

Abstract

A sequence consisting of Lewis acid-catalyzed Diels–Alder reaction on a 2-halocyclohexenone, followed by reductive alkylation provides a route to trans-fused octalinones bearing angular methyl groups with functionality corresponding to that which would have been possible from trans-directed Diels–Alder reaction.
The concept of prioritized strategic bond disconnection (SBD), so well articulated by the Corey school, is clearly the most powerful resource yet devised in guiding retrosynthetic analysis (RSA)1 en route to complex targets. Recently, we have suggested an approach to RSA which is complementary to the paradigm of strategic bond disconnection. We refer to this mode of retrosynthesis as pattern recognition analysis (PRA).2 Here, the thought exercise focuses on identifying domains within the target. Particularly valuable is the identification of patterns which are accessible by doable chemistry. Indeed, the discernment of patterns around which a synthesis might be organized may well provoke the discovery of new reactions or reaction sequences to enable creation of the molecular architecture inherent in the patterns.
One of the pillars of PRA is the notion of building cis-fused ring systems via the Diels–Alder (DA) reaction.3 In PRA, the DA cycloaddition is more than a specific reaction type. More broadly, it really implies an overall logic, encompassing a variety of post-DA forays. The DA reaction has been a central implement in the field of chemical synthesis, both in the construction of relatively small molecules as well as for rather complex targets. The scope of PRA in synthetic planning would be much enhanced if the overall logic of the DA reaction could be marshalled to produce trans-fused structures. In principle, the ideal solution to this challenge would be to, somehow, change the stereochemical course of the DA reaction such that the cycloaddition event is antarafacial4 (Figure 1, 2). While speculations about such a fascinating possibility continue, 5 we have been unable to generate any ideas of value along these lines.
Figure 1.

Figure 2.

The next best approach would be to conduct suprafacial DA reactions, with the important caveat that one of the junction substituents may be replaced by another group with overall inversion of configuration (3→5). In that case, a trans junction would become available from the initial DA-derived cis junction. In intermolecular DA reactions, the junction substituents of the product arise from the dienophile. Hence, this type of exercise would require the use of appropriate dienophiles, carrying substituents amenable to inversion of configuration in the context of a hindered ring junction.6
We recently reported instances wherein this line of conjecture could be reduced to practice, though in simplified form.7 The key discovery was that in a cis-decalinoid-like structure (3b), free radical–mediated reduction of a bridgehead C-nitro function can be achieved with inversion of configuration (cf. 3b→5b). While this feasibility demonstration was certainly encouraging, all of the functionality had come from the diene component. As dienophiles, we had exploited nitrocycloalkenes. For wider impact it would be helpful if the trans-Diels–Alder plan could be implemented with valuable functionality from the dienophile, which persists in the final trans product.
At the planning stage, it had been hoped that the radical generated upon reductive cleavage (3→4) could be trapped with appropriate carbon-centered coupling partners so as to produce junction carbon–carbon bonds in the ultimate transfused system (5a). Unfortunately, a variety of projected methods (Keck allylations,8 Giese conjugate additions and trapping with captodative agents9) failed, presumably for reasons of high steric hindrance. Hence, we could only create a trans-fused product bearing a hydrogen in place of the cis-disposed nitro group. Another problem to be overcome was that nitrocycloalkenes are rather sluggish dienophiles. Unfortunately, we have been unable to prepare a compound such as 2-nitrocyclohex-2-enone, which could well have been a much more reactive dienophile. Hence, we were unable to “deliver” a keto group to the final trans-decalinoid system.
The starting point for the chemistry described here was the finding that Lewis acid catalysis (cf. MeAlCl2) allows for rather efficient Diels–Alder of 2-halo-cyclohex-2-enones and cyclopent-2-enones (Figure 2, 6+7→8).10 The DA chemistry works with a range of dienes. In this way, there are generated halogen functions at the junction of cis-fused decalinoids (or hydrindanoids). We first turned to the possibility of radical mediated debromination (8→9). Parenthetically, it was of mechanistic interest to assess whether the striking results we had observed above could be extended to the bromine leaving group α- to a ketone. The results, shown in Table 1, are, indeed, quite encouraging.11 We note that the ratios of trans- to cis- isomer in these experiments are even higher than the corresponding ratios encountered in the reduction of the angular nitro functions with the same radical forming reagents. The structures were assigned by preparation of reference samples of cis-fused octalinones by Lewis acid catalyzed Diels–Alder reactions of cyclohex-2-enone itself with the appropriate dienes.11a,12 In each case, following debromination as described above, the minor product corresponded to the reference cis sample. Explication of the reasons for the higher levels of trans-selectivity observed here will require further study. In the context of the emergence of the trans-Diels–Alder “logic,” valuable functionality (a keto group) had been accommodated in the decalinoid product. It is well to note that a number of these transfused products, though very simple structures, were previously unknown in 2010 literature searches.
Table 1.
 | ||||
|---|---|---|---|---|
| entry | α-bromoketone | product | yield (%) | trans:cis |
| 1 |
 10 |
 11 |
95 | 25:1 |
| 2 |
 12 |
 13 |
97 | 16:1 |
| 3 |
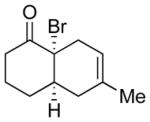 14 |
 15 |
87 | 18:1 |
| 4 |
 16 |
 17 |
99 | 18:1 |
| 5 |
 18 |
 19 |
95 | >30:1d |
Reaction conditions: To a mixture of α-bromoketone (1 equiv) and AIBN (0.3 equiv) in degassed benzene (0.1 M) was added tri-n-butyltin hydride (2 equiv) and the mixture was heated under reflux for 2 h.
Isolated yield.
Determined by 1H NMR analysis. The relative stereochemistry of the major isomer was confirmed by comparison with the corresponding cis-diastereomers, prepared through the CH3AlCl2-catalyzed Diels–Alder reaction between cyclohex-2-enone and the appropriately substituted 1,3-butadiene.
The corresponding cis isomer was not detected within the limits of 1H and 13C NMR.
Having accomplished the goal of synthesizing C1 keto functionalized trans-fused decalinoids via initiating Diels–Alder reaction, we addressed the matter of inserting a carbon-carbon bond at the junction position α- to the ketone in the ultimate trans product. As earlier efforts to accomplish this goal via radical-mediated trapping experiments of the type described above had been unsuccessful (vide supra), our next attempt in this regard was that of reductive alkylation.
We began by studying zinc–induced debromination of the α-bromoketone, hoping to generate a zinc enolate, which might be trapped by suitable carbon–based electrophiles.14 However, alkylation of a relatively reactive zinc enolate15 with methyl iodide could not be realized. We next explored the possibility of a more reactive lithium enolate as the reagent. Debromination was conducted with lithium naphthalide.16
It will be recognized that here, we were attempting what was known in classical natural products chemistry as angular alkylation. To generate the junction enolate, at the time of the historic Johnson studies, it was necessary to insulate the α′ position from enolization (cf. 20).11b,17 Subsequently, House and Trost studied angular alkylation on the simple enolate 21, derived from the non-specifically generated enol acetate. In the decalinoid series, products were either primarily cis-fused or modestly trans-selective.18 However, unlike the cases cited above, in our systems, the α′ position would uniquely contain both a free α-methylene group and a double bond parallel to the junction in an octalin framework. We first investigated the direct methylation of two α-bromoketone substrates. With methyl iodide as the electrophile, reductive alkylation of substrate 10 did, indeed occur, but the trans:cis ratio was, at best, 3:1. In the hydrindanoid series (cf. 27), the cis product was predominant (cis:trans = 4:1).11a These results were hardly in keeping with the goal of a stereoselective synthesis of trans-fused bicyclic systems.
We next explored the use of iodomethyl phenyl sulfide as the alkylating agent. This electrophile had been introduced by Trost in a totally different context.19 Happily, its use resulted in dramatic improvements in selectivity. As shown in Table 2, promising stereoselectivity was accomplished even in the case of the hydrindenone (entry 6). In each case, the phenylthiomethyl group can be converted to the methyl functionality through a high yielding Raney nickel reduction.
Table 2.
Methylation of α-Bromoketones.
 | ||||
|---|---|---|---|---|
| entry | α-bromoketone | product | Step A | Step B |
| yield trans:cis | yield trans:cis | |||
| 1a |
 10 |
 22 |
87% 12:1 dr |
80% 12:1 dr |
| 2a |
 12 |
 23 |
57% 7:1 dr |
78% 7:1 dr |
| 3b |
 14 |
 24 |
88% 10:1 dr |
90% 10:1 dr |
| 4a |
 16 |
 25 |
52% 12:1 dr |
90% 12:1 dr |
| 5b |
 18 |
 26 |
89% >30:1 drc |
95% >30:1 drc |
| 6a |
 27 |
 28 |
42% 3:1 drd |
70% >30:1 drc |
Solvent: DME.
Solvent: THF.
The cis isomer was not detected by NMR.
The cis isomer was isolated in 15% yield.
At this stage it is difficult to know whether the superior results achieved with iodomethyl phenyl sulfide are primarily due to its greater steric demands, or suggest a subtle electronic effect in the nature of the alkylation event. Preliminary experiments were done in this regard. Thus, the relative rates of alkylation of iodomethyl phenyl sulfide and methyl iodide were actually quite comparable in competitive experiments.20
We established the stereostructures of our products by preparing the authentic cis-fused compounds by Lewis acid-catalyzed Diels–Alder cycloaddition of the appropriate diene with the corresponding α-methylated cyclenones. In this way we could rigorously assign the structures of the minor products to the cis-isomer.11c Accordingly, the major products are established as trans. Again, it is instructive to note that these simple trans-fused bicyclic structures, carrying an angular methyl group, were previously unknown. In fact, on reflection, it would not have been a straightforward matter to synthesize such compounds by methodology previous to that which is described here. This type of trans-Diels–Alder paradigm increases the range of synthetically accessible structures with consequences for library construction, as well as for the synthesis of intermediate structures en route to complex targets. A view of the direction in which this type of chemistry can lead was gained by studying the product of trans-piperylene with α-bromocyclohexenone. The major course of this reaction, though not by an impressive ratio (3:1), is that of endo addition.10 Nonetheless, it produced adequate amounts of exo compound for separate study in an alkylation experiment. This led to a highly stereoselective formation of the trans-fused thiophenyl compound (30)11c and, by Raney nickel, the reduction to the angular methyl compound (31, Figure 4).21
Figure 4.

In addition to serving as precursors for achieving angular methylation, the phenylthiomethyl groups which appear in the post-Diels–Alder/alkylation sequence can be exploited in a variety of other ways. Thus, dihydroxylation of compound 23 with osmium tetroxide occurs with remarkably poor stereoselection. By contrast, dihydroxylation of 32 was accomplished with very high margins of stereoselectivity. Subsequent acetylation, diastereoselective reduction of the carbonyl group, and desulfurization gave rise to the previously encountered compound 34 (Figure 5).
Figure 5.

A particularly fascinating application of the angular thio-containing functionality arose from the prior oxidation to the corresponding sulfoxide. For instance, a diastereomeric mixture of sulfoxides was prepared from the corresponding phenythiomethyl intermediate. One of them has been obtained in crystalline form and its stereochemistry is known (35). Anticipating a Pummerer rearrangement, this compound was treated with trifluoroacetic anhydride. In fact, this reaction gave rise to 37, whose structure was surmised from spectral and mechanistic considerations, and verified by a crystallographic determination.11c Presently, we cannot assert the mechanism of this reaction in detail. Qualitatively, it seems that an electrophilically activated sulfoxide could well undergo heterolysis with capture of the thionium group by, ultimately, some equivalent of hydration and cyclization to afford the observed α-epoxide. The scope and mechanism of this very surprising, but good-yielding reaction are currently matters of great importance in our laboratory.
In summary, the research described above generates what we call trans-Diels–Alder equivalents endowed with exploitable functionality. Application of these newly acquired capabilities, as well as expansion of the trans-Diels–Alder logic are ongoing matters in our laboratory.
Supplementary Material
Figure 3.

Acknowledgments
This paper is dedicated to Professor Jahyo Kang (Sogang University) in celebration of his 60th birthday. We thank Rebecca Wilson for editorial advice; Aaron Sattler and Wesley Sattler (Parkin group, Columbia) for X-ray experiments (NSF, CHE-0619638). Support was provided by the NIH (HL25848 to SJD).
Footnotes
Supporting Information Available Experimental procedures, copies of spectral data, and characterization data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Corey EJ. Pure Appl Chem. 1967:19–37. [Google Scholar]; (b) Corey EJ, Wipke WT. Science. 1969;166:178–192. doi: 10.1126/science.166.3902.178. [DOI] [PubMed] [Google Scholar]; (c) Corey EJ. Angew Chem, Int Ed. 1991;30:455–612. [Google Scholar]; (d) Corey EJ, Chelg X. The Logic of Chemical Synthesis. Wiley-VCH; New York: 1995. [Google Scholar]
- 2.Wilson RM, Danishefsky SJ. J Org Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- 3.(a) Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chem, Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; (b) Danishefsky S. Acc Chem Res. 1981;14:400–406. [Google Scholar]
- 4.Woodward RB, Hoffmann R. Angew Chem Int Ed. 1969;8:781–932. [Google Scholar]
- 5.For earlier attempts from other laboratories to accomplish this objective, see references in the Supporting Information.
- 6.Following completion of these studies, we noted an extremely elegant demonstration of this idea via the use of a hindered aluminum enolate, as described by Yamamoto and co-workers, which produced a strong trans:cis ratio even with a 6:5 junction, with methyl or ethyl alkylating agents. See: Shibatomi K, Futatsugi K, Kobayashi F, Iwasa S, Yamamoto H. J Am Chem Soc. 2010;132:5625–5627. doi: 10.1021/ja1018628.
- 7.(a) Kim WH, Lee JH, Danishefsky SJ. J Am Chem Soc. 2009;131:12576–12578. doi: 10.1021/ja9058926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim WH, Lee JH, Aussedat B, Danishefsky SJ. Tetrahedron. 2010;66:6391–6398. doi: 10.1016/j.tet.2010.04.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Keck GE, Yates JB. J Am Chem Soc. 1982;104:5829–5831. [Google Scholar]; (b) Keck GE, Enholm EJ, Yates JB, Wiley MR. Tetrahedron. 1985;41:4079–4094. [Google Scholar]
- 9.Viehe HG, Janousek Z, Merényi R, Stella L. Acc Chem Res. 1985;18:148–154. and pertinent references cited therein. [Google Scholar]
- 10.Lee JH, Kim WH, Danishefsky SJ. Tetrahedron Lett. 2010;51:4653–4654. doi: 10.1016/j.tetlet.2010.06.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.See Supporting Information for (a) experimental details and characterization; (b) further references; (c) X-ray confirmation data.
- 12.Preparation of reference cis-fused octalinones: Lee JH, Kim WH, Danishefsky SJ. Tetrahedron Lett. 2009;50:5482–5484. doi: 10.1016/j.tetlet.2009.07.068.
- 13.The same reaction, conducted on bromohydrindenone 27, gave primarily cis fusion (see Supporting Information).
- 14.Spencer TA, Britton RW, Watt DS. J Am Chem Soc. 1967;89:5727–5729. [Google Scholar]
- 15.(a) Knochel P, Yeh MCP, Berk SC, Talbert J. J Org Chem. 1998;53:2390–2392. [Google Scholar]; (b) Zhu L, Wehmeyer RM, Rieke RD. J Org Chem. 1991;56:1445–1453. [Google Scholar]
- 16.Ebert GW, Rieke RD. J Org Chem. 1988;53:4482–4488. [Google Scholar]
- 17.The best of these was the furfurylidene group: Johnson WS, Allen DS., Jr J Am Chem Soc. 1957;79:1261–1262.
- 18.House HO, Trost BM. J Org Chem. 1965;30:2502–2512. [Google Scholar]
- 19.(a) Trost BM, Kunz RA. J Org Chem. 1974;39:2648–2650. [Google Scholar]; (b) Trost BM, King SA. J Am Chem Soc. 1990;112:408–422. [Google Scholar]
- 20.Under otherwise identical conditions, quenching lithium enolate derived from 10 in DME with a 1:1 mixture of MeI and PhSCH2I (5.0 equiv total) gave a 1:1.5 mixture of 22 and the corresponding phenylthiomethylated adduct in 51% isolated yield.
- 21.Interestingly, in the endo isomer, angular methylation delivers cis-fused product. Effects of neighboring stereocenters on angular alkylation are being studied in detail.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.