entrya | Ligand | k1 ([M]/min) | k2 ([M]/min) | k1 / k2 |
|---|---|---|---|---|
| 1 | --- | 1.3 × 10−4 | 3.5 × 10−4 | 0.37 |
| 2 | Boc-Val-OH | 4.7 × 10−3 | 5.2 × 10−3 | 0.90 |
| 3 | Ac-Ile-OH | 7.6 × 10−3 | 5.5 × 10−3 | 1.38 |
See Supporting Information for experimental details.
Next, we sought to determine whether the differences in these relative rate profiles could be attributed to mechanistic changes in C–H cleavage and also to test the hypothesis that the ligand-induced rate increases were a result of acceleration in the C–H cleavage step. We prepared deuterium-labeled compound 12, a representative electron-rich substrate, and used it to perform intermolecular KIE experiments (Table 12). In the absence of ligand, we observed a large KIE of 6.1, suggesting that cleavage of the C–H bond is the rate-limiting step. In the presence of Boc-Val-OH, though the absolute reaction rates of both 2 and 12 were found to be substantially greater, the measured KIE was roughly the same (5.5), suggesting that C–H cleavage was still the rate-limiting step. Interestingly, with Ac-Ile-OH, the ligand that gives the fastest overall rate, the KIE had markedly decreased to 1.7, suggesting that the rate of C–H cleavage had increased to the point where it was no longer the slow step in the catalytic cycle. In this case, the moderate KIE (between 1 and 2) is consistent with C–H cleavage taking place before the rate-limiting step.
Table 12.
Intermolecular KIE experiments.a

| Entry | Ligand | kH / kD |
|---|---|---|
| 1 | --- | 6.1 |
| 2 | Boc-Val-OH | 5.5 |
| 3 | Ac-Ile-OH | 1.7 |
See Supporting Information for experimental details.
Taken together, the data from the competition experiments and the KIE experiments paint an interesting picture for the operative mechanisms in these systems. Our tentative hypothesis is that in the absence of amino acid ligands, among the three possible pathways (H–J, Scheme 10), the reaction proceeds through an electrophilic palladation mechanism (H). At first glance, the large KIE of 6.1 (Entry 1, Table 12) would seem to contradict this proposal since electrophilic aromatic substitution reactions (including electrophilic palladation processes) often have small KIE values.62 In these cases, deprotonation is assumed to be fast relative to formation of the arenium species. However, in cases where the rate of deprotonation is slow, larger KIE values have been observed.49c,63 Thus if we follow this logic and invoke an electrophilic palladation mechanism, then with electron-rich substrates, such as 2, palladation to generate the putative Wheland intermediate is fast, and intramolecular deprotonation by internally bound acetate is the rate-limiting step, which is consistent with the large KIE of 6.1 for electron-rich substrates (2 and 12). In contrast, with electron-poor substrates, such as 1, palladation to form the Wheland intermediate is less favorable, and thus becomes rate-limiting, in accordance with the results of the competition experiments.
Scheme 10.
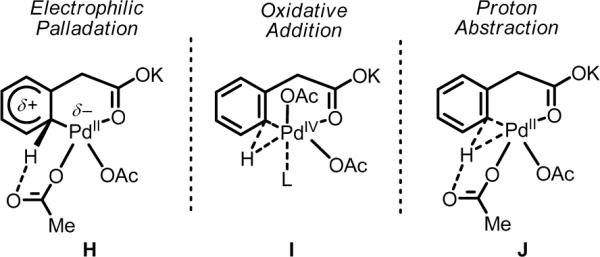
Possible mechanisms for C–H cleavage without ligands: electrophilic palladation (H), oxidative addition (I), and proton abstraction (J).
With the amino acid ligands, several possibilities exist (K–P, Scheme 11), again falling within the three general classifications described above. It is important to recognize that when Ac-Ile-OH is used, the KIE data suggests that C–H cleavage is not rate-limiting; thus the results of the competition experiments (Table 10, Entry 3 and Table 11, Entry 3) need to be interpreted cautiously because the relative rate changes reflect an elementary step other than C–H cleavage. Nevertheless, the high levels of reactivity with both electron-poor and electron-rich substrates would seem to argue against an electrophilic palladation mechanism (K and L). Furthermore, in the case of Boc-Val-OH the KIE data indicates that C–H cleavage is still rate-limiting, yet in competition experiments (Table 10, Entry 2 and Table 11, Entry 2) electron-poor and electron-rich substrates were found to react with similar rates, suggesting that electrophilic palladation (which we hypothesize is operative in the ligand-free conditions) is not operative. Though we cannot rule out the possibility that Boc-Val-OH and Ac-Ile-OH promote different mechanisms, we suspect that they coordinate to Pd(II) in an identical bidentate fashion and thus affect C–H cleavage similarly. Because previous computational studies have not found oxidative addition mechanisms to be energetically favorable (M),60f our current understanding would support one of the proton abstraction mechanisms (N–P). Here, the weak and reversible bidentate coordination of the amino acid ligand likely plays a crucial role in facilitating the initial agostic interaction between Pd(II) and the C–H bond and then in shuttling the hydrogen atom to an internal (O and P) or external (N) base. Across the various ligands that enhanced reactivity in these studies (Tables 1–3), it is possible that multiple C–H cleavage mechanisms are at play simultaneously (or that the operative mechanism is substrate-dependent).
Scheme 11.

Possible mechanisms for C–H cleavage: (K and L) electrophilic palladation, (M) oxidative addition, (N–P) and proton abstraction.
As mentioned above, our selection of Boc-Val-OH and Ac-Ile-OH for these mechanistic studies was motivated by the finding that these ligands were highly reactive in our early work45 and present work, respectively. Valine and isoleucine–derived ligands were found to show similar levels of activity when Boc-protected (Table 1, comparing Entries 3 and 4) and Ac-protected (Table 3, comparing Entries 1 and 5), consistent with the fact that their side chains differ only by a remotely located methyl group. Thus, the origin of the observed differences in catalysis between Boc-Val-OH and Ac-Ile-OH likely originate primarily from the different steric and electron properties of the N-protecting groups.
A detailed analysis of how the amino acid ligands affect the transition state energies of various C–H cleavage processes remains a point to be clarified by computational studies. Given the dramatic effect that small perturbations in the structure of the amino acid side chain and the N-protecting group have on the observed reactivity and on the competition and KIE studies, the precise role of the ligands is likely quite complex. Here, we put forward general mechanistic models to guide future analysis (Scheme 11). An important question that remains to be addressed is what coordination mode the amino acid ligand adopts with Pd(II) in the transition state. In our previous work using mono-N-protected amino acid ligands for enantioselective C–H activation,43–44 our working stereomodel suggests that the ligand is coordinated in a bidentate fashion throughout the C–H cleavage process. However, if we invoke a bidentate coordination mode for the ligand (L–O), Pd(II) will be coordinatively saturated when bound to the carboxylate directing group and the C–H bond (through an agostic interaction) in the pre-transition state. This implies that traditional intramolecular deprotonation by bound acetate (A, C, and D, Scheme 7) is no longer viable since the bound acetate must be displaced in order for the C–H agostic interaction to occur. Thus, in this case, deprotonation is likely occurring either by an external base (N) or by one of the basic groups from the ligand (L, O, and P). On the other hand, the amino acid ligand could adopt a monodentate coordination mode in the transition state (K and P), in which case, the models discussed in Scheme 7 could still be applicable.
An overall proposal for the catalytic cycle is depicted in Scheme 12. Following coordination of the substrate to Pd(II), carboxylate-directed C–H cleavage takes place to generate the reactive cyclopalladated intermediate. Coordination of an olefin followed by 1,2-migratory cleavage effects formation of the new C–C bond. β-Hydride elimination and reductive elimination give the desired product with concomitant formation of a Pd(0) species, which can be reoxidized by molecular oxygen46,47,64 to regenerate Pd(II) and close the catalytic cycle.
Scheme 12.
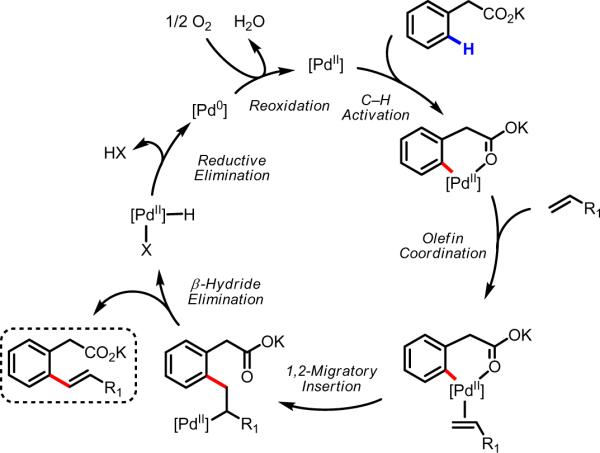
Proposed catalytic cycle.
3. Conclusion
We have developed an improved protocol for aerobic Pd(II)-catalyzed C–H olefination of phenylacetic acid substrates through the discovery of a novel ligand, Ac-Ile-OH, which is capable of accelerating the reaction. This ligand offers dramatically improved substrate scope, reaction rate, and catalyst lifetime. Catalyst loadings as low as 0.2 mol% and reaction times as fast as 20 minutes were demonstrated. Moreover, the reaction could be run using air as the terminal oxidant. We disclosed evidence based on initial rate studies, reactivity trends, competition experiments, and KIEs that suggests that the increased reaction rates stem from acceleration in the C–H cleavage step. Furthermore, this data points to a change in mechanism of C–H cleavage from electrophilic palladation to proton abstraction or oxidative addition when amino acid ligands are added to the reaction. Efforts are currently underway in our laboratory to examine whether amino acid ligands are competent in accelerating Pd(II)-mediated C–H cleavage with other directing groups. Our preliminary results concerning ligand-enabled C–H olefination of phenethyl alcohol substrates are promising in this respect.49g Additionally, if the observed rate enhancement truly stems from acceleration in the C–H cleavage step, as we now presume, other Pd(II)-catalyzed C–H functionalization reactions should, in principle, benefit from amino acid ligands. This hypothesis is also being actively investigated by our group at the present time.
4. Experimental Section
4.1. General Information
Unless otherwise noted, all materials were used as received from commercial sources without further purification. The phenylacetic acid substrates and olefin coupling partners were purchased from Acros, Sigma-Aldrich, TCI and Alfa-Aesar and were used as received. 2-(Trifluoromethyl)phenylacetic acid (1) was purchased from TCI; samples of 1 from other commercial sources were found to give irreproducible results. 1,4-Benzoquinone (BQ) was sublimed prior to used. Freshly distilled methyl vinyl ketone was used in the synthesis of 1g. Commercially available organic acid ligands were purchased from Acros, Sigma Aldrich, and Alfa Aesar. In the optimization studies 5a was used as a ligand; its synthesis is described herein. Commercially available amino acid ligands were purchased from Bachem, EMD, or Novabiochem. L4 was prepared according to a method developed by Burgess.65 L6 was prepared according to a literature procedure.66 All others were prepared following literature precedent.43,67 Palladium acetate and potassium hydrogen carbonate were purchased from Sigma-Aldrich and Fisher, respectively, and were used without further purification. All reactions were run on hot plates with oil baths calibrated to an external thermometer. Prior to beginning an experiment, the hot plate was turned on, and the oil bath was allowed to equilibrate to the desired temperature for 30 minutes. Infrared spectra were recorded on a Perkin Elmer FT-IR Spectrometer. 1H and 13C NMR spectra were recorded on Varian Mercury (300 MHz and 75 MHz, respectively), Varian Inova (400 MHz and 100 MHz, respectively) and Bruker DRX (500 MHz and 125 MHz, respectively) instruments internally referenced to SiMe4 or chloroform signals. The following abbreviations (or combinations thereof) were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and a = apparent. High resolution mass spectra were recorded at the Center for Mass Spectrometry, The Scripps Research Institute.
4.2 General procedure for determining the initial reaction rate for C–H olefination of 1 (or 2) under different conditions
Four different reaction conditions were examined: (1) without Boc-Val-OH, without BQ, (2) without Boc-Val-OH, with BQ, (3) with Boc-Val-OH, without BQ, (4) with Boc-Val-OH, with BQ. To establish the initial rate under each of the conditions, four parallel reactions were set up simultaneously. Four 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) were obtained, each equipped with a magnetic stir bar. Each tube was charged with 1 (or 2) (0.5 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), KHCO3 (100.1 mg, 1.0 mmol), BQ (2.7 mg, 0.025 mmol) (when used), Boc-Val-OH (10.9 mg, 0.05 mmol) (when used), ethyl acrylate (106 μL, 1.0 mmol), and t-AmylOH (2.5 mL). The reaction tubes were capped, then evacuated briefly under high vacuum and charged with O2 (1 atm, balloon) (×3). The reaction mixtures were stirred at room temperature for 5 min, then at 90 °C for the appropriate time. At regular intervals (every 5 minutes, or every 30 minutes), one of the reactions would be removed from the hot plate and cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL) and diethyl ether (10 mL) were then added. A small aliquot of the organic phase was taken, concentrated in vacuo, and analyzed by 1H NMR. The conversion was determined by integration of the benzylic methylene proton signals, which appear as singlets (approximately 3.87 ppm for 1, 4.01 ppm for 1a, 3.67 ppm for 2, and 3.84 ppm for 2a). For each condition, this process was repeated three times. The resulting data was plotted, and linear regression established the initial rate. Representative data for the determination of one initial rate are shown in Figure S1.
4.3 General procedure for mono-N-protected L-valine ligands43,67
A 500 mL round bottom flask equipped with a magnetic stir bar was charged with distilled H2O (100 mL) and NaOH (100 mmol, 4.0 g). The resulting solution was cooled to 0 °C in an ice bath. L-valine (35 mmol, 4.1 g) was added, and the solution was stirred until it was homogeneous. The flask was equipped with an addition funnel. The corresponding carbonyl chloride (45.5 mmol) in 1,4-dioxane (40 mL) was added dropwise. The reaction mixture was then allowed to stir at room temperature overnight. The following morning, the solution was extracted with Et2O (3 × 50 mL), and the organic layers were discarded. The aqueous layer was again cooled to 0 °C in an ice bath, and concentrated HCl was added dropwise until the pH had reached 2 (as observed by pH paper). The aqueous solution was extracted with Et2O (3 × 100 mL). The organic layers were combined, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give the crude product. The pure product was obtained following recrystallization from Et2O/hexanes or column chromatography using 15:1 DCM:MeOH as the solvent system. For a general depiction of this procedure, see Scheme S1.
4.4 Ligand optimization for Pd(II)-catalyzed olefination with 2-(Trifluoromethyl)phenylacetic acid (1)
A 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar was charged with 1 (102.1 mg, 0.5 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), KHCO3 (100.1 mg, 1.0 mmol), ligand (0.05 mmol), ethyl acrylate (106 μL, 1.0 mmol), and t-AmylOH (2.5 mL). The reaction tube was capped, then evacuated briefly under high vacuum and charged with O2 (1 atm, balloon) (×3). The reaction mixture was stirred at room temperature for 5 min, then at 90 °C for the appropriate time. The reaction vessel was removed from the oil bath and immediately cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL) and diethyl ether (10 mL) were added. A small aliquot of the organic phase was taken, concentrated in vacuo, and analyzed by 1H NMR. The conversion was determined by integration of the benzylic methylene proton signals, which appear as singlets (approximately 3.87 ppm for 1 and 4.01 ppm for 1a). When indicated, the reactions were performed in triplicates, and the values shown represent the average result from the three experiments. In our initial efforts, we measured the conversion after 2 h (Table S1), but we observed quantitative conversion for many ligands. We then adjusted our assay and examined the conversion after 20 min (Tables S2 and S3).
4.5 Additional optimization for Pd(II)-catalyzed olefination with 2-(Trifluoromethyl)phenylacetic acid (1)
The effects of ligand loading (Table S4), olefin loading (Table S5), and reaction temperature (Tables S6–S10) were explored using Ac-Ile-OH as the ligand. A 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar was charged with 1 (102.1 mg, 0.5 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), KHCO3 (100.1 mg, 1.0 mmol), Ac-Ile-OH, ethyl acrylate, and t-AmylOH (2.5 mL). The reaction tube was capped, then evacuated briefly under high vacuum and charged with O2 (1 atm, balloon) (×3). The reaction mixture was stirred at room temperature for 5 min, then at the appropriate temperature for the indicated time. The reaction vessel was then cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL) and diethyl ether (10 mL) were added. A small aliquot of the organic phase was taken, concentrated in vacuo, and analyzed by 1H NMR. The conversion was determined by integration of the benzylic methylene proton signals, which appear as singlets (approximately 3.87 ppm for 1 and 4.01 ppm for 1a). When indicated, the reactions were performed in triplicates, and the values shown represent the average result from the three experiments.
4.5 General procedure for Pd(II)-catalyzed ortho-C–H olefination of phenylacetic acids 1–10
A 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar was charged with the phenylacetic acid starting material (1–10) (0.5 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), KHCO3 (100.1 mg, 1.0 mmol), ligand (0.05 mmol), the olefin coupling partner (1.0 mmol), and t-AmylOH (2.5 mL). The reaction tube was capped, then evacuated briefly under high vacuum and charged with O2 (1 atm, balloon) (×3). The reaction mixture was stirred at room temperature for 5 min, then at 90 °C for 2 h (longer, when noted). The reaction vessel was then cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL), and the mixture was extracted with EtOAc (3 × 20 mL). The organic layers were combined, dried over anhydrous Na2SO4, filtered, and concentrate in vacuo. The resulting residue was purified by silica gel flash column chromatography using 3:1 hexanes:EtOAc (with 3% HOAc) as the eluent. For a general depiction of this procedure, see Scheme S2.
4.6 Procedure for intermolecular competition experiments between compounds 1 and 2
A 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar was charged with 1 (51.1 mg, 0.25 mmol), 2 (37.6 mg, 0.25 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), KHCO3 (100.1 mg, 1.0 mmol), ligand (0.05 mmol), ethyl acrylate (106 μL, 1.0 mmol), and t-AmylOH (2.5 mL). The reaction tube was capped, then evacuated briefly under high vacuum and charged with O2 (1 atm, balloon) (×3). The reaction mixture was stirred at room temperature for 5 min, then at 90 °C for the appropriate time. The reaction vessel was then cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL) and diethyl ether (10 mL) were added. A small aliquot of the organic phase was taken, concentrated in vacuo, and analyzed by 1H NMR. The conversion was determined by integration of the benzylic methylene proton signals, which appear as singlets (approximately 3.87 ppm for 1, 4.01 ppm for 1a, 3.67 ppm for 2, and 3.84 ppm for 2a). The results are shown in Table S11.
4.7 Procedure for initial rate studies for single-component reactions of 1 and 2
The procedure for reactions run with and without Boc-Val-OH is described on page S-2. An identical protocol was followed to determine the initial rate in the presence of Ac-Ile-OH (8.7 mg, 0.05 mmol), with reactions stopped at 5 min, 7.5 min, 10 min, and 15 min. The reactions were repeated three times, and determination of the initial rate was performed using linear regression. The overall results are shown in Table S12.
4.8 Procedure for intermolecular kinetic isotope experiments between compounds 2 and 12
To a 20 mL scintillation vial were added, 2 (15.0 mg, 0.1 mmol), 12 (15.7 mg, 0.1 mmol), and CDCl3 (0.5 mL). The solution was stirred until homogenous, and a small aliquot was taken for 1H NMR analysis to ensure that the weighed quantities corresponded to a mixture with 50% ± 2% of each component. The solvent was removed in vacuo, and the mixture of 2 and 12 was transferred in t-AmylOH (1 mL) to a 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar. Pd(OAc)2 (2.2 mg, 0.01 mmol), KHCO3 (40.0 mg, 0.4 mmol), ligand (0.02 mmol), and ethyl acrylate (43 μL, 0.4 mmol) were then added. The reaction tube was capped, then evacuated briefly under high vacuum and charged with O2 (1 atm, balloon) (×3). The reaction mixture was stirred at room temperature for 5 min, then at 90 °C for the appropriate time. The reaction vessel was then cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL) and diethyl ether (10 mL) were added. A small aliquot of the organic phase was taken, concentrated in vacuo, and analyzed by 1H NMR. The conversion of 2a, X2a, was determined by integration of the methyl proton signals, which appear as singlets (approximately 2.32 ppm for 2 and 2.36 ppm for 2a). The total conversion Xtotal was determined by integration of the benzylic methylene group signals, which also appear as singlets (3.67 ppm for 2/12 and 3.84 ppm for 2a/12a). The conversion of 12a, X12a, could then be determined from the following formula:
The experiments were repeated three times without ligand, three times with Boc-Val-OH, and five times with Ac-Ile-OH. The results are shown in Table S13.
Supplementary Material
Acknowledgements
We gratefully acknowledge The Scripps Research Institute (TSRI), the NIH (NIGMS, 1 R01 GM084019-01A1), the NSF (NSF CHE-1011898), Amgen, and Eli Lilly for financial support. We thank the A. P. Sloan Foundation for a fellowship (J.-Q.Y.), and we are thankful for predoctoral fellowships from the China Scholarship Council (D.-H.W.), as well as the NSF, the DOD, TSRI and the Skaggs-Oxford Scholarship program (K.M.E.).
Footnotes
Supporting Information Avialable. Detailed experimental procedures, characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For the initial reports of the Mizoroki–Heck reaction, see: Mizoroki T, Mori K, Ozaki A. Bull. Chem. Soc. Jpn. 1971;44:581.. Heck RF, Nolley JP., Jr. J. Org. Chem. 1972;37:2320..; For reviews, see: Heck RF. Acc. Chem. Res. 1979;12:146.. Heck RF. Org. React. 1982;27:345.. Tsuji J. Palladium Reagents and Catalysts. Wiley; Chichester, UK: 1995. Chapter 4.. Bräse S, de Meijere A. Chapter 5. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; New York: 2004. . Dounay AB, Overman LE. Chem. Rev. 2003;103:2945. doi: 10.1021/cr020039h..
- (2).For the first reports of Pd(0)-catalyzed aryl halide/alkyne coupling, see: Cassar L. J. Organomet. Chem. 1975;93:253.. Dieck AH, Heck RF. J. Organomet. Chem. 1975;93:259..; For an improved protocol in the presence of Cu(I) salts (the Sonogashira reaction), see: Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467..
- (3).For the initial reports of the Ni-catalyzed Kumada–Corriu coupling reaction, see: Corriu RJP, Masse JP. J. Chem. Soc., Chem. Commun. 1972:144a.. Tamao K, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:4374..; For the first Pd(0)-catalyzed reaction, see: Yamamura M, Moritani I, Murahashi S-I. J. Organomet. Chem. 1975;91:C39–C42..
- (4).For initial report of the Negishi coupling reaction, see: Negishi E, King AO, Okukado N. J. Org. Chem. 1977;42:1821..; For a review, see: Negishi E. Acc. Chem. Res. 1982;15:340..
- (5).For initial reports of the Stille coupling reaction, see: Milstein D, Stille JK. J. Am. Chem. Soc. 1978;100:3636.. Milstein D, Stille JK. J. Am. Chem. Soc. 1979;101:4992..
- (6).For initial reports of the Suzuki–Miyaura coupling reaction, see: Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437.. Miyaura N, Suzuki A. J. Chem. Soc., Chem. Commun. 1979:866..; For a review, see: Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457..
- (7).For the initial report of the Hiyama coupling reaction, see: Hatanaka Y, Hiyama T. J. Org. Chem. 1988;53:918..; For fluoride-free coupling of silanols (the Hiyama–Denmark reaction), see: Denmark SE, Sweis RF. J. Am. Chem. Soc. 2001;123:6439. doi: 10.1021/ja016021q..; For a review, see: Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486. doi: 10.1021/ar800037p..
- (8).For early reviews of the Tsuji–Trost reaction, see: Trost BM. Acc. Chem. Res. 1980;13:385.. Tsuji J. Tetrahedron. 1986;42:4361..
- (9).For initial reports of the Buchwald–Hartwig amination reaction, see: Paul F, Patt J, Hartwig JF. J. Am. Chem. Soc. 1994;116:5969.. Guram AS, Buchwald SL. J. Am. Chem. Soc. 1994;116:7901..
- (10).For the initial report of Miyaura borylation, see: Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508..
- (11).For initial reports of Buchwald–Hartwig etherification, see: Palucki M, Wolfe JP, Buchwald SL. J. Am. Chem. Soc. 1996;118:10333.. Mann G, Hartwig JF. J. Am. Chem. Soc. 1996;118:13109.. Aranyos A, Old DW, Kiyomori A, Wolfe JP, Sadighi JP, Buchwald SL. J. Am. Chem. Soc. 1999;121:4369.. Mann G, Incarvito C, Rheingold AL, Hartwig JF. J. Am. Chem. Soc. 1999;121:3224..
- (12).For the first report of Pd(0)-catalyzed aryl halide/fluoride coupling, see: Watson DA, Su M, Teverovskiy G, Zhang Y, Garcia-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239..
- (13).For a comprehensive review of cyclopalladation and the reactivity of palladacycles with different nucleophiles and electrophiles, see: Ryabov AD. Synthesis. 1985:233..
- (14).For reviews of Pd-catalyzed C–H activation reactions, see: Jia C, Kitamura T, Fujiwara Y. Acc. Chem. Res. 2001;34:633. doi: 10.1021/ar000209h.. Satoh T, Miura M. Chem. Lett. 2007;36:200.. Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173. doi: 10.1039/b606984n.. Campeau L-C, Stuart DR, Fagnou K. Aldrichim. Acta. 2007;40:35.. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760.. Li B-J, Yang S-D, Shi Z-J. Synlett. 2008:949.. Kakiuchi F, Kochi T. Synthesis. 2008:3013.. Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058.. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273.. Ackermann L, Vicente R, Kapdi AR. Angew. Chem., Int. Ed. 2009;48:9792. doi: 10.1002/anie.200902996.. Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e..
- (15).For diastereoselective C–H iodination and acetoxylation using Pd(II)/Pd(IV) catalysis from our group, see: Giri R, Chen X, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884.. Giri R, Liang J, Lei J-G, Li J-J, Wang D-H, Chen X, Naggar IC, Guo C, Foxman BM, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:7420. doi: 10.1002/anie.200502767..
- (16).For C–H halogenation using Pd(II)/Pd(IV) catalysis from our group, see: Mei T-S, Giri R, Maugel N, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:5215. doi: 10.1002/anie.200705613..
- (17).For C–H fluorination using Pd(II)/Pd(IV) catalysis from our group, see: Wang X, Mei T-S, Yu J-Q. J. Am. Chem. Soc. 2009;131:7520. doi: 10.1021/ja901352k..
- (18).For C–H amination using Pd(II)/Pd(IV) catalysis from our group, see: Wasa M, Yu J-Q. J. Am. Chem. Soc. 2008;130:14058. doi: 10.1021/ja807129e..
- (19).For an example of C–H alkylation using Pd(II)/Pd(IV) catalysis from our group, see: Zhang Y-H, Shi B-F, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:6097. doi: 10.1002/anie.200902262..
- (20).For early reports of C–H iodination using Pd(II)/(IV) catalysis, see: Fahey DR. J. Organomet. Chem. 1971;27:283.. Andrienko OS, Goncharov VS, Raida VS. Russ. J. Org. Chem. 1996;32:89..
- (21).For early reports of C–H acetoxylation using Pd(II)/(IV) catalysis, see: Henry PM. J. Org. Chem. 1971;36:1886.. Yoneyama T, Crabtree RH. J. Mol. Catal. A: Chem. 1996;108:35..
- (22).For C–H acetoxylation using Pd(II)/(IV) catalysis, see: Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja031543m.. Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542. doi: 10.1021/ja046831c..
- (23).For early reports of C–H alkylation using Pd(II)/Pd(IV) catalysis, see: Tremont SJ, Rahman HU. J. Am. Chem. Soc. 1984;106:5759.. McCallum JS, Gasdaska JR, Liebeskind LS, Tremont SJ. Tetrahedron Lett. 1989;30:4085..
- (24).For domino reactions involving C–H activation/C–C bond formation along a Pd(0)/Pd(II)/Pd(IV) catalytic cycle, see: Catellani M, Frignani F, Rangoni A. Angew. Chem., Int. Ed. 1997;36:119.. Mauleón P, Alonso I, Carretero JC. Angew. Chem., Int. Ed. 2001;40:1291. doi: 10.1002/1521-3773(20010401)40:7<1291::aid-anie1291>3.0.co;2-9..; For recent developments using Pd(0)/Pd(II)/Pd(IV) catalysis, see: Mariampillai B, Alliot J, Li M, Lautens M. J. Am. Chem. Soc. 2007;129:15372. doi: 10.1021/ja075599i.. Gericke KM, Chai DI, Bieler N, Lautens M. Angew. Chem., Int. Ed. 2009;48:1447. doi: 10.1002/anie.200805512..
- (25).For C–H arylation reactions using Pd(II)/(IV) catalysis, see: Xia M, Chen ZC. Synth. Commun. 2000;30:531.. Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f.. Shabashov D, Daugulis O. Org. Lett. 2005;7:3657. doi: 10.1021/ol051255q.. Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f.. Deprez NR, Kalyani D, Krause A, Sanford MS. J. Am. Chem. Soc. 2006;128:4972. doi: 10.1021/ja060809x.. Reddy BVS, Reddy LR, Corey EJ. Org. Lett. 2006;8:3391. doi: 10.1021/ol061389j.. Lazareva A, Daugulis O. Org. Lett. 2006;8:5211. doi: 10.1021/ol061919b.. Shabashov D, Daugulis O. J. Am. Chem. Soc. 2010;132:3965. doi: 10.1021/ja910900p..
- (26).For C–H fluorination using Pd(II)/(IV) catalysis, see: Hull KL, Anani WQ, Sanford MS. J. Am. Chem. Soc. 2006;128:7134. doi: 10.1021/ja061943k..
- (27).For C–H halogenation using Pd(II)/(IV) catalysis, see: Kalyani D, Dick AR, Anani WQ, Sanford MS. Org. Lett. 2006;8:2523. doi: 10.1021/ol060747f..
- (28).For C–H amination using Pd(II)/(IV) catalysis, see: Jordan-Hore JA, Johansson CCC, Gulias M, Beck EM, Gaunt MJ. J. Am. Chem. Soc. 2008;130:16184. doi: 10.1021/ja806543s..
- (29).For C–H sulfonylation using Pd(II)/(IV) catalysis, see: Zhao X, Dimitrijević E, Dong VM. J. Am. Chem. Soc. 2009;131:3466. doi: 10.1021/ja900200g..
- (30).For evidence and discussion of Pd(III) intermediates in C–H activation reactions, see: Powers DC, Ritter T. Nature Chem. 2009;1:302. doi: 10.1038/nchem.246.. Powers DC, Geibel MAL, Klein JEMN, Ritter T. J. Am. Chem. Soc. 2009;131:17050. doi: 10.1021/ja906935c.. Deprez NR, Sanford MS. J. Am. Chem. Soc. 2009;131:11234. doi: 10.1021/ja904116k..; For pioneering inorganic studies of Pd(III) species, see: Cotton FA, Koshevoy IO, Lahuerta P, Murillo CA, Sanaú M, Ubeda MA, Zhao Q. J. Am. Chem. Soc. 2006;128:13674. doi: 10.1021/ja0656595..; For a related study concerning Pt(III) intermediates, see: Whitfield SR, Sanford MS. Organometallic. 2008;27:1683..; For a recent inorganic study, see: Khusnutdinova JR, Rath NP, Mirica LM. J. Am. Chem. Soc. 2010;132:7303. doi: 10.1021/ja103001g..
- (31).For an example of C–H amination where Pd(III) is implicated in the catalytic cycle, see: Mei T-S, Wang X, Yu J-Q. J. Am. Chem. Soc. 2009;131:10806. doi: 10.1021/ja904709b..
- (32).For a reviews of organopalladium(IV) chemistry, see: Yu J-Q, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k.. Muñiz K. Angew. Chem., Int. Ed. 2009;121:9576.. Xu L-M, Li B-J, Yang Z, Shi Z-J. Chem. Soc. Rev. 2010;39:712. doi: 10.1039/b809912j.. Sehnal P, Taylor RJK, Fairlamb IJS. Chem. Rev. 2010;110:824. doi: 10.1021/cr9003242..
- (33).For the initial reports of Pd(II)-mediated arene C–H olefination, see: Moritani I, Fujiwara Y. Tetrahedron Lett. 1967;8:1119.. Fujiwara Y, Moritani I, Matsuda M, Teranishi S. Tetrahedron Lett. 1968;9:633..
- (34).For selected examples of subsequent work to improve the catalytic turnover and expand the substrate scope of Pd(II)-mediated C–H olefination, see: Fujiwara Y, Moritani I, Danno S, Asano R, Teranishi S. J. Am. Chem. Soc. 1969;91:7166. doi: 10.1021/ja01053a047.. Asano R, Moritani I, Fujiwara Y, Teranishi S. J. Chem. Soc. D: Chem. Commun. 1970:1293. doi: 10.1021/ja01053a047.. Fujiwara Y, Asano R, Moritani I, Teranishi S. J. Org. Chem. 1976;41:1681. doi: 10.1021/ja01053a047.. Tsuji J, Nagashima H. Tetrahedron. 1984;40:2699.. Jia C, Lu W, Kitamura T, Fujiwara Y. Org. Lett. 1999;1:2097. doi: 10.1021/ol006156l.. Jia C, Piao D, Oyamada J, Lu W, Kitamura T, Fujiwara Y. Science. 2000;287:1992. doi: 10.1126/science.287.5460.1992.. Yokota T, Tani M, Sakaguchi S, Ishii Y. J. Am. Chem. Soc. 2003;125:1476. doi: 10.1021/ja028903a..
- (35).For an example of Pd(II)-catalyzed C–H olefination using haloolefins, see: Zaitsev VG, Daugulis O. J. Am. Chem. Soc. 2005;127:4156. doi: 10.1021/ja050366h..
- (36).For C–H amination using Pd(II)/Pd(0) catalysis, see: Tsang WCP, Zheng N, Buchwald SL. J. Am. Chem. Soc. 2005;127:14560. doi: 10.1021/ja055353i.. Yamamoto M, Matsubara S. Chem. Lett. 2007;36:172..
- (37).For the first reports of Pd(II)/Pd(0) C–H activation/C–C cross coupling, see: Chen X, Li J-J, Hao X-S, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:78. doi: 10.1021/ja0570943.. Chen X, Goodhue CE, Yu J-Q. J. Am. Chem. Soc. 2006;128:12634. doi: 10.1021/ja0646747..
- (38).For subsequent reports from our laboratory, see: Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614.. Wang D-H, Wasa M, Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:7190. doi: 10.1021/ja801355s.. Wang D-H, Mei T-S, Yu J-Q. J. Am. Chem. Soc. 2008;130:17676. doi: 10.1021/ja806681z..
- (39).For examples of Pd(II)/Pd(0) C–H activation/C–C cross coupling from other groups, see: Yang S, Li B, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:6066. doi: 10.1021/ja070767s.. Yang S-D, Sun C-L, Fang Z, Li B-J, Li Y-Z, Shi Z-J. Angew. Chem., Int. Ed. 2008;47:1473. doi: 10.1002/anie.200704619.. Nishikata T, Abela AR, Huang S, Lipshutz BH. J. Am. Chem. Soc. 2010;132:4978. doi: 10.1021/ja910973a..
- (40).For an example of Pd(II)-catalyzed C–H alkynylation using haloalkynes, see: Tobisu M, Ano Y, Chatani N. Org. Lett. 2009;11:3250. doi: 10.1021/ol901049r..; For previous work on Pd(0)-catalyzed C–H alkynylation using haloalkynes, see: Seregin IV, Ryabova V, Gevorgyan V. J. Am. Chem. Soc. 2007;129:7742. doi: 10.1021/ja072718l.. Gu Y, Wang X-M. Tetrahedron Lett. 2009;50:763..; For Pd(II)-catalyzed intermolecular annulation reactions with alkynes via C–H activation, see: Yamashita M, Hirano K, Satoh T, Miura M. Org. Lett. 2009;11:2337. doi: 10.1021/ol900736s.. Shi Z, Zhang C, Li S, Pan D, Ding S, Cui Y, Jiao N. Angew. Chem., Int. Ed. 2009;48:4572. doi: 10.1002/anie.200901484..
- (41).For reviews detailing the importance of ligand design in Pd(0) chemistry, see: Littke AF, Fu GC. Angew. Chem., Int. Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.. Hartwig JF. Acc. Chem. Res. 2008;41:1534. doi: 10.1021/ar800098p.. Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461. doi: 10.1021/ar800036s..
- (42).For a reviews of Pd(0)-catalyzed C–C bond–forming reactions in total synthesis, see: Chemler SR, Trauner D, Danishefsky SJ. Angew. Chem., Int. Ed. 2001;40:4544. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n.. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem., Int. Ed. 2005;44:4442. doi: 10.1002/anie.200500368..
- (43).For the first example of Pd(II)-catalyzed enantioselective C–H activation/C–C bond formation controlled by mono-N-protected amino acid ligands, see: Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:4882. doi: 10.1002/anie.200801030..
- (44).For the lone example of Pd(II)-catalyzed enantioselective C–H activation/olefination controlled by mono-N-protected amino acid ligands, see: Shi B-F, Zhang Y-H, Lam JK, Wang D-H, Yu J-Q. J. Am. Chem. Soc. 2010;132:460. doi: 10.1021/ja909571z..
- (45).Wang D-H, Engle KM, Shi B-F, Yu J-Q. Science. 2010;327:315. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).For the first example of aerobic Pd(II)-catalyzed C–H olefination, see: Shue RS. J. Chem. Soc. D: Chem. Commun. 1971:1510..
- (47).For exceptionally efficient catalysis (TON > 750) in aerobic Pd(II)-catalyzed C–H olefination using arene substrates as solvent, see: Dams M, De Vos DE, Celen S, Jacobs PA. Angew. Chem., Int. Ed. 2003;42:3512. doi: 10.1002/anie.200351524..
- (48).For examples of non-directed Pd(II)-catalyzed C–H olefination with nitrogen-containing heterocycles, see: Fujiwara Y, Maruyama O, Yoshidomi M, Taniguchi H. J. Org. Chem. 1981;46:851.. Itahara T, Ikeda M, Sakakibara T. J. Chem. Soc., Perkin Trans. 1983;1:1361.. Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2003;125:9578. doi: 10.1021/ja035054y.. Beccalli EM, Broggini G. Tetrahedron Lett. 2003;44:1919.. Ma S, Yu S. Tetrahedron Lett. 2004;45:8419.. Liu C, Widenhoefer RA. J. Am. Chem. Soc. 2004;126:10250. doi: 10.1021/ja046810i.. Grimster NP, Gauntlett C, Godfrey CRA, Gaunt MJ. Angew. Chem., Int. Ed. 2005;44:3125. doi: 10.1002/anie.200500468..
- (49).For examples of Pd(II)-catalyzed C–H olefination using substrates containing directing groups, see: Miura M, Tsuda T, Satoh T, Nomura M. Chem. Lett. 1997;26:1103.. Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J. Org. Chem. 1998;63:5211.. Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586. doi: 10.1021/ja0176907.. Cai G, Fu Y, Li Y, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:7666. doi: 10.1021/ja070588a.. Houlden CE, Bailey CD, Ford JG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. J. Am. Chem. Soc. 2008;130:10066. doi: 10.1021/ja803397y.. Li J-J, Mei T-S, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:6452. doi: 10.1002/anie.200802187.. Lu Y, Wang D-H, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:5916. doi: 10.1021/ja101909t. Nishikata T, Lipshutz BH. Org. Lett. 2010;12:1972. doi: 10.1021/ol100331h.. Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2010 ASAP.
- (50).(a) Capito E, Brown JM, Ricci A. Chem. Commun. 2005:1854. doi: 10.1039/b417035k. [DOI] [PubMed] [Google Scholar]; (b) Maehara A, Tsurugi H, Satoh T, Miura M. Org. Lett. 2008;10:1159. doi: 10.1021/ol8000602. [DOI] [PubMed] [Google Scholar]; (c) García-Rubia A, Arrayás RG, Carretero JC. Angew. Chem., Int. Ed. 2009;48:6511. doi: 10.1002/anie.200902802. [DOI] [PubMed] [Google Scholar]
- (51).Cho SH, Hwang SJ, Chang S. J. Am. Chem. Soc. 2008;130:9254. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]
- (52).For an example of ligand-promoted Pd(II)-catalyzed C–H olefination with electron-deficient arenes, see: Zhang Y-H, Shi B-F, Yu J-Q. J. Am. Chem. Soc. 2009;131:5072. doi: 10.1021/ja900327e..
- (53).For the first example of Pd(II)-catalyzed C(sp3)–H olefination, see: Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:3680. doi: 10.1021/ja1010866..
- (54).For examples of Pd(II)-mediated C–H olefination in total synthesis, see: Trost BM, Godleski SA, Genêt JP. J. Am. Chem. Soc. 1978;100:3930.. Cushing TD, Sanz-Cervera JF, Williams RM. J. Am. Chem. Soc. 1993;115:9323.. Baran PS, Corey EJ. J. Am. Chem. Soc. 2002;124:7904. doi: 10.1021/ja026663t.. Garg NK, Caspi DD, Stoltz BM. J. Am. Chem. Soc. 2004;126:9552. doi: 10.1021/ja046695b.. Beck EM, Hatley R, Gaunt MJ. Angew. Chem., Int. Ed. 2008;47:3004. doi: 10.1002/anie.200705005.. Bowie AL, Jr., Trauner D. J. Org. Chem. 2009;74:1581. doi: 10.1021/jo801791j..
- (55).For examples of Pd(II)-catalyzed C–H activation/enantioselective olefination controlled by external ligands, see: Mikami K, Hatano M, Terada M. Chem. Lett. 1999;28:55.. Schiffner JA, Machotta AB, Oestreich M. Synlett. 2008:2271.. Schiffner JA, Wöste TH, Oestreich M. Eur. J. Org. Chem. 2010:174..
- (56).For examples of arene/olefin coupling reactions using metals other than palladium, see: Lewis LN, Smith JF. J. Am. Chem. Soc. 1986;108:2728.. Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Nature. 1993;366:529.. Matsubara T, Koga N, Musaev DG, Morokuma K. J. Am. Chem. Soc. 1998;120:12692.. Lenges CP, Brookhart M. J. Am. Chem. Soc. 1999;121:6616.. Matsumoto T, Taube DJ, Periana RA, Taube H, Yoshida H. J. Am. Chem. Soc. 2000;122:7414.. Jun C-H, Hong J-B, Kim Y-H, Chung K-H. Angew. Chem., Int. Ed. 2000;39:3440. doi: 10.1002/1521-3773(20001002)39:19<3440::aid-anie3440>3.0.co;2-1.. Weissman H, Song X, Milstein D. J. Am. Chem. Soc. 2001;123:337. doi: 10.1021/ja003361n.. Thalji RK, Ellman JA, Bergman RG. J. Am. Chem. Soc. 2004;126:7192. doi: 10.1021/ja0394986.. Luedtke AT, Goldberg KI. Angew. Chem., Int. Ed. 2008;47:7694. doi: 10.1002/anie.200800524.. Tsuchikama K, Kasagawa M, Hashimoto Y-K, Endo K, Shibata T. J. Organomet. Chem. 2008;693:3939..
- (57).Berrisford DJ, Bolm C, Sharpless KB. Angew. Chem., Int. Ed. 1995;34:1059. [Google Scholar]
- (58).(a) Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. Angew. Chem., Int. Ed. 1995;34:259. [Google Scholar]
- (59).(a) Anastas PT, Williamson TC. Green Chemistry. American Chemical Society; Washington, DC: 1996. Chapter 1. [Google Scholar]; (b) Clark JH. Green Chem. 1999;1:1. [Google Scholar]
- (60).For studies concerning the mechanistic aspects C–H cleavage by Pd(II), see: Ryabov AD, Sakodinskaya IK, Yatsimirsky AK. J. Chem. Soc. Dalton Trans. 1985:2629.. Canty AJ, van Koten G. Acc. Chem. Res. 1995;28:406.. Gomez M, Granell J, Martinez M. Organometallics. 1997;16:2539.. Gómez M, Granell J, Martinez M. J. Chem. Soc., Dalton Trans. 1998:37.. Biswas B, Sugimoto M, Sakaki S. Organometallics. 2000;19:3895.. Davies DL, Donald SMA, Macgregor SA. J. Am. Chem. Soc. 2005;127:13754. doi: 10.1021/ja052047w.. Tunge JA, Foresee LN. Organometallics. 2005;24:6440..
- (61).For conceptually related studies concerning C–H activation with Pd(0)-PR3/Ar-X catalytic systems, see: Mota AJ, Dedieu A, Bour C, Suffert J. J. Am. Chem. Soc. 2005;127:7171. doi: 10.1021/ja050453+.. García-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2006;128:1066. doi: 10.1021/ja056165v.. Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l.. Pinto A, Neuville L, Retailleau P, Zhu J. Org. Lett. 2006;8:4927. doi: 10.1021/ol062022h.. García-Cuadrado D, de Mendoza P, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2007;129:6880. doi: 10.1021/ja071034a.. Gorelsky SI, Lapointe D, Fagnou K. J. Am. Chem. Soc. 2008;130:10848. doi: 10.1021/ja802533u..
- (62).Zollinger H. Adv. Phys. Org. Chem. 1964;2:162. [Google Scholar]
- (63).For early studies where large KIE values (k1/k2 ≥ 5) were observed in reactions thought to proceed through electrophilic Pd(II)-mediated C–H cleavage, see Davidson JM, Triggs C. J. Chem. Soc. A. 1968:1324.. Shue RS. J. Am. Chem. Soc. 1971;93:7116..; For a related arene mercuration reaction, see: Kresge AJ, Brennan JF. J. Org. Chem. 1967;32:752..; For discussion of KIE values in Pd(II)-mediated C–H cleavage reactions, see Lane BS, Brown MA, Sames D. J. Am. Chem. Soc. 2005;127:8050. doi: 10.1021/ja043273t.. Campeau L-C, Parisien M, Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581. doi: 10.1021/ja055819x..
- (64).For reviews of Pd(II) oxidation chemistry, see: Stahl SS. Angew. Chem., Int. Ed. 2004;43:3400. doi: 10.1002/anie.200300630.. Sigman MS, Schultz MJ. Org. Biomol. Chem. 2004;2:2551. doi: 10.1039/B409127M.. Stoltz BM. Lett. 2004;33:362..
- (65).Moye-Sherman A, Jin S, Ham I, Lim D, Scholtz J, Burgess K. J. Am. Chem. Soc. 1998;120:9435. [Google Scholar]
- (66).Pradeille N, Zerbe O, Möhle K, Linden A, Heimgartner H. Chem. Biodivers. 2005;2:1127. doi: 10.1002/cbdv.200590084. [DOI] [PubMed] [Google Scholar]
- (67).Buckley TF, Rapport H. J. Am. Chem. Soc. 1981;103:6157. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.